Stereodynamic Investigation of Labile Stereogenic Centres in Dihydroartemisinin


Abstract
:
{kind=link}
{kind=link}
{kind=link}
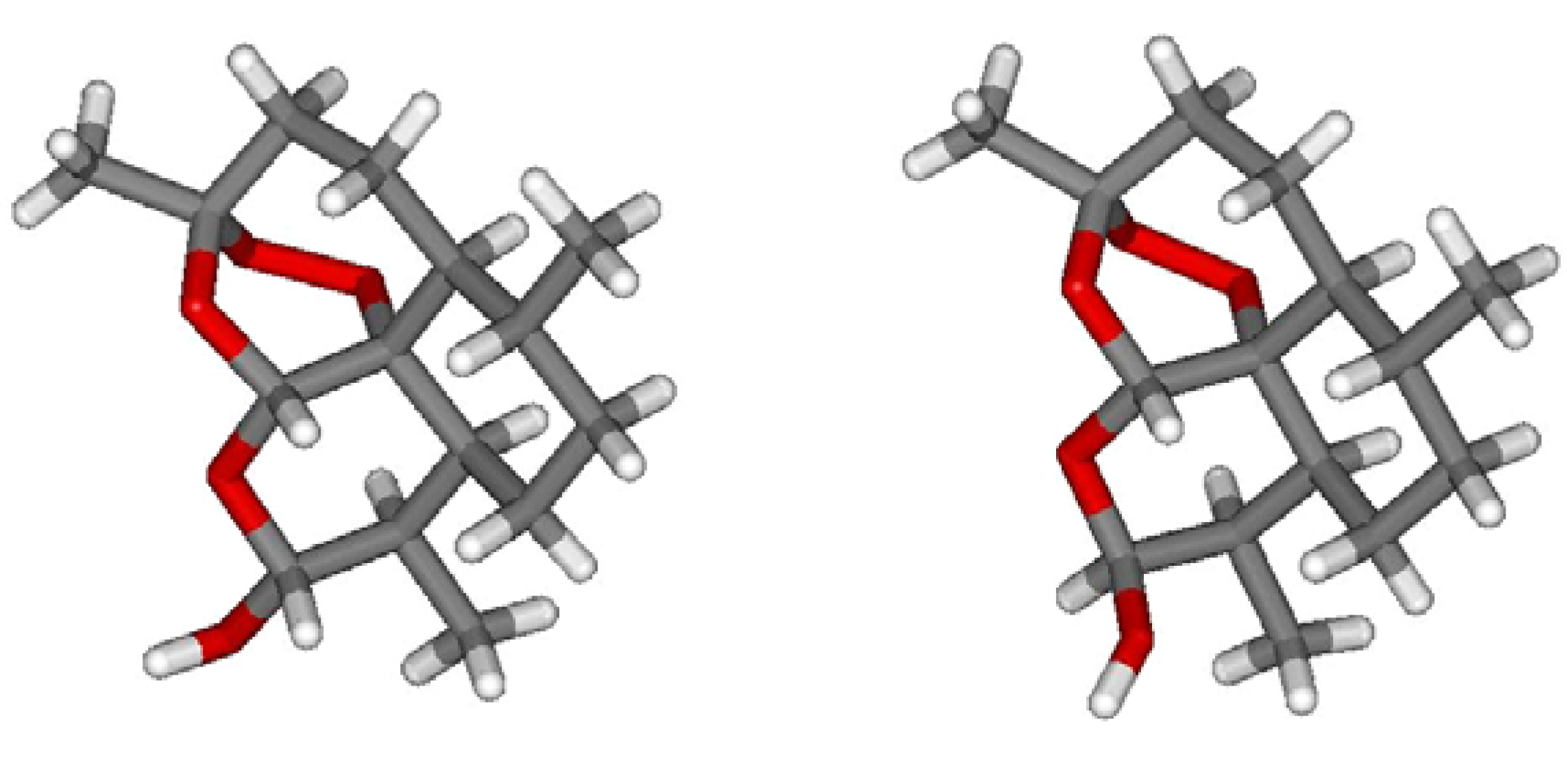{kind=link}
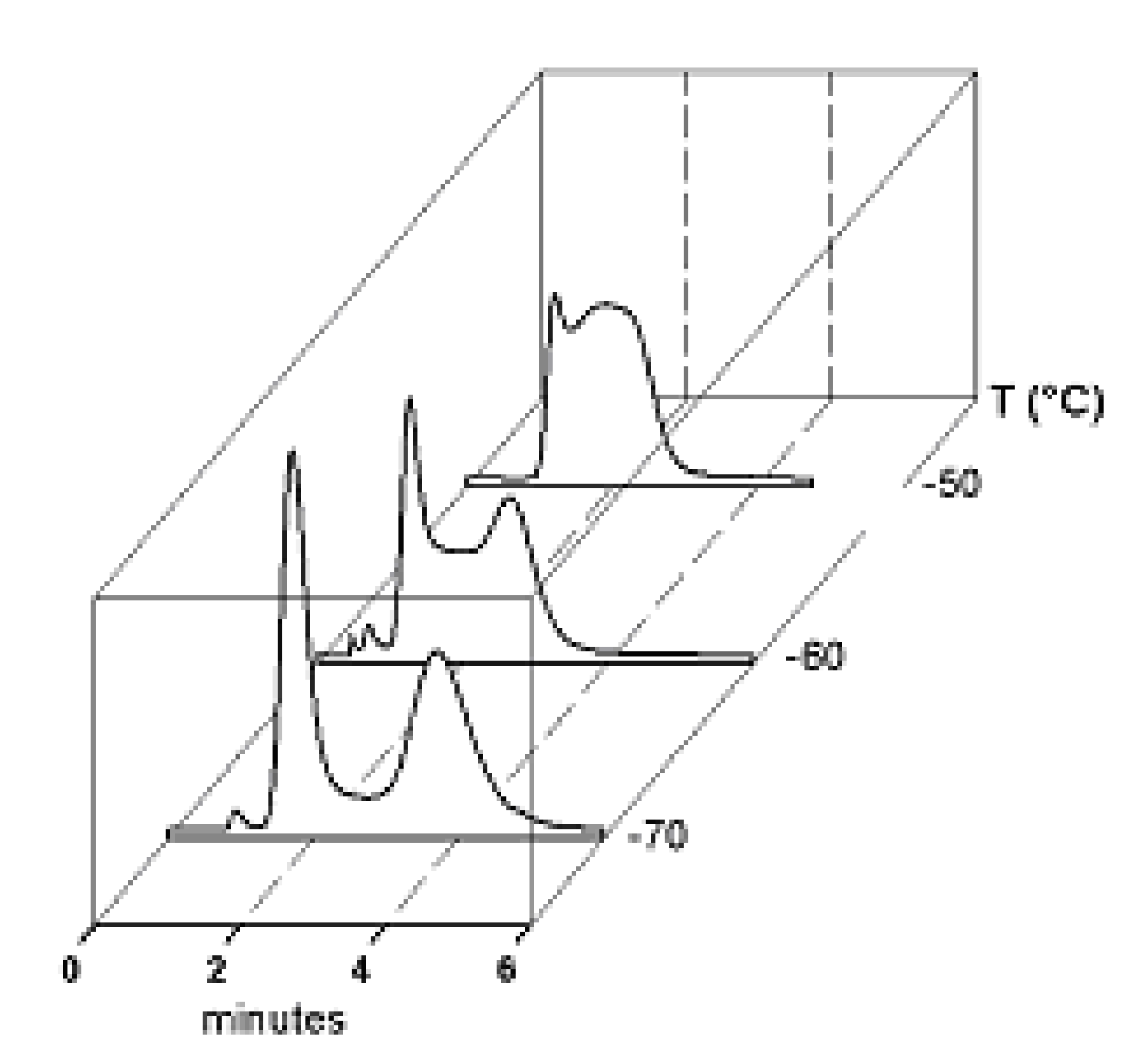{kind=link}
