Enhanced Reactivity of [Hydroxy(tosyloxy)iodo]benzene in Fluoroalcohol Media. Efficient Direct Synthesis of Thienyl(aryl)iodonium Salts

Abstract
:1. Introduction
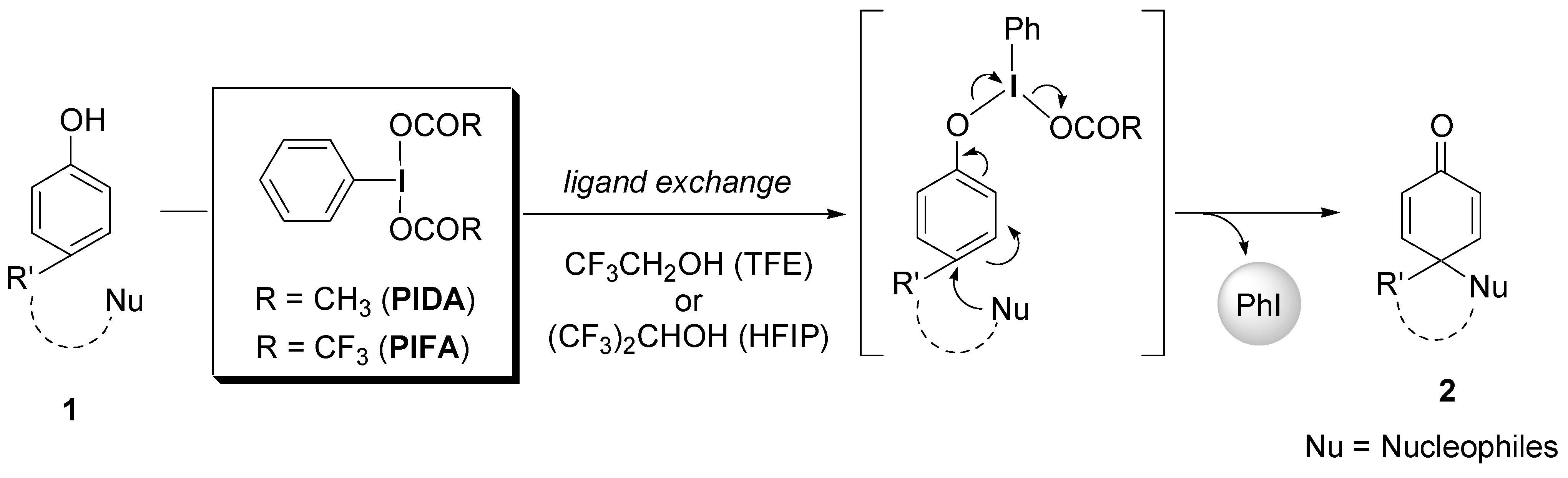
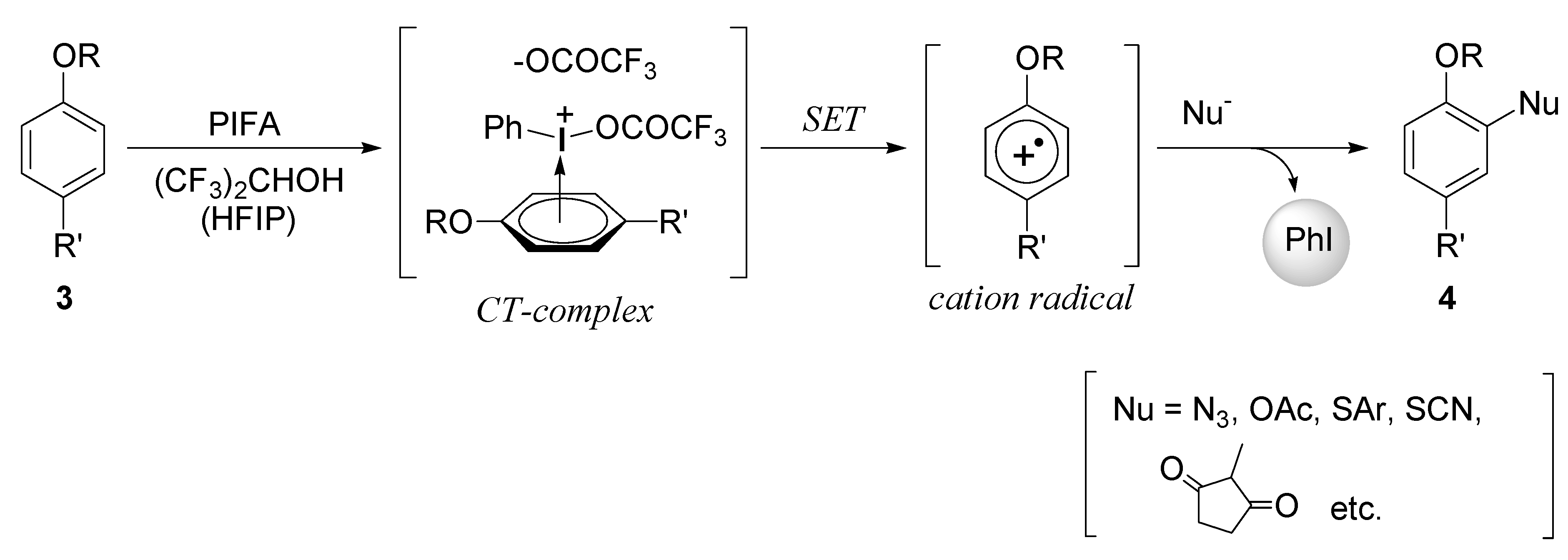
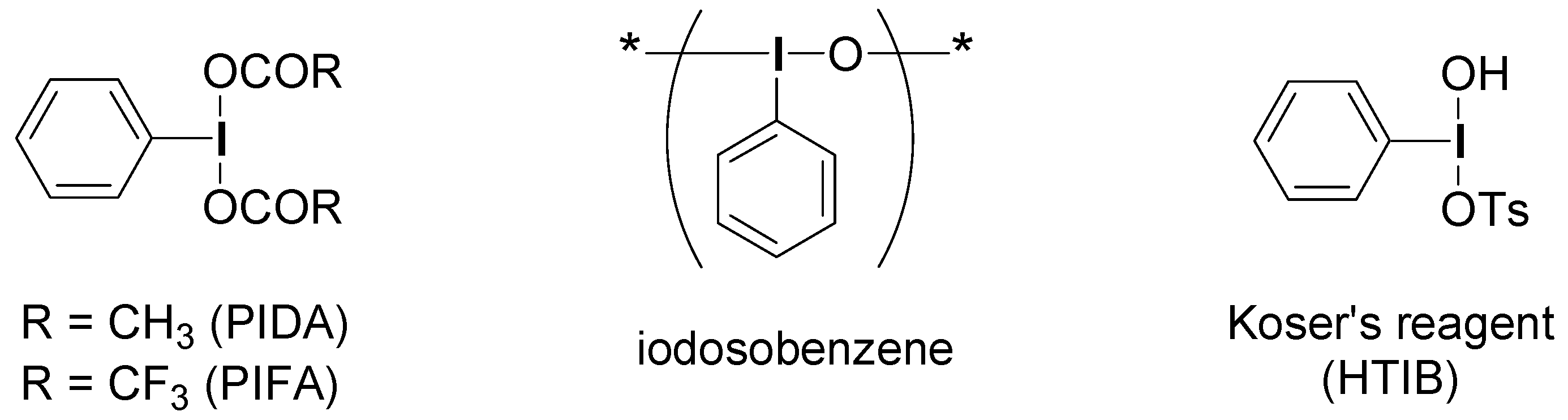
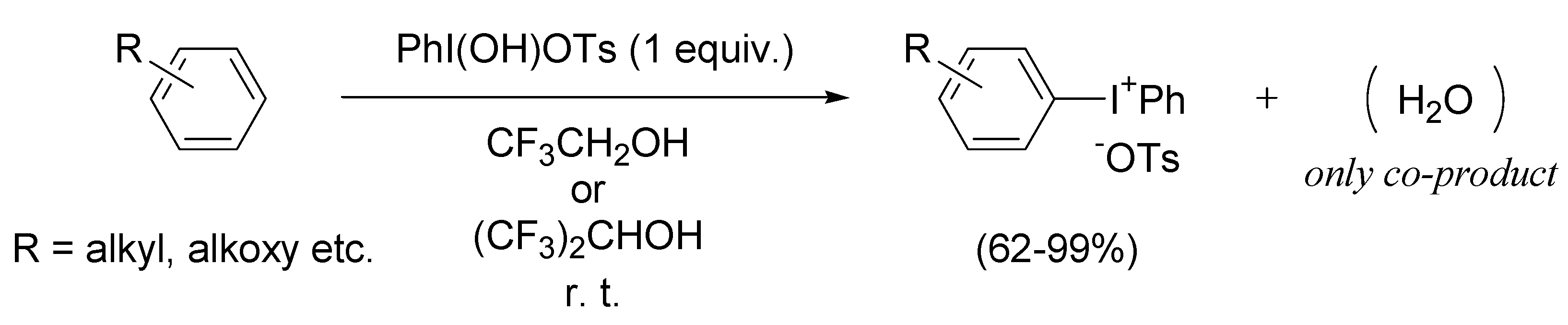
2. Results and Discussion
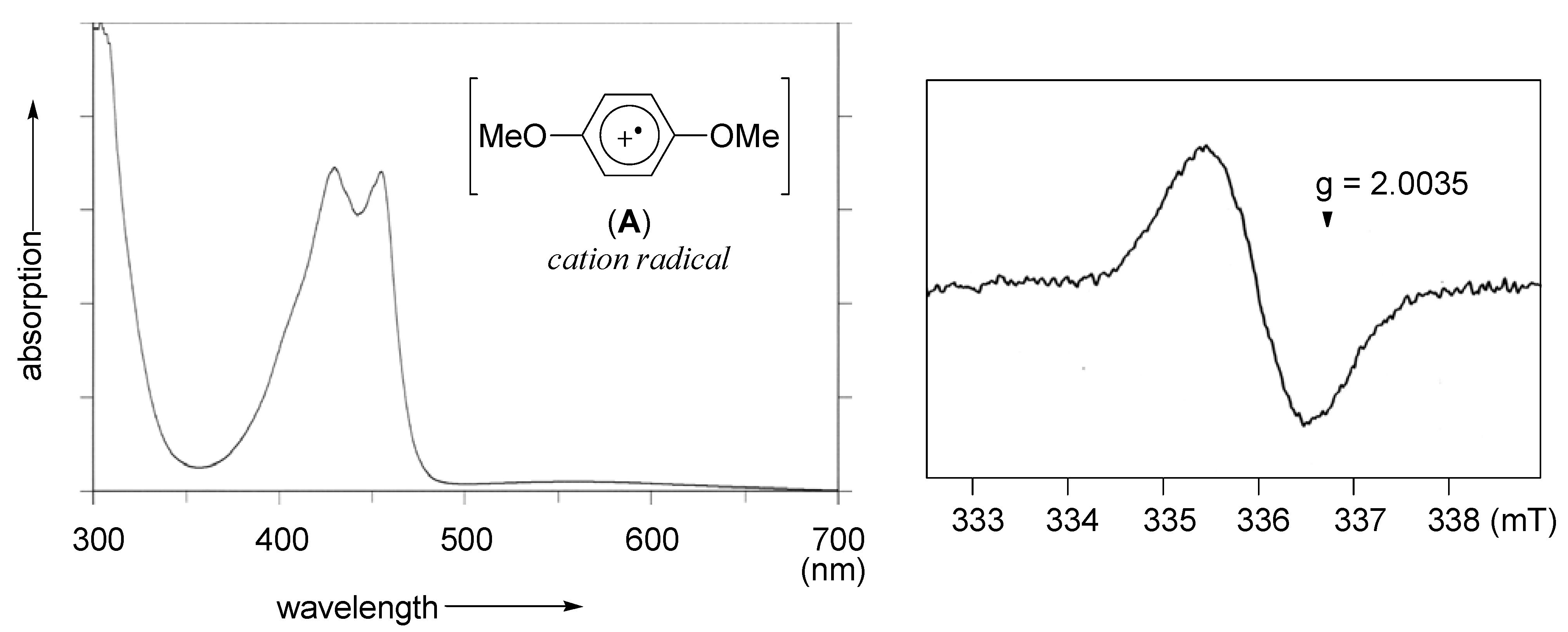
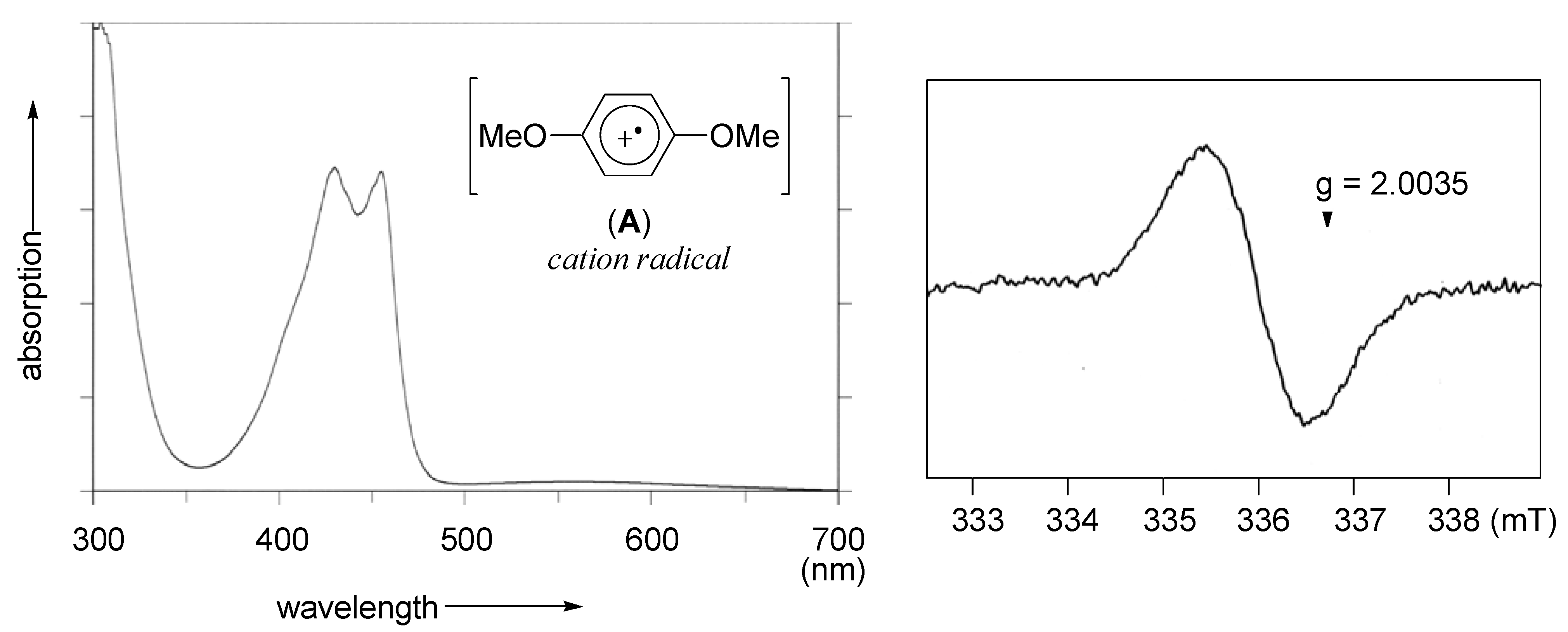
2.1. Generation of aromatic cation radicals of phenyl ethers by HTIB
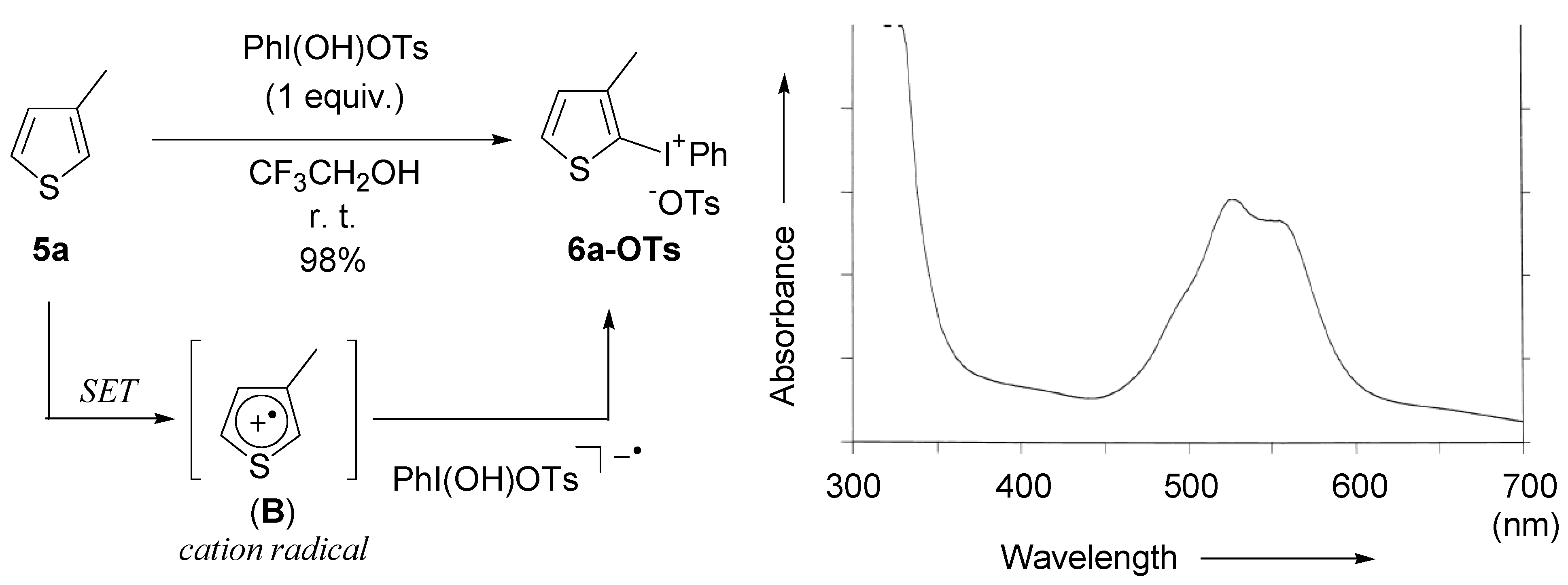
2.2. Application to the synthesis of thienyliodonium(III) salts

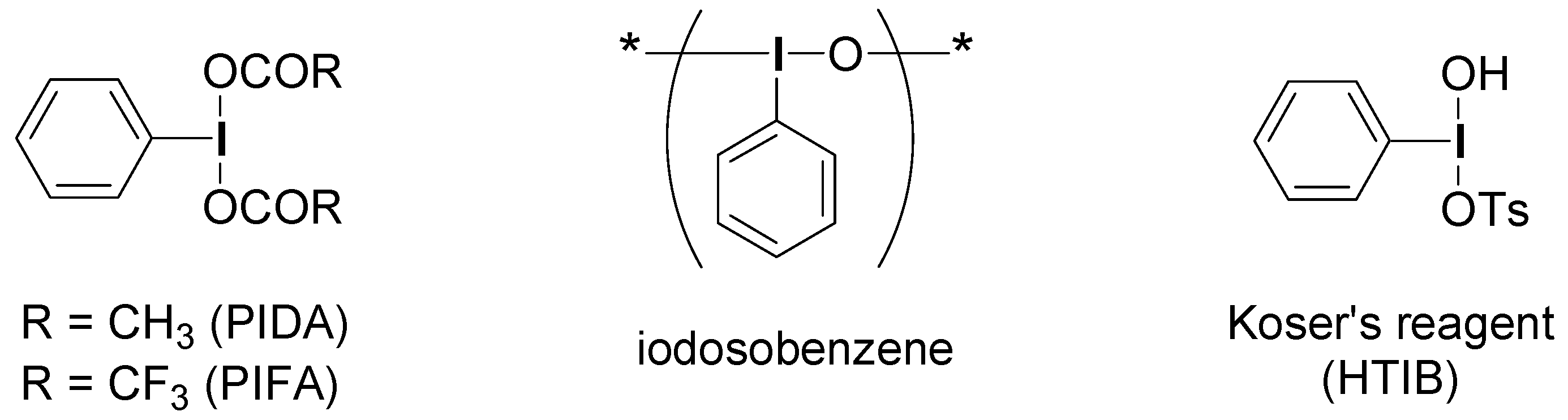{kind=link}
{kind=link}
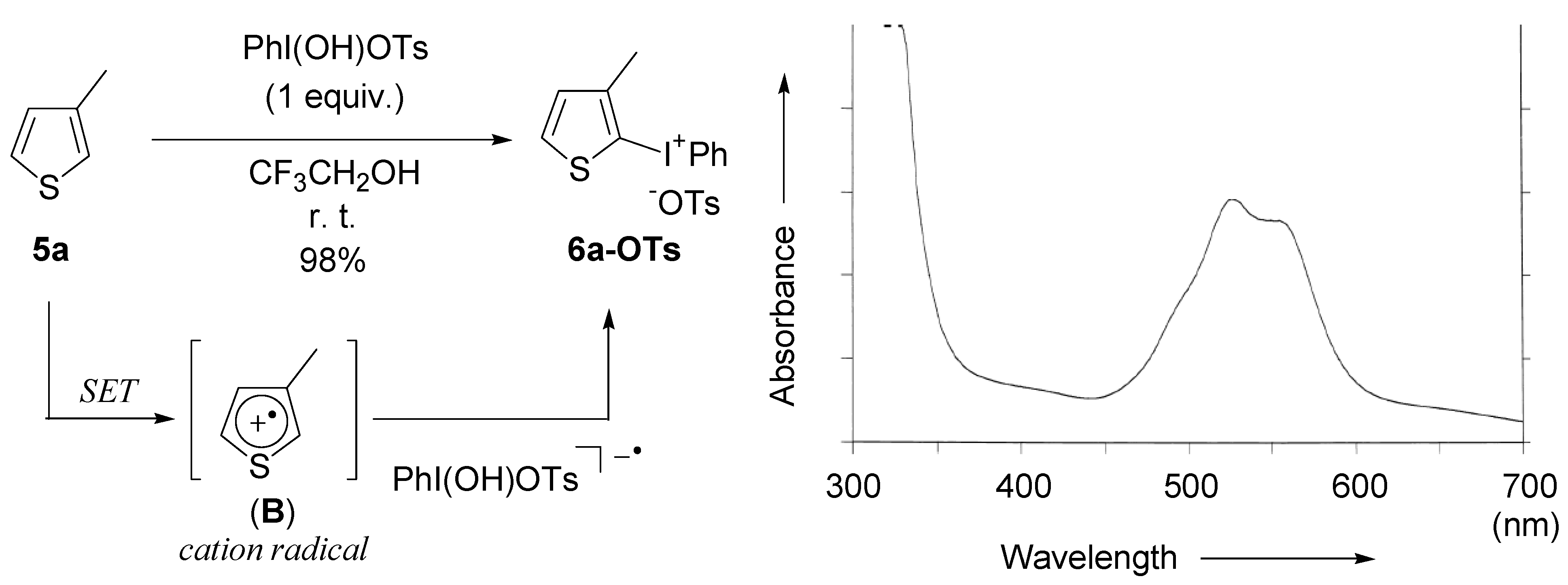{kind=link}
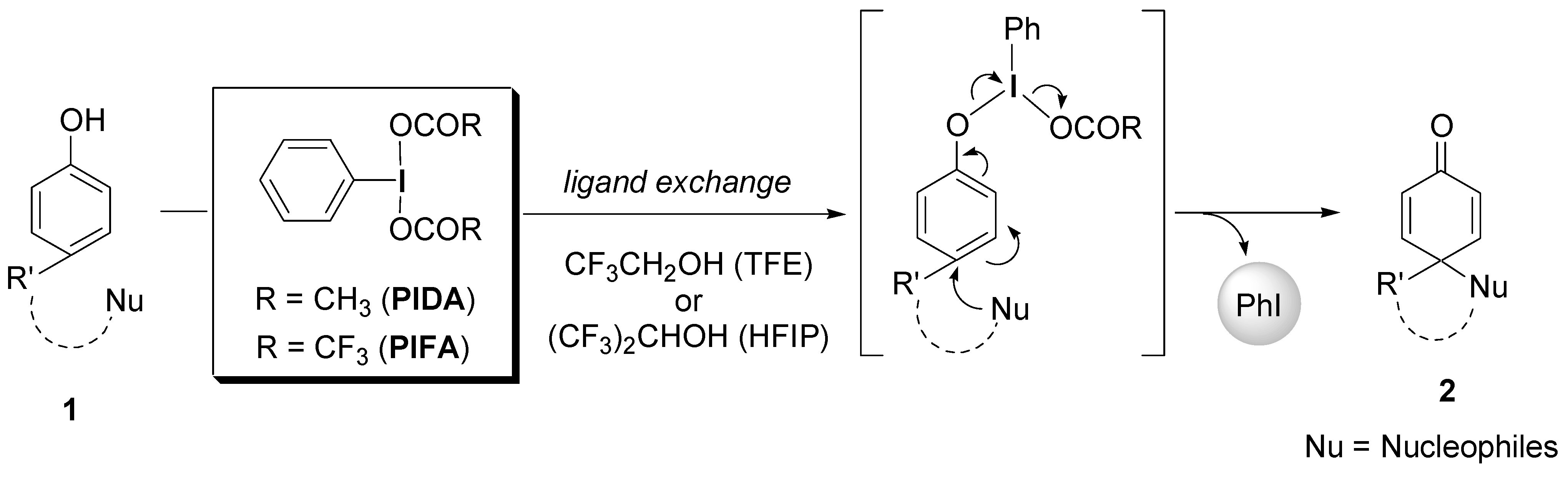{kind=link}
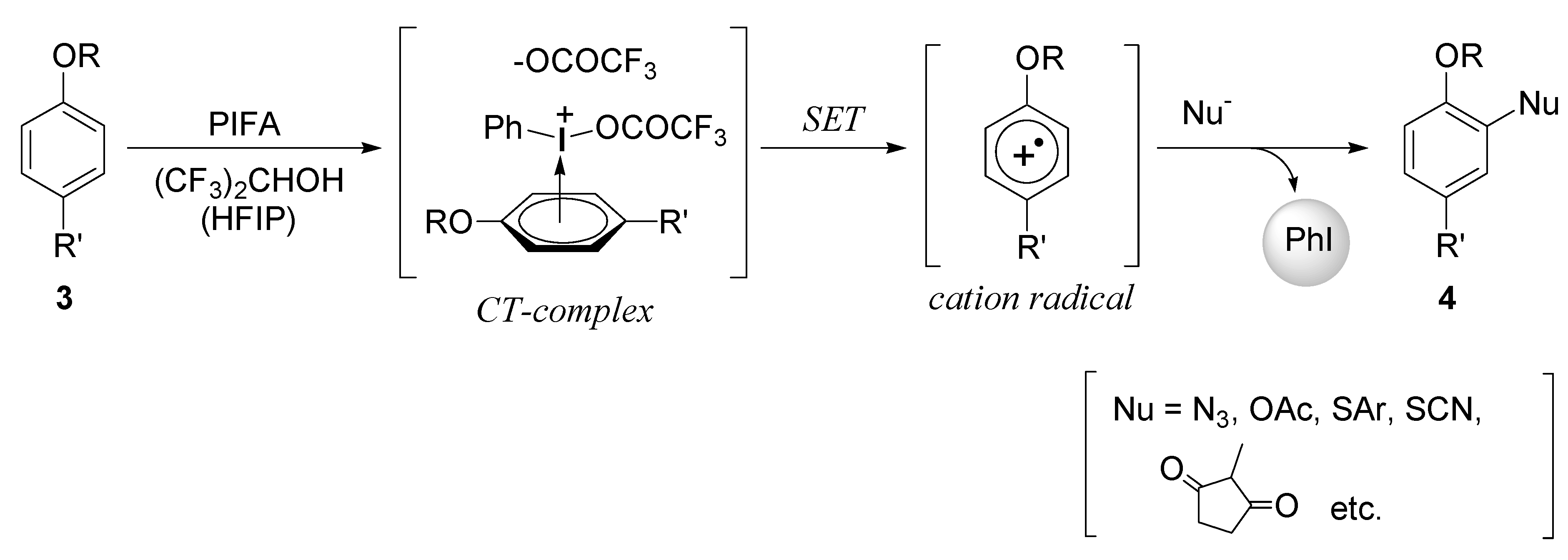{kind=link}
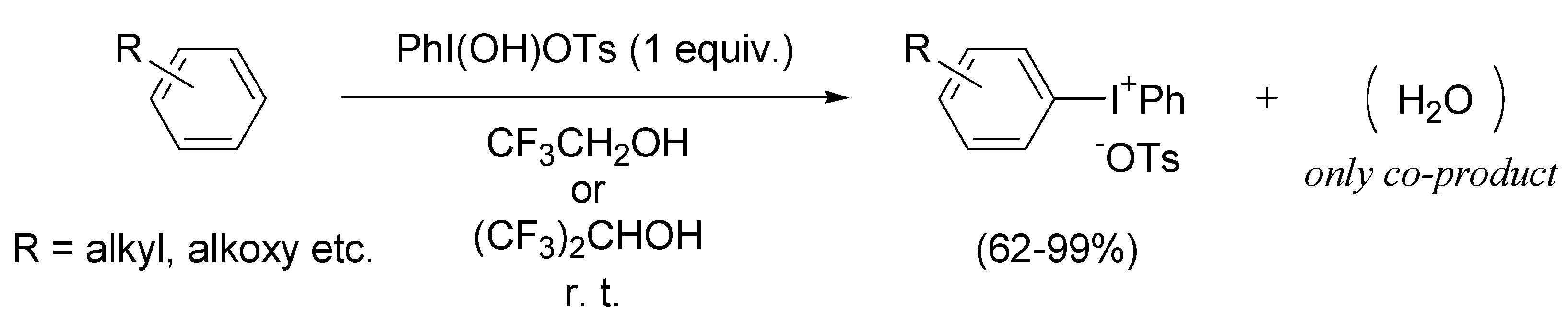{kind=link}
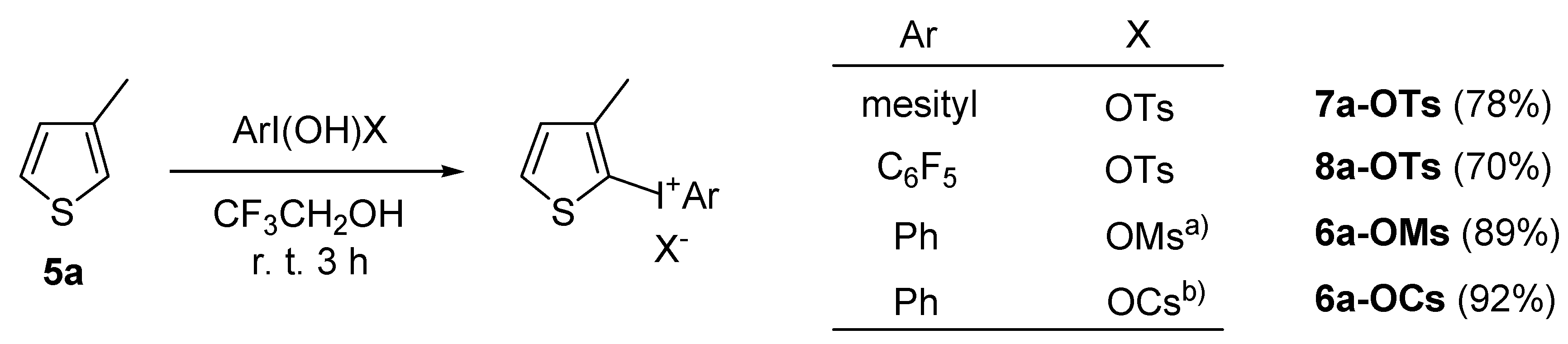{kind=link}
| Entry | Thiophene | Thienyliodonium salt | Yield (%)b |
|---|---|---|---|
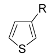 | 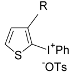 | ||
| 1 | R = Me ( 5a) | 6a-OTs | 98 |
| 2 | R = Hexyl ( 5b) | 6b-OTs | 84 |
| 3 | R = i-Buyl (5c) | 6c-OTs | 93 |
| 4 | R = c-Hex (5d) | 6d-OTs | 74 |
| 5 | R = OMe ( 5e) | 6e-OTs | 89 |
| 6 | R = Br ( 5f) | 6f-OTs | 95 |
| 7 | R = Ph ( 5g) | 6g-OTs | 88 |
| 8 | R = 4-MeO2CC6H4 ( 5h) | 6h-OTs | 98 |
| 9 | 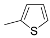 | 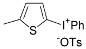 | |
| ( 5i) | (6i-OTs) | 93 | |
| 10 |  | 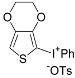 | 94 |
| ( 5j) | (6j-OTs) | ||
| 11 |  | 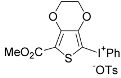 | 88 |
| ( 5k) | (6k-OTs) | ||
| 12 | 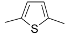 | 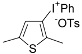 | 62 |
| ( 5l) | (6l-OTs) | ||
| 13 | 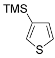 | 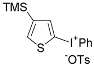 | 71 |
| ( 5m) | (6m-OTs) | ||
| 14 |  | 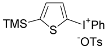 | 91c |
| ( 5n) | (6n-OTs) |
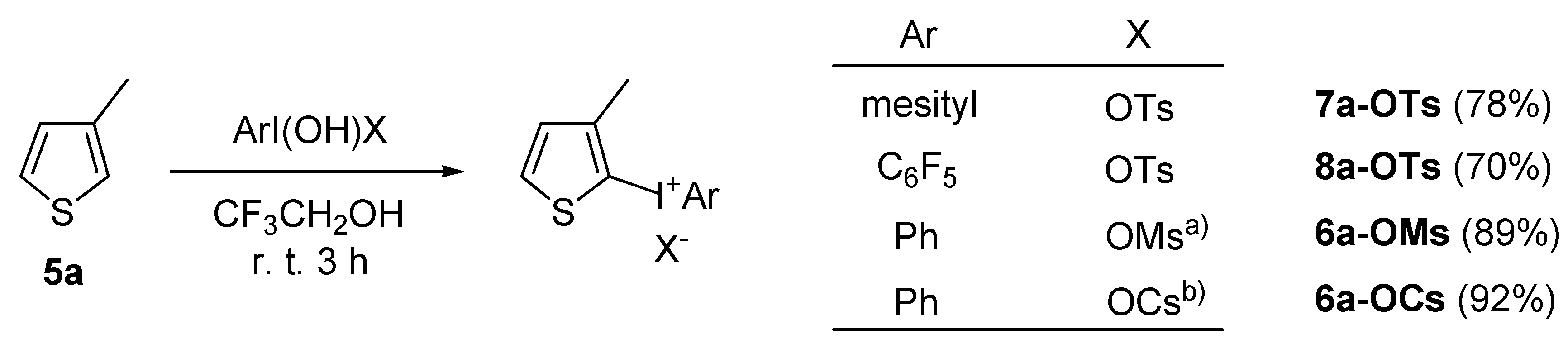
3. Conclusions
4. Experimental
4.1. Measurement of UV-VIS absorption spectrum
4.2. Measurement of electron spin resonance spectrum
4.3. General procedure for the preparation of diaryliodonium salts
Acknowledgments
- Sample Availability: Samples of the products are available from the authors.
References
- Varvoglis, A. Hypervalent Iodine in Organic Synthesis; Academic Press: San Diego, CA, USA, 1997. [Google Scholar]
- Ochiai, M. In Chemistry of Hypervalent Compounds; Akiba, K., Ed.; Wiley-VCH: New York, NY, USA, 1999; Chapter 12. [Google Scholar]
- Zhdankin, V.V.; Stang, P.J. Recent developments in the chemistry of polyvalent iodine compounds. Chem. Rev. 2002, 102, 2523–2584. [Google Scholar] [CrossRef]
- Wirth, T. Hypervalent Iodine Chemistry; Springer-Verlag: Berlin, Germany, 2003. [Google Scholar]
- Tohma, H.; Kita, Y. Hypervalent iodine reagents for the oxidation of alcohols and their application to complex molecule synthesis. Adv. Synth. Catal. 2004, 346, 111–124. [Google Scholar] [CrossRef]
- Moriarty, R.M. Organohypervalent iodine: Development, applications, and future directions. J. Org. Chem. 2005, 70, 2893–2903. [Google Scholar] [CrossRef]
- Wirth, T. Hypervalent iodine chemistry in synthesis: Scope and new directions. Angew. Chem. Int. Ed. 2005, 44, 3656–3665. [Google Scholar] [CrossRef]