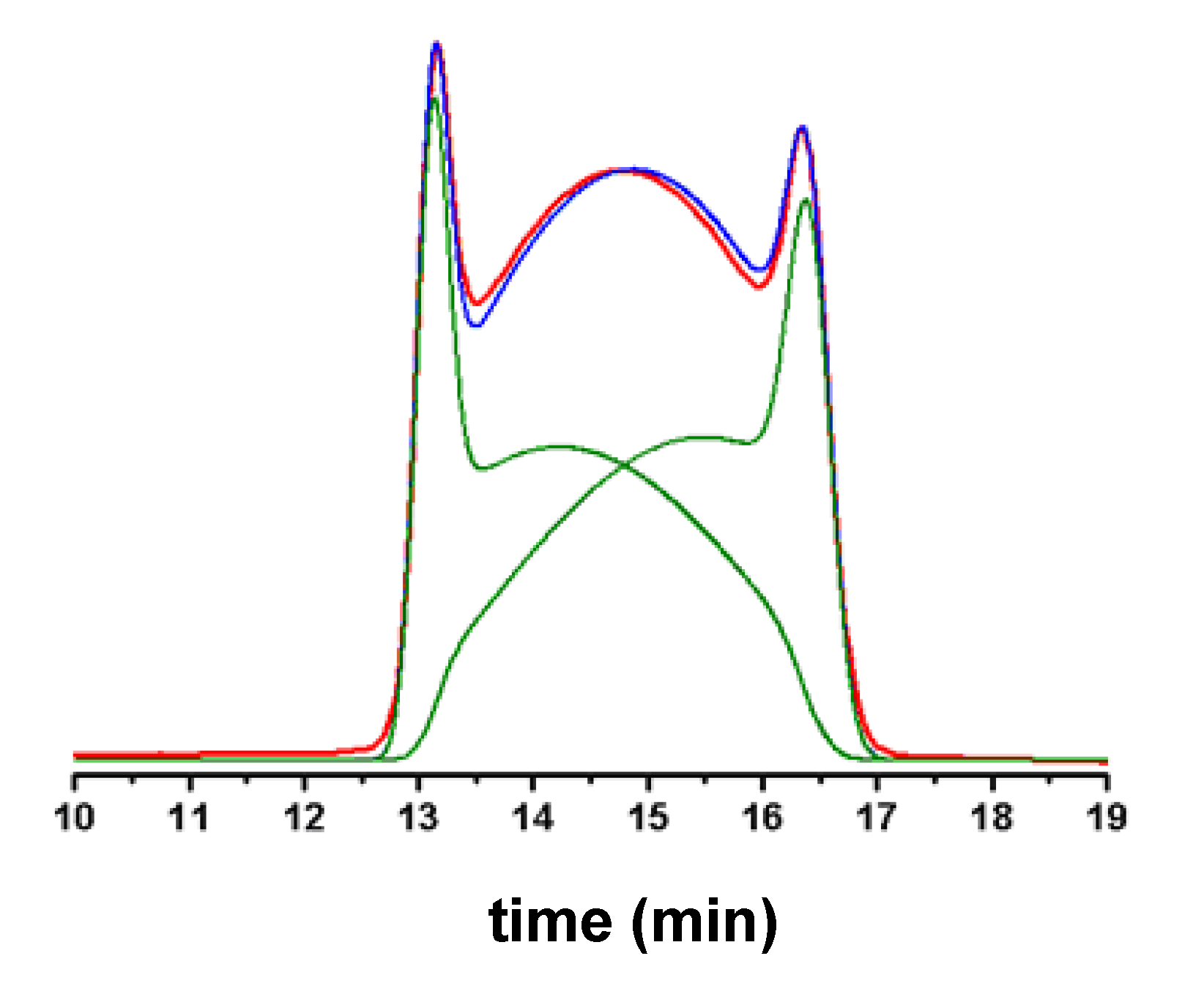{kind=link}
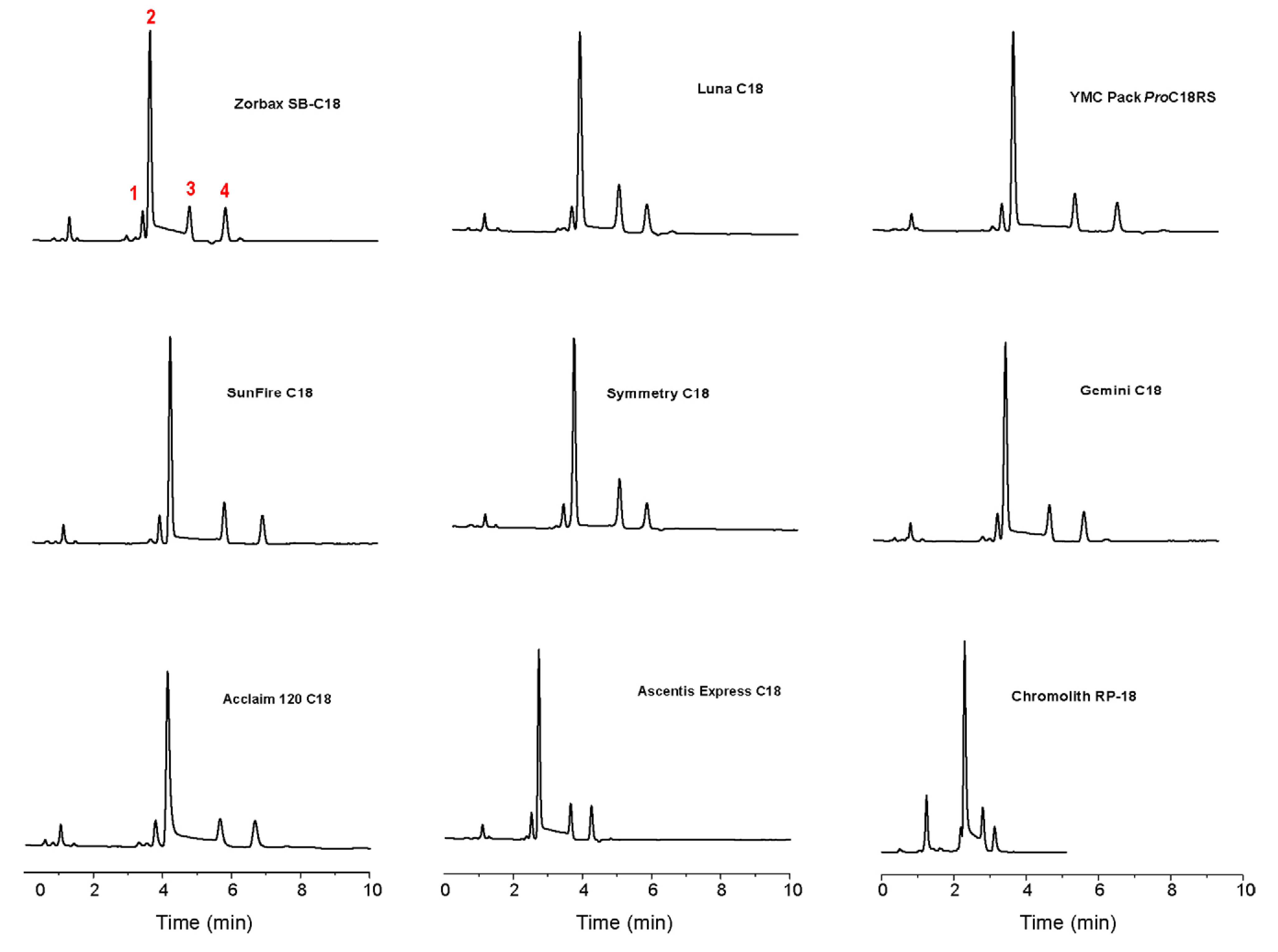{kind=link}
{kind=link}
{kind=link}
{kind=link}
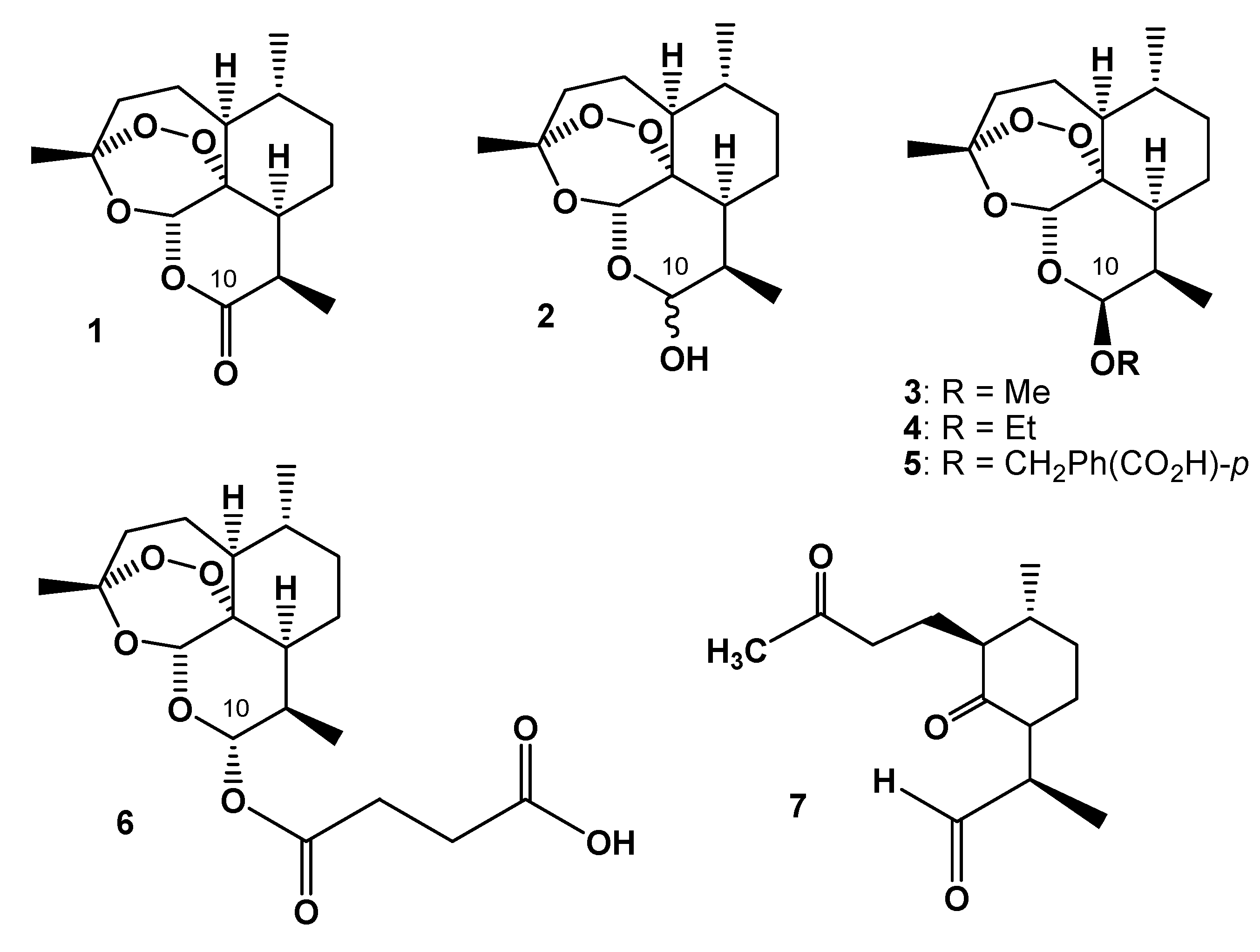