- Zhdankin, V.V.; Stang, P.J. Chemistry of polyvalent iodine. Chem. Rev. 2008, 108, 5299–5358. [Google Scholar] [CrossRef]
- Dohi, T.; Kita, Y. Hypervalent iodine reagents as a new entrance to organocatalysts. Chem. Commun. 2009, 2073–2085. [Google Scholar] [CrossRef]
- Tamura, Y.; Yakura, T.; Haruta, J.; Kita, Y. Hypervalent iodine oxidation of p-alkoxyphenols and related compounds: A general route to p-benzoquinone monoacetals and spiro lactones. J. Org. Chem. 1987, 52, 3927–3930. [Google Scholar] [CrossRef]
- Kita, Y.; Tohma, H.; Kikuchi, K.; Inagaki, M.; Yakura, T. Hypervalent iodine oxidation of N-acyltyramines: synthesis of quinol ethers, spirohexadienones, and hexahydroindol-6-ones. J. Org. Chem. 1991, 56, 435–438. [Google Scholar] [CrossRef]
- Tamura, Y.; Yakura, T.; Tohma, H.; Kikuchi, K.; Kita, Y. Hypervalent iodine oxidation of p-alkoxy- and related phenols: A facile and efficient synthesis of p-quinones. Synthesis 1989, 126–127. [Google Scholar]
- Kita, Y.; Yakura, T.; Tohma, H.; Kikuchi, K.; Tamura, Y. A synthetic approach to discorhabdin alkaloids: Hypervalent iodine oxidation of p-substituted phenol derivatives to azacarbocyclic spirodienones. Tetrahedron Lett. 1989, 30, 1119–1120. [Google Scholar] [CrossRef]
- Kita, Y.; Tohma, H.; Inagaki, M.; Hatanaka, K.; Kikuchi, K.; Yakura, T. ypervalent iodine oxidation of O-silylated phenol derivatives to azacarbocyclic spirodienones; synthetic approach to the anticancer marine alkaloid, discorhabdin C. Tetrahedron Lett. 1991, 32, 2035–2038. [Google Scholar]
- Kita, Y.; Takada, T.; Ibaraki, M.; Gyoten, M.; Mihara, S.; Fujita, S.; Tohma, H. An intramolecular cyclization of phenol derivatives bearing aminoquinones using a hypervalent iodine reagent. J. Org. Chem. 1996, 61, 223–227. [Google Scholar] [CrossRef]
- Kita, Y.; Arisawa, M.; Gyoten, M.; Nakajima, M.; Hamada, R.; Tohma, H.; Takada, T. Oxidative intramolecular phenolic coupling reaction induced by a hypervalentiodine(III)reagent:Leading to galanthamine-type Amaryllidaceae alkaloids. J. Org. Chem. 1998, 63, 6625–6633. [Google Scholar] [CrossRef]
- Kita, Y.; Tohma, H.; Inagaki, M.; Hatanaka, K.; Yakura, T. A novel oxidative azidation of aromatic compounds with hypervalent iodine reagent, phenyliodine(III) bis(trifluoroacetate) (PIFA) and trimethylsilyl azide. Tetrahedron Lett. 1991, 32, 4321–4324. [Google Scholar]
- Kita, Y.; Tohma, H.; Hatanaka, K.; Takada, T.; Fujita, S.; Mitoh, S.; Sakurai, H.; Oka, S. Hypervalent iodine-induced nucleophilic substitution of para-substituted phenol ethers. Generation of cation radicals as reactive intermediates. J. Am. Chem. Soc. 1994, 116, 3684–3691. [Google Scholar] [CrossRef]
- Kita, Y.; Takada, T.; Mihara, S.; Whelan, B. A.; Tohma, H. Novel and direct nucleophilic sulfenylation and thiocyanation of phenol ethers using a hypervalent iodine(III) reagent. J. Org. Chem. 1995, 60, 7144–7148. [Google Scholar] [CrossRef]
- Arisawa, M.; Ramesh, N. G.; Nakajima, M.; Tohma, H.; Kita, Y. Hypervalent iodine(III)-induced intramolecular cyclization of α-(aryl)alkyl-β-dicarbonyl compounds: A convenient synthesis of benzannulated and spirobenzannulated compounds. J. Org. Chem. 2001, 66, 59–65. [Google Scholar] [CrossRef]
- Tohma, H.; Iwata, M.; Maegawa, T.; Kiyono, Y.; Maruyama, A.; Kita, Y. A novel and direct synthesis of alkylated 2,2'-bithiophene derivatives using a combination of hypervalent iodine(III) reagent and BF3·Et2O. Org. Biomol. Chem. 2003, 1, 1647–1649. [Google Scholar] [CrossRef]