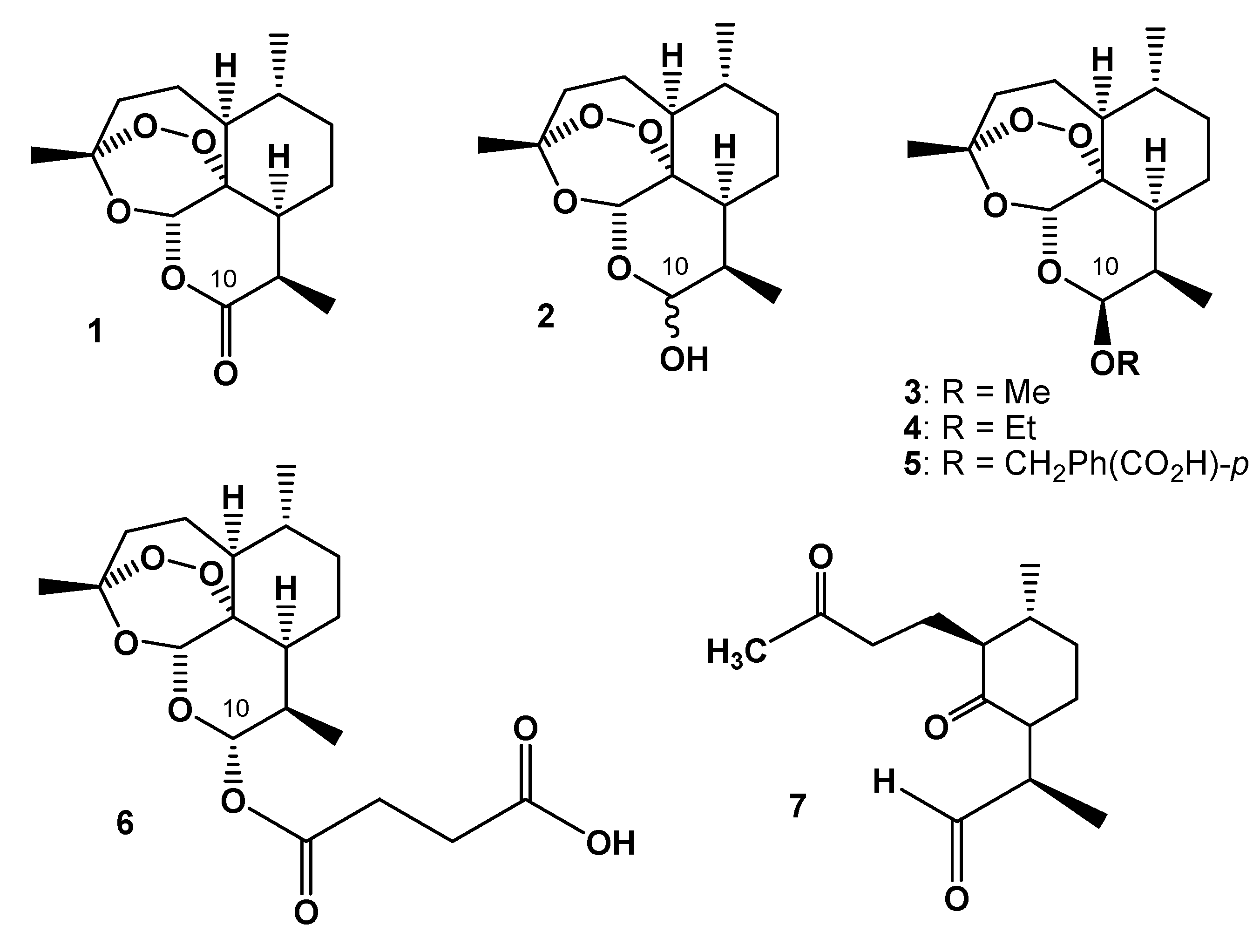
1. Introduction
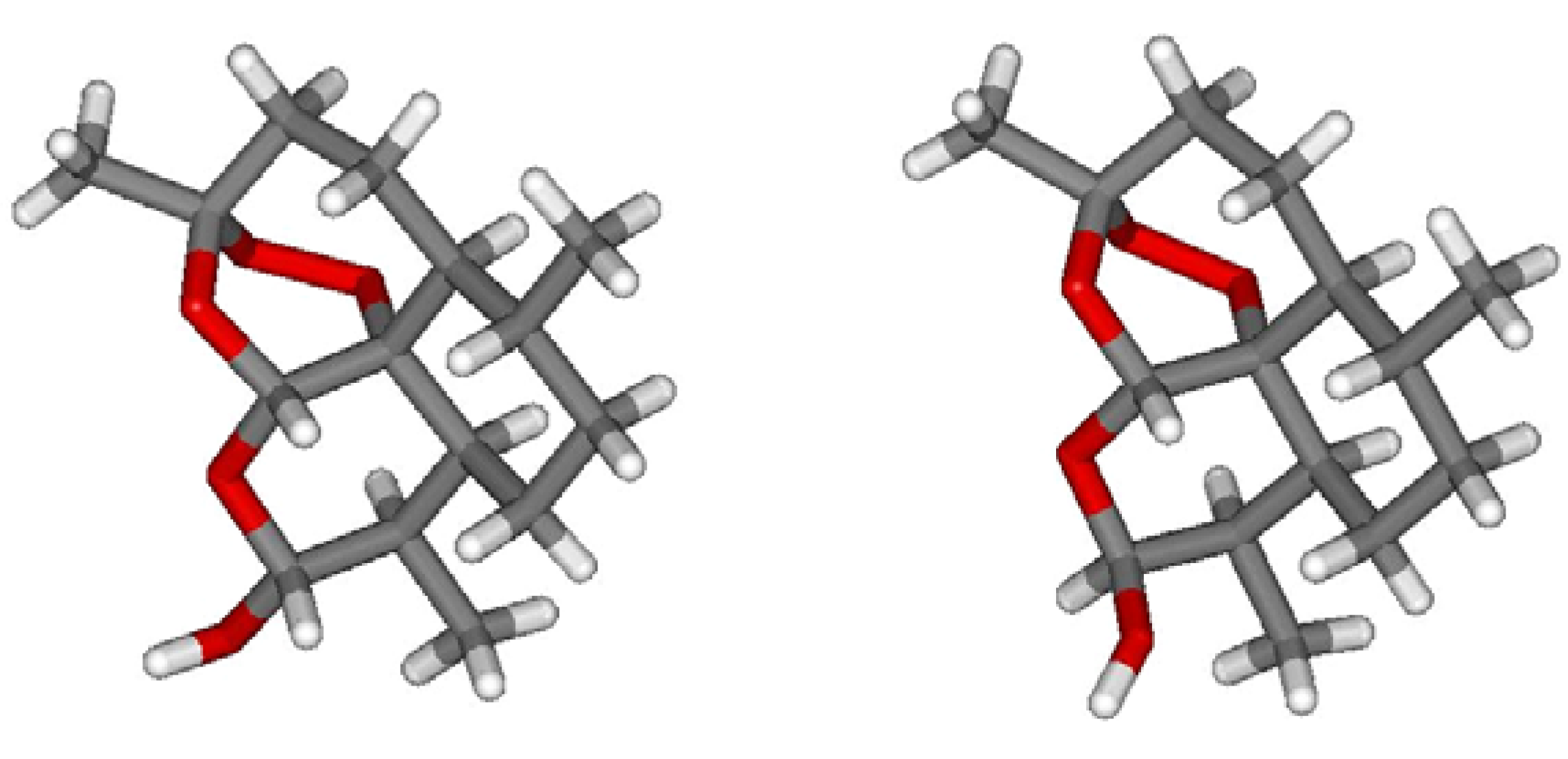
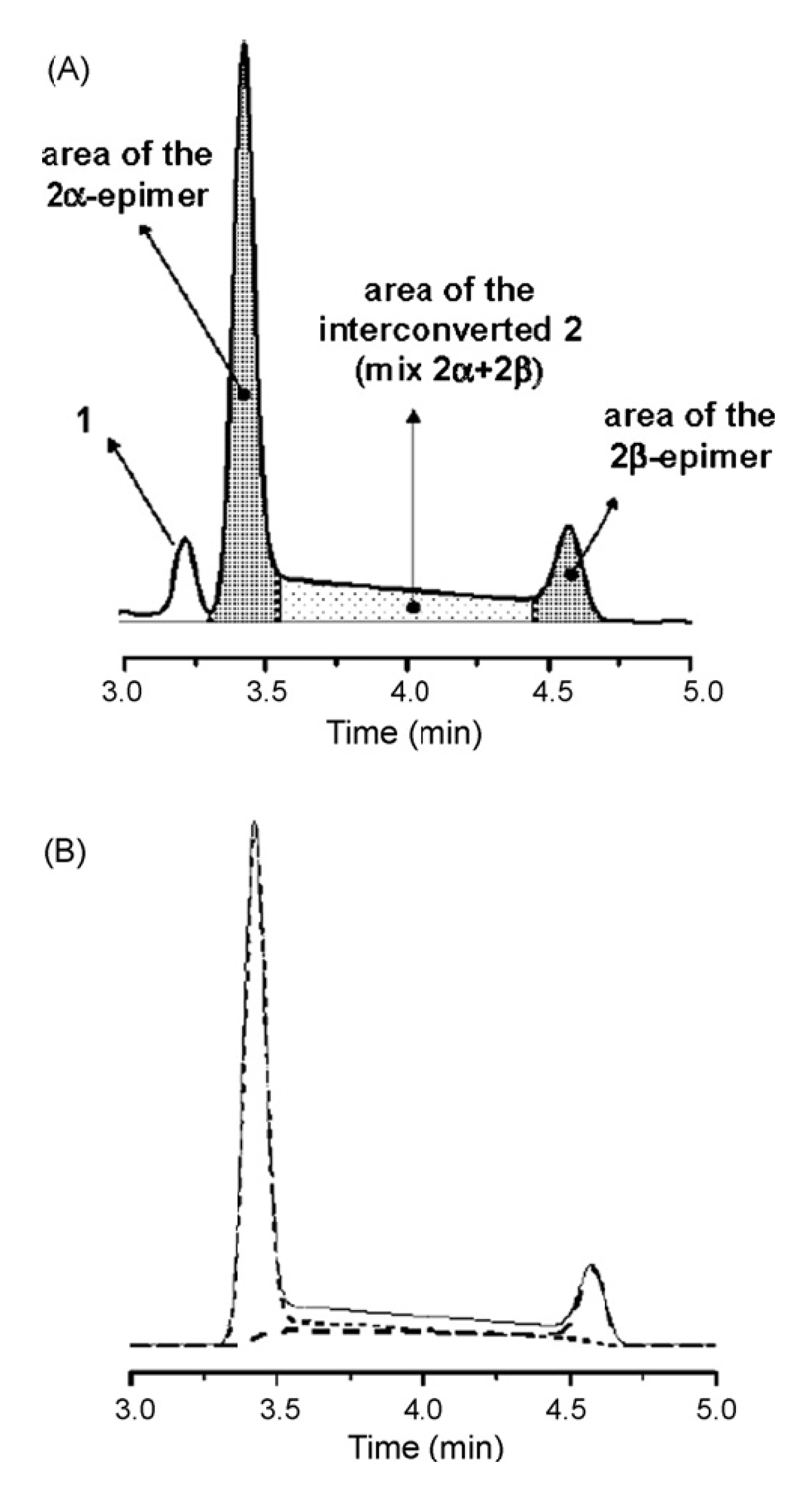
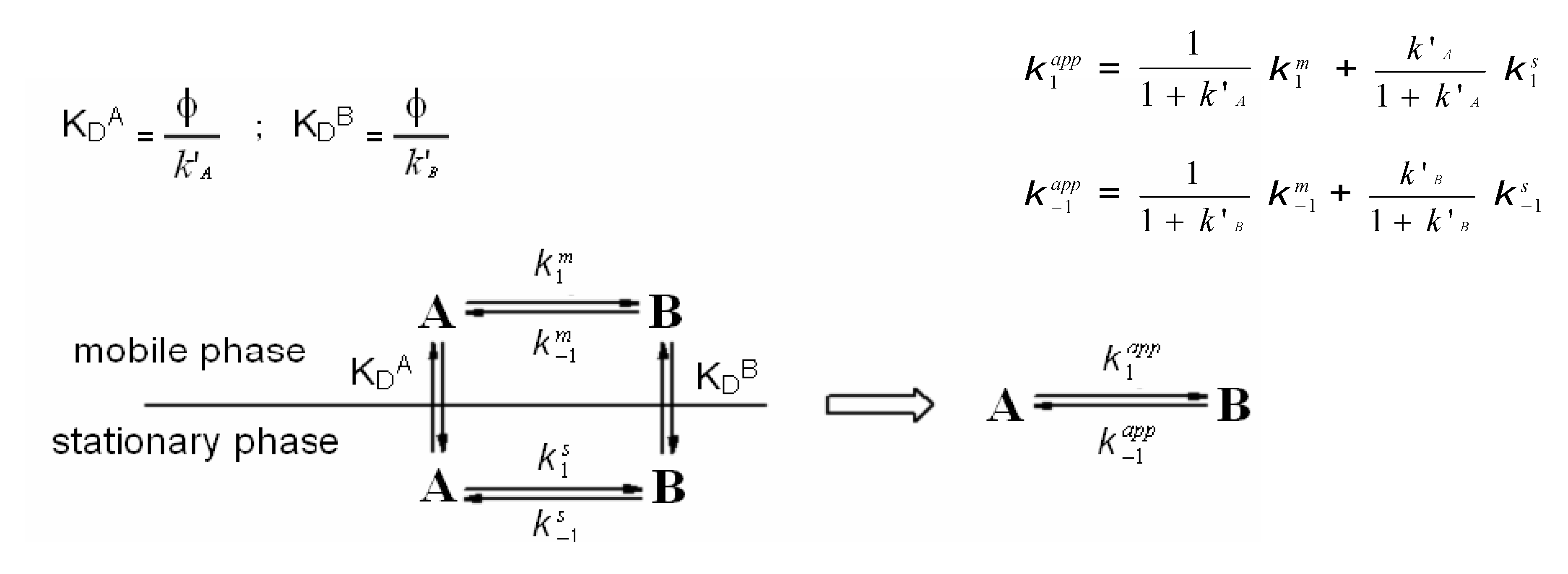
2. On-Column Interconversions
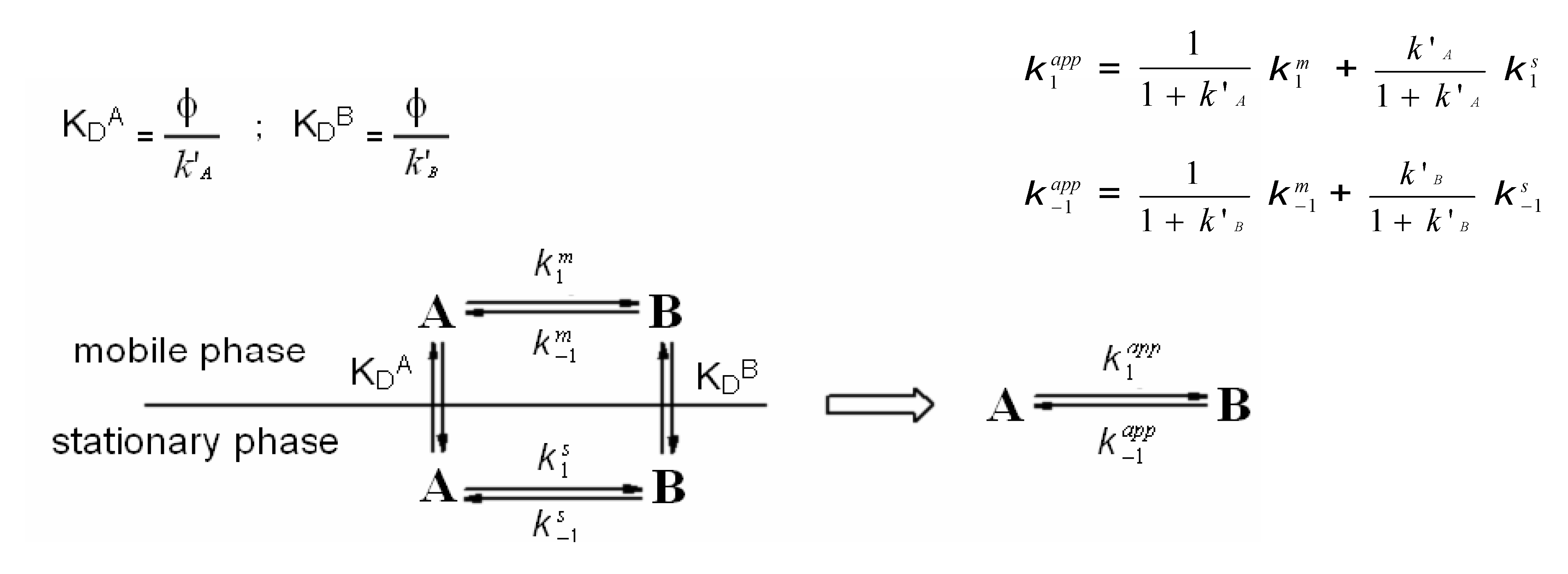
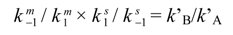
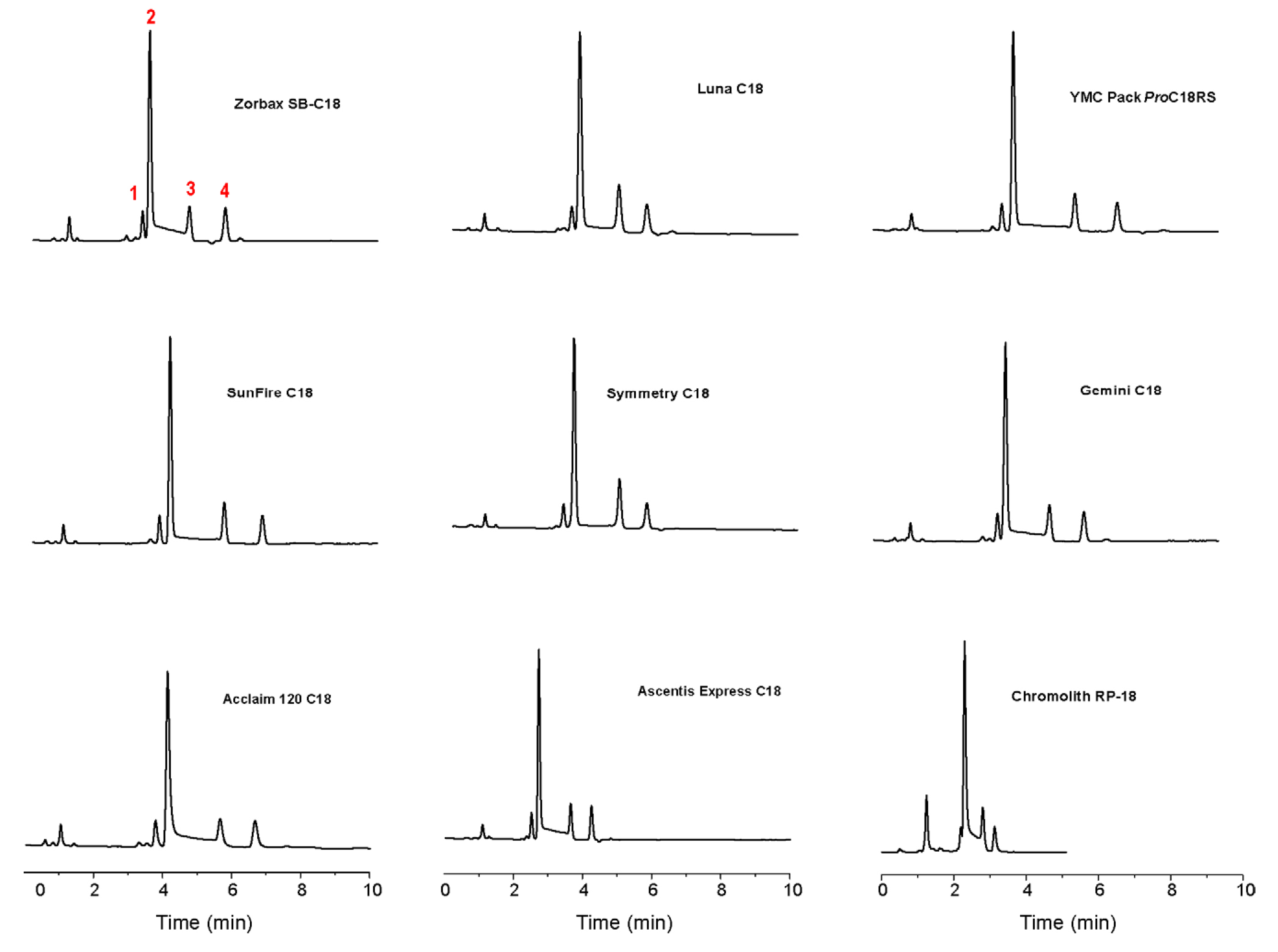
3. HPLC Analytical Methods for DHA
4. Pharmacopoeia Guidelines on Antimalarial drugs