- Dohi, T.; Morimoto, K.; Kiyono, Y.; Maruyama, A.; Tohma, H.; Kita, Y. The synthesis of head-to-tail (H-T) dimers of 3-substituted thiophenes by the hypervalent iodine(III)-induced oxidative biaryl coupling reaction. Chem. Commun. 2005, 2930–2932. [Google Scholar]
- Dohi, T.; Morimoto, K.; Kiyono, Y.; Tohma, H.; Kita, Y. Novel and direct oxidative cyanation reactions of heteroaromatic compounds mediated by ahypervalent iodine(III) reagent. Org. Lett. 2005, 7, 537–540. [Google Scholar] [CrossRef]
- Dohi, T.; Morimoto, K.; Takenaga, N.; Goto, A.; Maruyama, A.; Kiyono, Y.; Tohma, H.; Kita, Y. Direct cyanation of heteroaromatic compounds mediated by hypervalent iodine(III) reagents: In situ generation of PhI(III)-CN species and their cyano transfer. J. Org. Chem. 2007, 72, 109–116. [Google Scholar] [CrossRef]
- Kita, Y.; Takada, T.; Tohma, H. Hypervalent iodine reagents in organic synthesis: Nucleophilic substitution of p-substituted phenol ethers. Pure Appl. Chem. 1996, 68, 627–630. [Google Scholar] [CrossRef]
- Eberson, L.; Hartshorn, M.P.; Persson, O.; Radner, F. Making radical cations live longer. Chem. Commun. 1996, 2105–2112. [Google Scholar]
- Bégué, J.-P.; Bonnet-delpon, D.; Crousse, B. Fluorinated alcohols: A new medium for selective and clean reaction. Synlett 2004, 18–29. [Google Scholar]
- Shuklov, I.A.; Dubrovina, N.V.; Boerner, A. Fluorinated alcohols as solvents, cosolvents and additives in homogeneous catalysis. Synthesis 2007, 2925–2943. [Google Scholar]
- Koser, G.F.; Telu, S.; Laali, K.K. Oxidative-substitution reactions of polycyclic aromatic hydrocarbons with iodine(III) sulfonate reagents. Tetrahedron Lett. 2006, 47, 7011–7015. [Google Scholar]