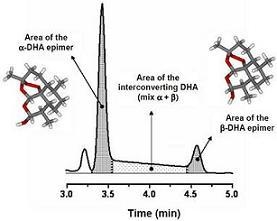
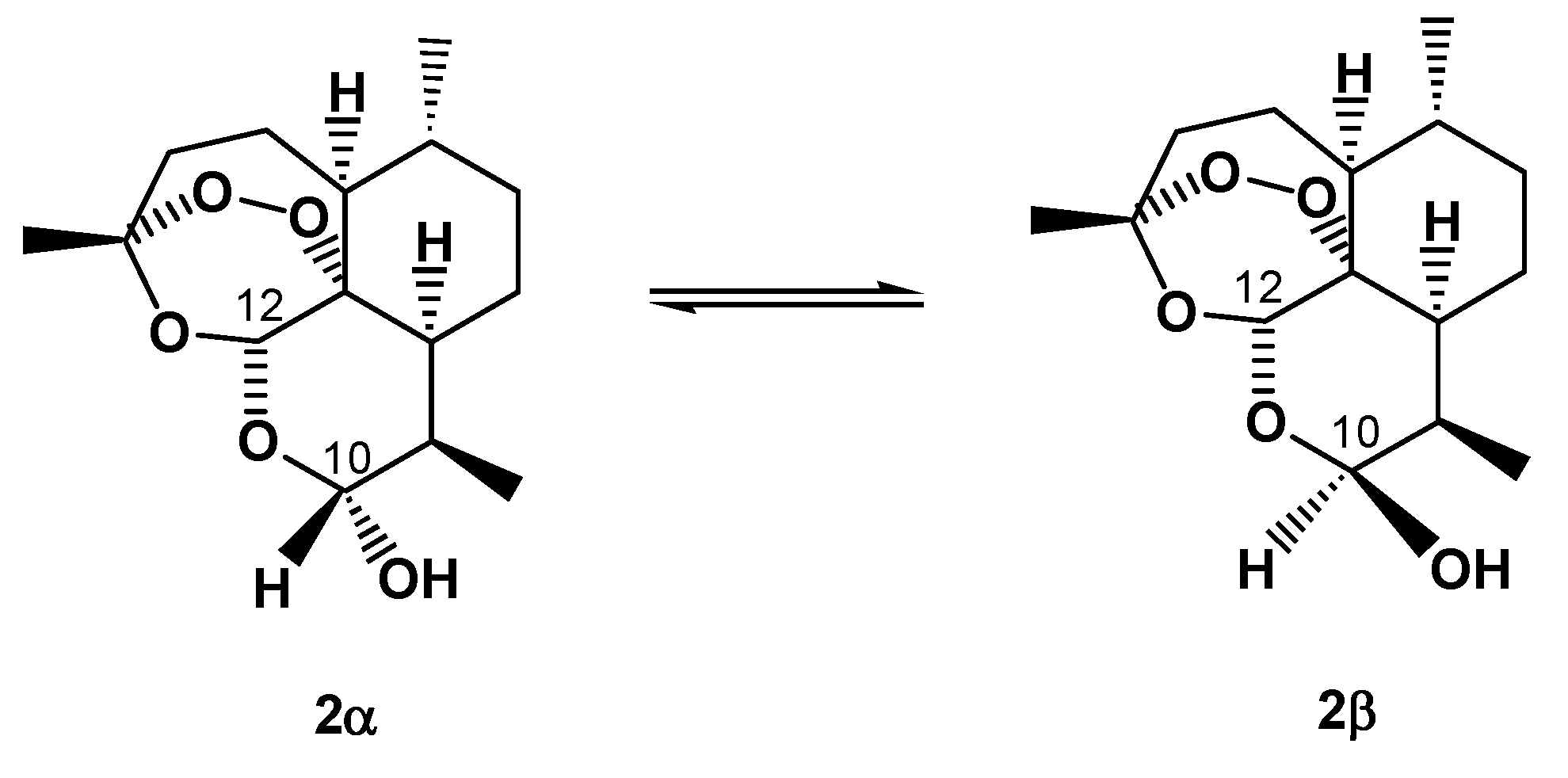
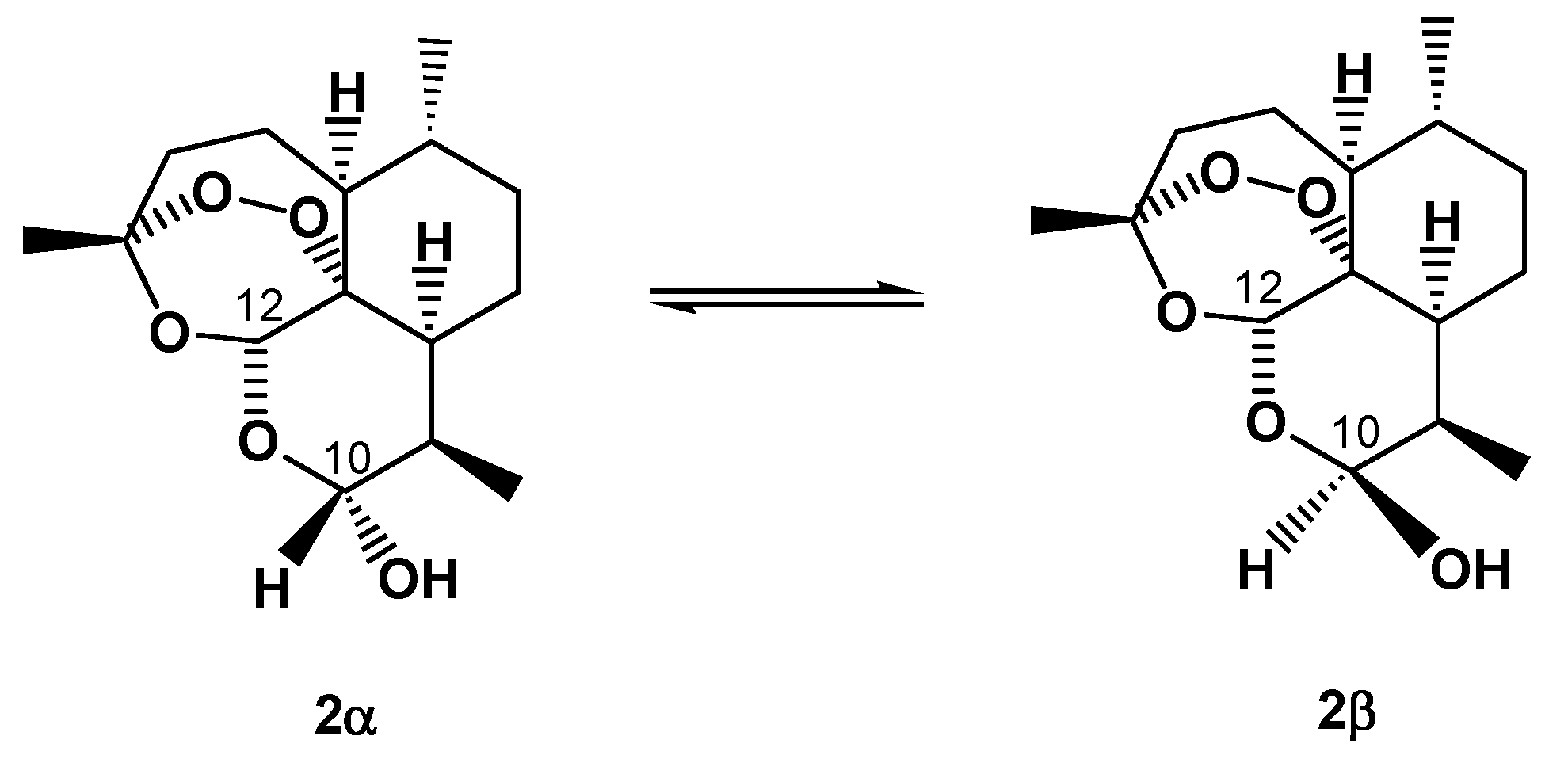
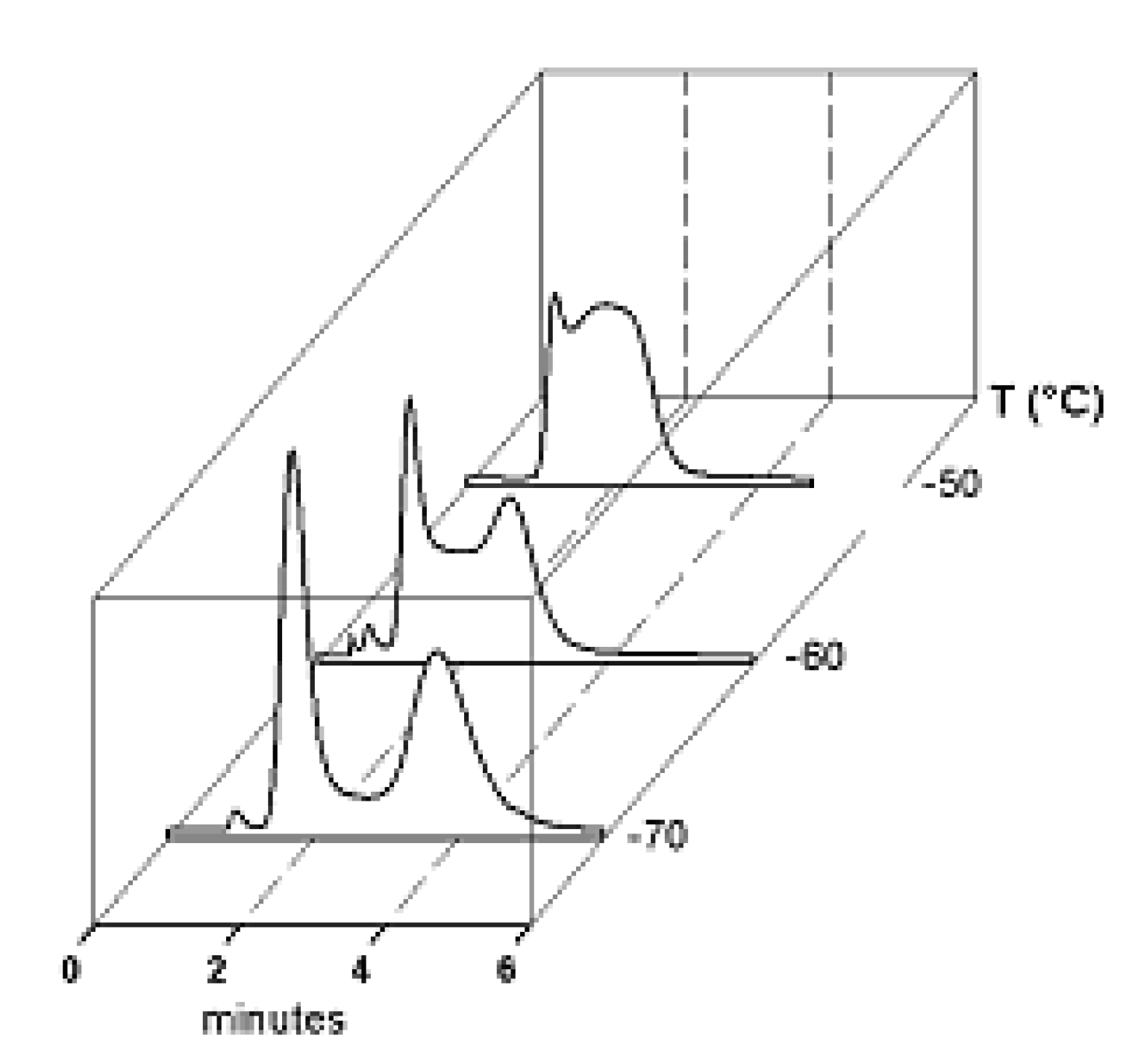
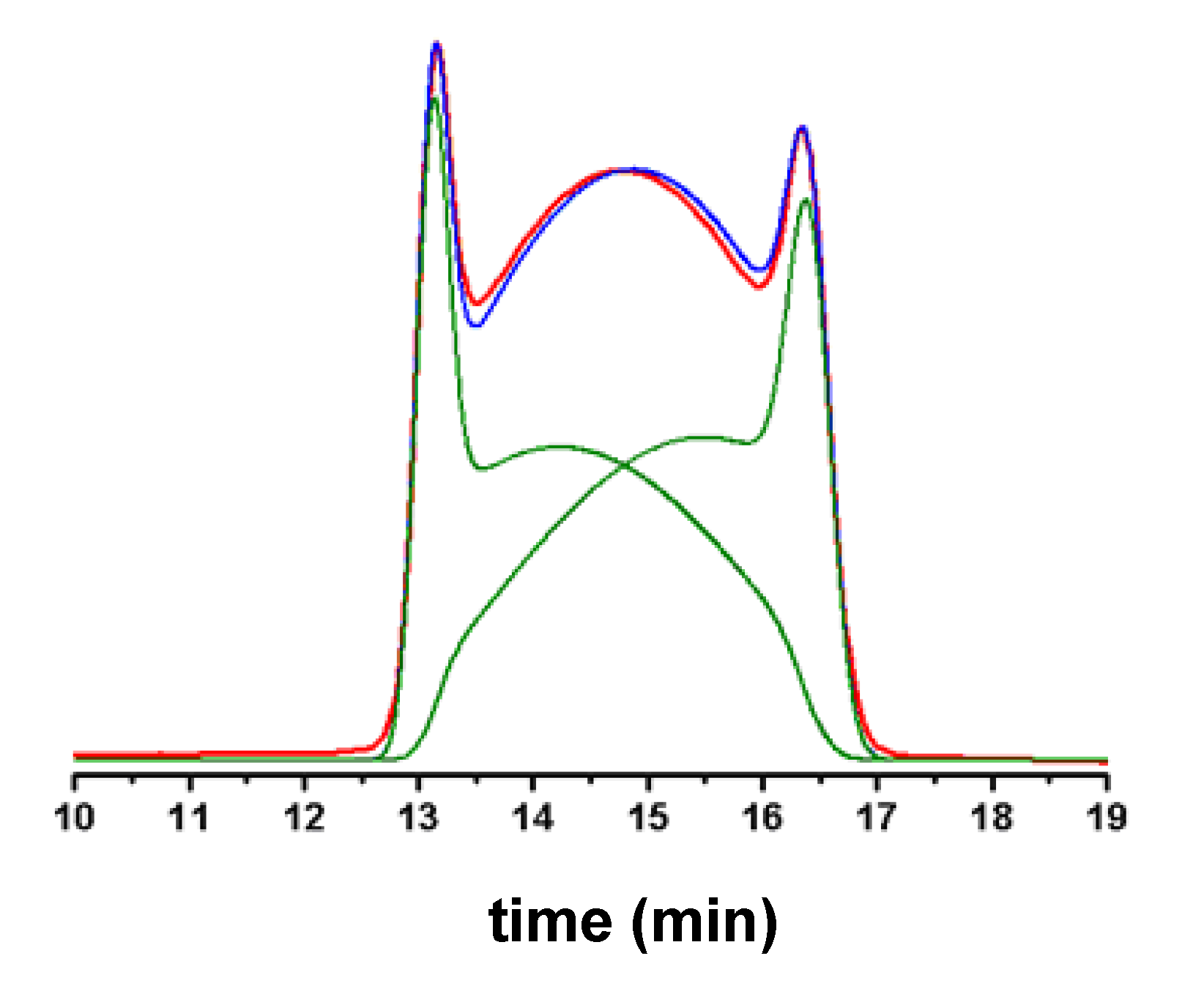
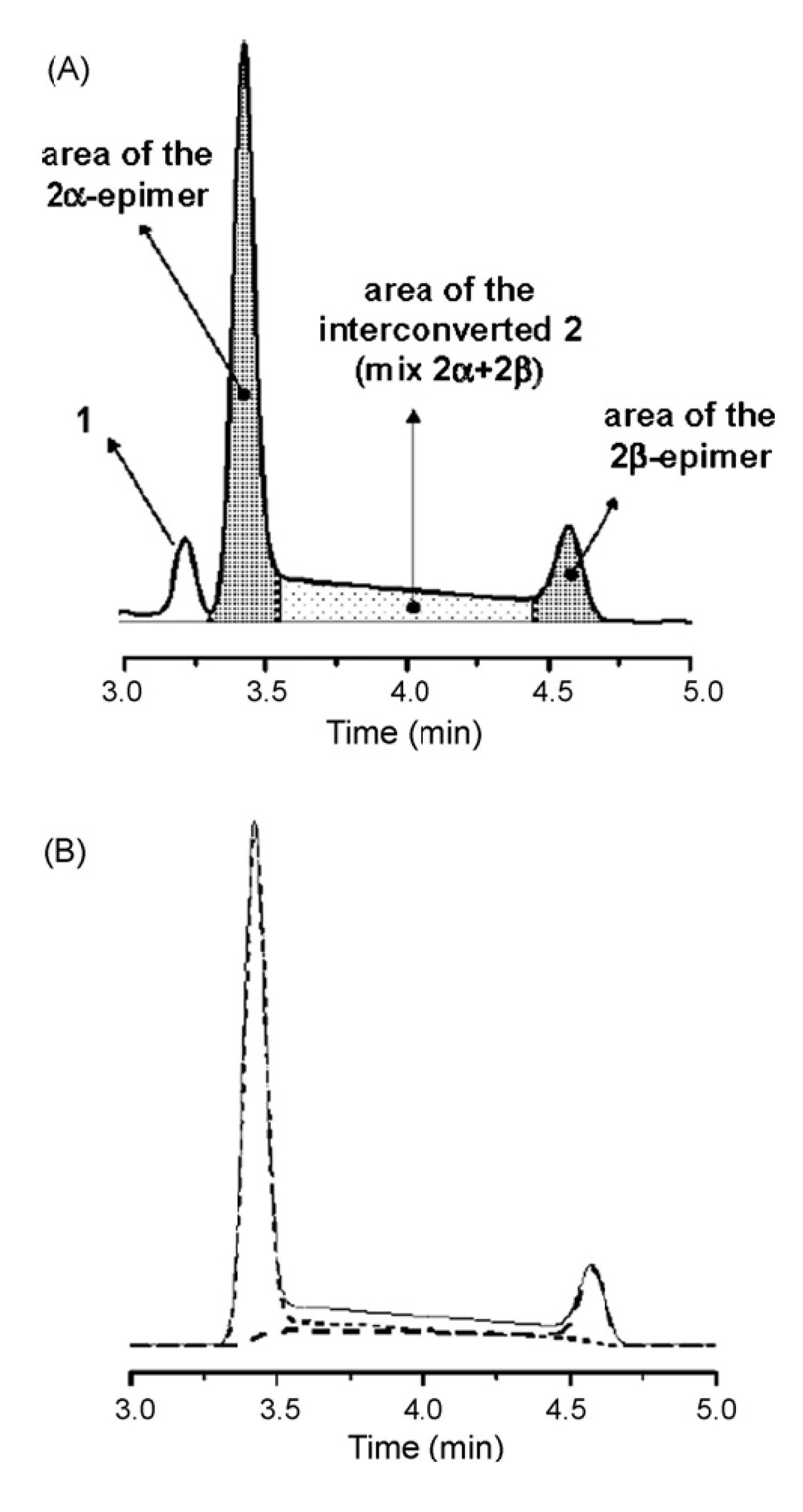