- Dohi, T.; Ito, M.; Morimoto, K.; Minamitsuji, Y.; Takenaga, N.; Kita, Y. Versatile direct dehydrative approach for diaryliodonium(III) salts in fluoroalcohol media. Chem. Commun. 2007, 4152–4154. [Google Scholar]
- Frechet, J.M.J. The photogeneration of acid and base within polymer coatings: Approaches to polymer curing and imaging. Pure Appl. Chem. 1992, 64, 1239–1248, and references therein. [Google Scholar]
- Gallop, P.M.; Paz, M.A.; Flückiger, R.; Stang, P.J.; Zhdankin, V.V.; Tykwinski, R. Highly effective PQQ inhibition by alkynyl and aryl mono- and diiodonium salts. J. Am. Chem. Soc. 1993, 115, 11702–11704. [Google Scholar]
- Múller, U. Photochemistry of lipophilic iodonium salts. Trends Photochem. Photobiol. 1999, 5, 117–138. [Google Scholar]
- Bykowski, D.; McDonald, R.; Hinkle, R.J.; Tykwinski, R.R. Structural and electronic characteristics of thienyl(aryl)iodonium triflates. J. Org. Chem. 2002, 67, 2798–2804. [Google Scholar] [CrossRef]
- Martin-Santamaría, S.; Carroll, M.A.; Carroll, C.M.; Carter, C.D.; Pike, V.W.; Rzepa, H.S.; Widdowson, D.A. Fluoridation of heteroaromatic iodonium salts - experimental evidencesupporting theoretical prediction of the selectivity of the process. Chem. Commun. 2000, 649–650. [Google Scholar]
- Ross, T.L.; Ermert, J.; Hocke, C.; Coenen, H.H. Nucleophilic 18F-Fluorination of Heteroaromatic Iodonium Salts with No-Carrier-Added [18F]Fluoride. J. Am. Chem. Soc. 2007, 129, 8018–8025. [Google Scholar]
- Kita, Y.; Morimoto, K.; Ito, M.; Ogawa, C.; Goto, A.; Dohi, T. Metal-free oxidative cross-coupling of unfunctionalized aromatic compounds. J. Am. Chem. Soc. 2009, 131, 1668–1669. [Google Scholar]
- Beringer, F.M.; Bachofner, H.E.; Falk, R.A.; Leff, M. Diaryliodonium salts. VII. Di-2-thienyl- and phenyl-2-thienyliodonium salts. J. Am. Chem. Soc. 1958, 80, 4279–4281. [Google Scholar]
- Margida, A.J.; Koser, G.F. Direct condensation of [hydroxy(tosyloxy)iodo]arenes with thiophenes. A convenient, mild synthesis of aryl(2-thienyl)iodonium tosylates. J. Org. Chem. 1984, 49, 3643–3646. [Google Scholar]
- Yamada, Y.; Okawara, M. Synthesis of substituted phenyl-2-thienyliodonium halides and their prolysis reactions. Bull. Chem. Soc. Jpn. 1972, 45, 2515–2519. [Google Scholar] [CrossRef]
- Zhu, M.; Jalalian, N.; Olofsson, B. One-pot synthesis of diaryliodonium salts using toluenesulfonic acid. A fast entry to electron-rich diaryliodonium tosylates and triflates. Synlett 2008, 592–596. [Google Scholar]
- For a recent review, see: Merritt, A.E.; Olofsson, B. Diaryliodonium salts: A journey from obscurity to fame. Angew. Chem. Int. Ed. 2009, 48, 9052–9070.
- Hoier, S.N.; Park, S.M. In situ spectroelectrochemical and kinetic studies on poly(3-methylthiophene) growth. J. Electrochem. Soc. 1993, 140, 2454–2463. [Google Scholar] [CrossRef]
- Dian, G.; Barbey, G. Electrochemical synthesis of polythiophenes and polyselenophenes. Synth. Met. 1986, 13, 281–289. [Google Scholar] [CrossRef]
- Koser, G.F. [Hydroxy(tosyloxy)iodo]benzene and closely related iodanes: The second stage of development. Aldrichimica Acta 2001, 34, 89–102. [Google Scholar]
- Koser, G.F.; Wettach, R.H. [Hydroxy(tosyloxy)iodo]benzene, a versatile reagent for the mild oxidation of aryl iodides at the iodine atom by ligand transfer. J. Org. Chem. 1980, 45, 1542–1543. [Google Scholar] [CrossRef]