5. On-Column Epimerization of DHA
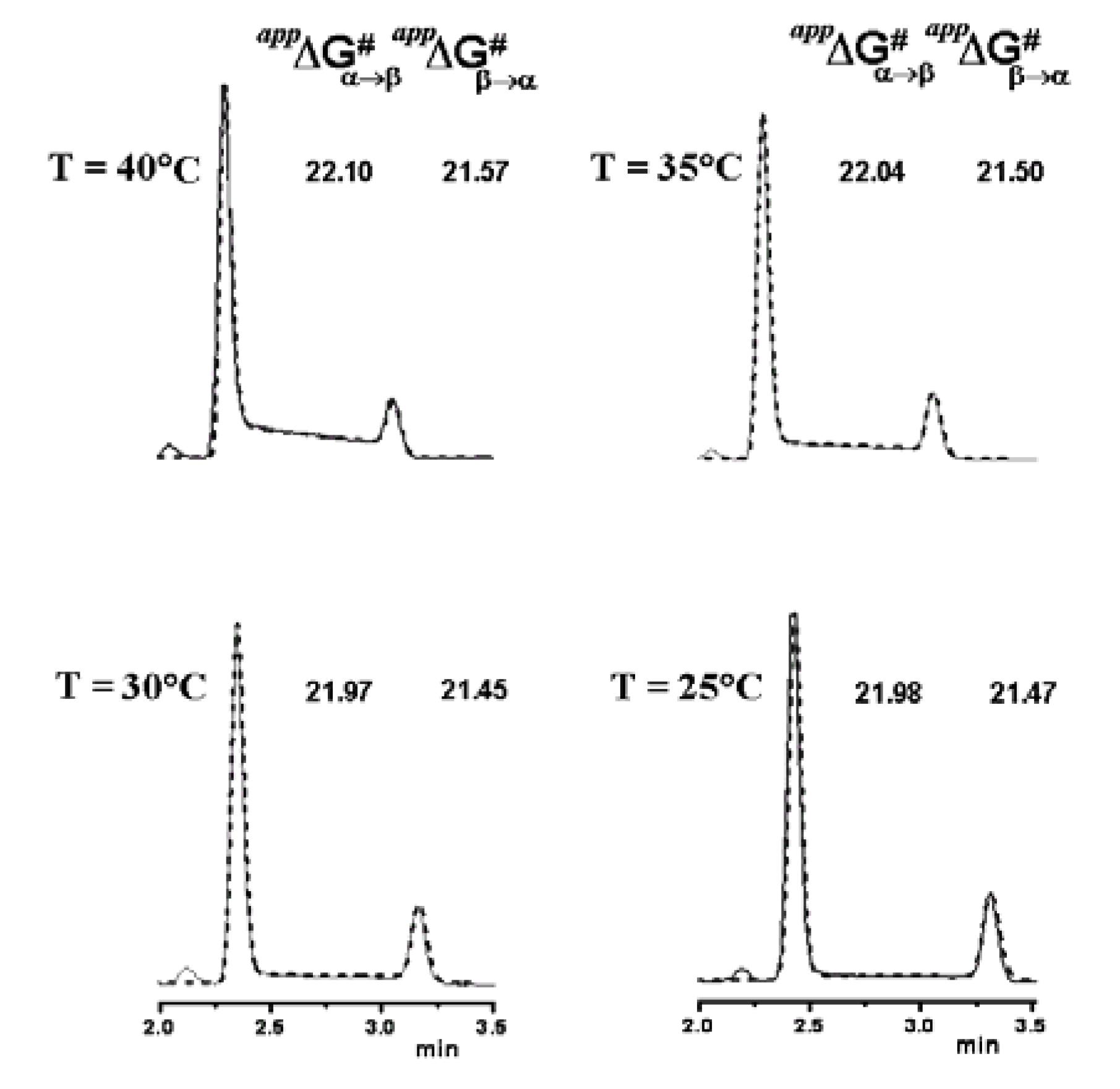
6. Conclusions
Acknowledgements
- Samples Availability: Samples of compounds 1, 2, and 7 are available from authors.