- Yusubov, M.S.; Wirth, T. Solvent-free reactions with hypervalent iodine reagents. Org. Lett. 2005, 7, 519–521. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Togo, H. Facile one-pot preparation of [hydroxy(sulfonyloxy)iodo]arenes from iodoarenes with MCPBA in the presence of sulfonic acids. Synlett 2005, 2486–2488. [Google Scholar]
- Blettner, C.G.; Koenig, W.A.; Stenzel, W.; Schotten, T. Poly(ethylene glycol)-supported liquid-phase synthesis of biaryls. Synlett 1998, 295–297. [Google Scholar]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ito, M.; Ogawa, C.; Yamaoka, N.; Fujioka, H.; Dohi, T.; Kita, Y. Enhanced Reactivity of [Hydroxy(tosyloxy)iodo]benzene in Fluoroalcohol Media. Efficient Direct Synthesis of Thienyl(aryl)iodonium Salts. Molecules 2010, 15, 1918-1931. https://doi.org/10.3390/molecules15031918
Ito M, Ogawa C, Yamaoka N, Fujioka H, Dohi T, Kita Y. Enhanced Reactivity of [Hydroxy(tosyloxy)iodo]benzene in Fluoroalcohol Media. Efficient Direct Synthesis of Thienyl(aryl)iodonium Salts. Molecules. 2010; 15(3):1918-1931. https://doi.org/10.3390/molecules15031918
Chicago/Turabian StyleIto, Motoki, Chieko Ogawa, Nobutaka Yamaoka, Hiromichi Fujioka, Toshifumi Dohi, and Yasuyuki Kita. 2010. "Enhanced Reactivity of [Hydroxy(tosyloxy)iodo]benzene in Fluoroalcohol Media. Efficient Direct Synthesis of Thienyl(aryl)iodonium Salts" Molecules 15, no. 3: 1918-1931. https://doi.org/10.3390/molecules15031918
APA StyleIto, M., Ogawa, C., Yamaoka, N., Fujioka, H., Dohi, T., & Kita, Y. (2010). Enhanced Reactivity of [Hydroxy(tosyloxy)iodo]benzene in Fluoroalcohol Media. Efficient Direct Synthesis of Thienyl(aryl)iodonium Salts. Molecules, 15(3), 1918-1931. https://doi.org/10.3390/molecules15031918