References and Notes
- Guidelines for the Treatment of Malaria; World Health Organization: Geneva, Switzerland, 2006.
- Klayman, D.L. Qinghaosu (artemisinin): An Anti-malarial Drug from China. Science 1985, 228, 1049–1055. [Google Scholar]
- Ashley, E.A.; White, N.J. Artemisinin-based Combinations. Curr. Opin. Infect. Dis. 2005, 18, 531–536. [Google Scholar] [CrossRef]
- O'Neill, P.M. The Therapeutic Potential of Semi-Synthetic Artemisinin and Synthetic Endoperoxide Antimalarial Agents. Expert Opin. Invest. Drugs 2005, 14, 1117–1128. [Google Scholar] [CrossRef]
- O’Neill, P.M.; Posner, G.H. A Medicinal Chemistry Perspective on Artemisinin and Related Endoperoxides. J. Med. Chem. 2004, 47, 2945–2964. [Google Scholar] [CrossRef]
- Li, Y.; Yu, P.-L.; Chen, Y.-X.; Li, L.-Q.; Gai, Y.-Z.; Wang, D.-S.; Zheng, Y.-P. Synthesis of some artemisinin derivatives. Chin. Sci. Bull. 1979, 24, 667–669. [Google Scholar]
- China Cooperative Research Group on Qinghaosu and its Derivatives as Antimalarials. The Chemistry and Synthesis of Qinghaosu Derivatives. J. Tradit. Chin. Med. 1982, 2, 9–16.
- Pathak, A.K.; Jain, D.C.; Sharma, R.P. 13C NMR Assignments of α- and β-dihydroartemisinin. Indian J. Chem., B: Org. Chem. Incl. Med. Chem. 1995, 34, 992–993. [Google Scholar]
- Haynes, R.K.; Chan, H.-W.; Cheung, M.-K.; Chung, S.T.; Lam, W.-L.; Tsang, H.W.; Voerste, A.; Williams, I.D. Stereoselective Preparation of 10α- and 10β-Aryl Derivatives of Dihydroartemisinin. Eur. J. Org. Chem. 2003, 2098–2114. [Google Scholar]
- Luo, X.; Yeh, H. J.C.; Brossi, A.; Flippen-Anderson, J.L.; Gilardi, R. The Chemistry of Drugs. Configurations of Antimalarials Derived from Qinghaosu: Dihydroqinghaosu, Artemether, and Artesunic Acid. Helv. Chim. Acta 1984, 67, 1515–1522. [Google Scholar]
- Haynes, R.K.; Chan, H.-W.; Cheung, M.-K.; Lam, W.-L.; Soo, M.-K.; Tsang, H.-W.; Voerste, A.; Williams, I.D. C-10 Ester and Ether Derivatives of Dihydroartemisinin 10-α Artesunate, Preparation of Authentic 10-β Artesunate, and of other Ester and Ether Derivatives Bearing Potential Aromatic Intercalating Groups at C-10. Eur. J. Org. Chem. 2002, 113–132. [Google Scholar]
- Haynes, R.K. From Artemisinin to New Artemisinin Antimalarials: Biosynthesis, Extraction, Old and New Derivatives, Stereochemistry and Medicinal Chemistry Requirements. Curr. Topics Med. Chem. 2006, 6, 509–537. [Google Scholar] [CrossRef]
- Maggs, J.L.; Madden, S.; Bishop, L.P.; O’Neill, P.M.; Park, K.B. The Rat Biliary Metabolites of Dihydroartemisinin, an Antimalarial Endoperoxide. Drug Metab. Dispos. 1997, 25, 1200–1204. [Google Scholar]
- Ramu, K.; Baker, J.K. Synthesis, Characterization and Antimalarial Activity of the Glucuronides of the Hydroxylated Metabolites of Arteether. J. Med. Chem. 1995, 38, 1911–1921. [Google Scholar] [CrossRef]
- Shishan, Z. Equilibrium between Alpha and Beta Isomers of Dihydroartemisinine and its Multiple Peaks in High Performance Liquid Chromatography. Chromatographia 1986, 22, 77–80. [Google Scholar] [CrossRef]
- Dynamic Nuclear Magnetic Resonance Spectroscopy; Jackman, L.M.; Cotton, F.A. (Eds.) Academic Press: New York, NY, USA, 1975.
- Kaplan, J.I.; Fraenkel, G. NMR of Chemically Exchanging Systems; Academic Press: New York, 1980. [Google Scholar]
- Sandström, J. Dynamic NMR Spectroscopy; Academic Press: London, UK, 1982. [Google Scholar]
- Oki, M. Applications of Dynamic NMR Spectroscopy to Organic Chemistry; VCH: Deerfield Beach, FL, USA, 1985. [Google Scholar]
- D’Acquarica, I.; Gasparrini, F.; Pierini, M.; Villani, C.; Zappia, G. Dynamic HPLC on Chiral Stationary Phases: A Powerful Tool for the Investigation of Stereomutation Processes. J. Sep. Sci. 2006, 29, 1508–1516. [Google Scholar] [CrossRef]
- Giddings, J.C. Kinetic Processes and Zone Diffusion in Chromatography. J. Chromatogr. 1960, 3, 443–453. [Google Scholar] [CrossRef]
- Kramer, R. Simultan-Reaktionsgaschromatographie Mit Reversibler Reaktion Erster Ordnung. I. J. Chromatogr. 1975, 107, 241–252. [Google Scholar] [CrossRef]
- Schurig, V.; Burkle, W. Extending the Scope of Enantiomer Resolution by Complexation Gas Chromatography. J. Am. Chem. Soc. 1982, 104, 7573–7580. [Google Scholar] [CrossRef]
- Burkle, W.; Karfunkel, H.; Schurig, V. Dynamic Phenomena during Enantiomer Resolution by Complexation Gas Chromatography. A Kinetic Study of Enantiomerization. J. Chromatogr. 1984, 288, 1–14. [Google Scholar] [CrossRef]
- Jacobson, J.; Melander, W.; Vaisnys, G.; Horváth, C. Kinetic Study on Cis-Trans Proline Isomerization by High-Performance Liquid Chromatography. J. Phys. Chem. 1984, 88, 4536–4542. [Google Scholar] [CrossRef]
- Veciana, J.; Crespo, M.I. Dynamic HPLC: A Method for Determining Rate Constants, Energy Barriers, and Equilibrium Constants of Molecular Dynamic Processes. Angew. Chem. Int. Ed. Engl. 1991, 30, 74–77. [Google Scholar] [CrossRef]
- Jung, M.; Schurig, V. Determination of Enantiomerization Barriers by Computer Simulation of Interconversion Profiles: Enantiomerization of Diaziridines during Chiral Inclusion Gas Chromatography. J. Am. Chem. Soc. 1992, 114, 529–534. [Google Scholar] [CrossRef]
- Cabrera, K.; Jung, M.; Fluck, M.; Schurig, V. Determination of Enantiomerization Barriers by Computer Simulation of Experimental Elution Profiles Obtained by High-Performance Liquid Chromatography on a Chiral Stationary Phase. J. Chromatogr. A 1996, 731, 315–321. [Google Scholar] [CrossRef]
- Oxelbark, J.; Allenmark, S. Enantiomerization Kinetics Studied by Dynamic Enantioselective Liquid Chromatography: Solvent, Temperature and Stationary Phase Effects on the Rate of N-Benzyl-1,3,2-Benzodithiazole 1-Oxide Enantiomer Interconversion. J. Chem. Soc. Perkin Trans. 1999, 2, 1587–1589. [Google Scholar]
- Trapp, O.; Schoetz, V.; Schurig, V. Determination of Enantiomerization Barriers by Dynamic and Stopped-Flow Chromatographic Methods. Chirality 2001, 13, 403–414. [Google Scholar] [CrossRef]
- Trapp, O.; Schurig, V. Approximation Function for the Direct Calculation of Rate Constants and Gibbs Activation Energies of Enantiomerization of Racemic Mixtures from Chromatographic Parameters in Dynamic Chromatography. J. Chromatogr. A 2001, 911, 167–175. [Google Scholar] [CrossRef]
- Kiesswetter, R.; Brandl, F.; Kastner-Pustet, N.; Mannschreck, A. Chiroptical Detection During Liquid Chromatography, 9. Deconvolution of Overlapping Peaks of Enantiomers and its Applications. Chirality 2003, 15, S40–S49. [Google Scholar] [CrossRef]
- Krupcik, J.; Oswald, P.; Májek, P.; Sandra, P.; Armstrong, D.W. Determination of the Interconversion Energy Barrier of Enantiomers by Separation Methods. J. Chromatogr. A 2003, 1000, 779–800. [Google Scholar]
- Wolf, C. Dynamic Stereochemistry of Chiral Compounds—Principles and Applications; RSC Publishing: Cambridge, UK, 2008. [Google Scholar]
- Trapp, O. Unified Equation for Access to Rate Constants of First-Order Reactions in Dynamic and On-Column Reaction Chromatography. Anal. Chem. 2006, 78, 189–198. [Google Scholar] [CrossRef]
- Gasparrini, F.; Lunazzi, L.; Misiti, D.; Villani, C. Organic Stereochemistry and Conformational Analysis from Enantioselective Chromatography and Dynamic Nuclear Magnetic Resonance Measurements. Acc. Chem. Res. 1995 28, 163–170.
- Dell’Erba, C.; Gasparrini, F.; Grilli, S.; Lunazzi, L.; Mazzanti, A.; Novi, M.; Pierini, M.; Tavani, C.; Villani, C. Conformational Studies by Dynamic NMR. 86. Structure, Stereodynamics, and Cryogenic Enantioseparation of the Stereolabile Isomers of O-Dinaphthylphenyl Derivatives. J. Org. Chem. 2002, 67, 1663–1668. [Google Scholar]
- Gasparrini, F.; Grilli, R.; Leardini, L.; Lunazzi, L.; Mazzanti, A.; Nanni, D.; Pierini, M.; Pinamonti, M. Conformational Studies by Dynamic NMR. 89. Stereomutation and Cryogenic Enantioseparation of Conformational Antipodes of Hindered Aryl Oximes. J. Org. Chem. 2002, 67, 3089–3095. [Google Scholar] [CrossRef]
- Dalla Cort, A.; Gasparrini, F.; Lunazzi, L.; Mandolini, L.; Mazzanti, A.; Pasquini, C.; Pierini, M.; Rompietti, R.; Schiaffino, L. Stereomutations of Atropisomers of Sterically Hindered Salophen Ligands. J. Org. Chem. 2005, 70, 8877–8883. [Google Scholar]
- Cirilli, R.; Ferretti, R.; La Torre, F.; Secci, D.; Bolasco, A.; Carradori, S.; Pierini, M. High-Performance Liquid Chromatographic Separation of Enantiomers and Diastereomers of 2-Methylcyclohexanone Thiosemicarbazone, and Determination of Absolute Configuration and Configurational Stability. J. Chromatogr. A 2007, 1172, 160–169. [Google Scholar]
- Cabri, W.; Ciogli, A.; D’Acquarica, I.; Di Mattia, M.; Galletti, B.; Gasparrini, F.; Giorgi, F.; Lalli, S.; Pierini, M.; Simone, P. On-column Epimerization of Dihydroartemisinin: An Effective Analytical Approach to Overcome the Shortcomings of the International Pharmacopoeia Monograph. J. Chromatogr. B 2008, 875, 180–191. [Google Scholar] [CrossRef]
- Zhou, Z.M.; Anders, J.C.; Chung, H.; Theoharides, A.D. Analysis of Artesunic Acid and Dihydroqinghaosu in Blood by High-Performance Liquid Chromatography with Reductive Electrochemical Detection. J. Chromatogr. 1987, 414, 77–90. [Google Scholar] [CrossRef]
- Batty, K.T.; Davies, T.M.E.; Thu, L.T.; Binh, T.Q.; Anh, T.K.; Ilett, K.F. Selective High-Performance Liquid Chromatographic Determination of Artesunate and α- and β-Dihydroartemisinin in Patients with Falciparum Malaria. J. Chromatogr. B 1996, 677, 345–350. [Google Scholar] [CrossRef]
- Batty, K.T.; Ilett, K.F.; Davis, T.M.E. Chemical Stability of Artesunate Injection and Proposal for its Administration by Intravenous Infusion. J. Pharm. Pharmacol. 1996, 48, 22–26. [Google Scholar]
- Navaratnam, V.; Mordi, M.N.; Mansor, S.M. Simultaneous Determination of Artesunic Acid and Dihydroartemisinin in Blood Plasma by High-Performance Liquid Chromatography For Application in Clinical Pharmacological Studies. J. Chromatogr. B 1997, 692, 157–162. [Google Scholar] [CrossRef]
- Karbwang, J.; Na-Bangchang, K.; Molunto, P.; Banmairuroi, V.; Congpuong, K. Determination of Artemether and its Major Metabolite, Dihydroartemisinin, in Plasma using High-Performance Liquid Chromatography with Electrochemical Detection. J. Chromatogr. B 1997, 690, 259–265. [Google Scholar] [CrossRef]
- Sandrenan, N.; Sioufi, A.; Godbillon, J.; Netterb, C.; Danker, M.; van Valkenburg, C. Determination of Artemether and its Metabolite, Dihydroartemisinin, in Plasma by High-Performance Liquid Chromatography and Electrochemical Detection in the Reductive Mode. J. Chromatogr. B 1997, 691, 145–153. [Google Scholar] [CrossRef]
- Avery, B.A.; Venkatesh, K.K.; Avery, M.A. Rapid Determination of Artemisinin and Related Analogues using High-Performance Liquid Chromatography and an Evaporative Light Scattering Detector. J. Chromatogr. B 1999, 730, 71–80. [Google Scholar] [CrossRef]
- Ortelli, D.; Rudaz, S.; Cognard, E.; Veuthey, J.L. Analysis of Dihydroartemisinin in Plasma by Liquid Chromatography-Mass Spectrometry. Chromatographia 2000, 52, 445–450. [Google Scholar] [CrossRef]
- Souppart, C.; Gauducheau, N.; Sandrenan, N.; Richard, F. Development and Validation of a High-Performance Liquid Chromatography-Mass Spectrometry Assay for the Determination of Artemether and its Metabolite Dihydroartemisinin in Human Plasma. J. Chromatogr. B 2002, 774, 195–203. [Google Scholar] [CrossRef]
- Sabarinath, S.; Rajanikanth, M.; Madhusudanan, K.P.; Gupta, R.C. A Sensitive and Selective Liquid Chromatographic/Electrospray Ionization Tandem Mass Spectrometric Assay for the Simultaneous Quantification of α-,β-Arteether and its Metabolite Dihydroartemisinin in Plasma, useful for Pharmacokinetic Studies. J. Mass Spectrom. 2003, 38, 732–742. [Google Scholar] [CrossRef]
- Rajanikanth, M.; Madhusudanan, K.P.; Gupta, R.C. An HPLC-MS Method for Simultaneous Estimation of α,β-Arteether and its Metabolite Dihydroartemisinin, in Rat Plasma for Application to Pharmacokinetic Study. Biomed. Chromatogr. 2003, 17, 440–446. [Google Scholar] [CrossRef]
- Kotecka, B.M.; Rieckmann, K.H.; Davis, T.M.E.; Batty, K.T.; Ilett, K.F. Comparison of Bioassay and High Performance Liquid Chromatographic Assay of Artesunate and Dihydroartemisinin in Plasma. Acta Trop. 2003, 87, 371–375. [Google Scholar] [CrossRef]
- Batty, K.T.; Ilett, K.F.; Davis, T.M.E. Protein Binding and Alpha:Beta Anomer Ratio of Dihydroartemisinin in Vivo. Br. J. Clin. Pharmacol. 2004, 57, 529–533. [Google Scholar] [CrossRef]
- Naik, H.; Murry, D.J.; Kirsch, L.E.; Fleckenstein, L. Development and Validation of a High-Performance Liquid Chromatography-Mass Spectroscopy Assay for Determination of Artesunate and Dihydroartemisinin in Human Plasma. J. Chromatogr. B 2005, 816, 233–242. [Google Scholar] [CrossRef]
- Lai, C.-S.; Nair, N.K.; Mansor, S.M.; Olliaro, P.L.; Navaratnam, V. An Analytical Method with A Single Extraction Procedure and Two Separate High Performance Liquid Chromatographic Systems for the Determination of Artesunate, Dihydroartemisinin and Mefloquine in Human Plasma for Application in Clinical Pharmacological Studies of the Drug Combination. J. Chromatogr. B 2007, 857, 308–314. [Google Scholar] [CrossRef]
- Liu, Y.; Zeng, X.; Deng, Y.; Wang, L.; Feng, Y.; Yang, L.; Zhou, D. Application of a Liquid Chromatography/Tandem Mass Spectrometry Method for the Pharmacokinetic Study of Dihydroartemisinin Injectable Powder in Healthy Chinese Subjects. J. Chromatogr. B 2009, 877, 465–470. [Google Scholar] [CrossRef]
- Cabri, W.; D’Acquarica, I.; Simone, P.; Di Iorio, M.; Di Mattia, M.; Gasparrini, F.; Giorgi, F.; Mazzanti, A.; Pierini, M.; Quaglia, M.; Villani, C. Stereolability of Dihydroartemisinin: a Full Thermodynamic and Kinetic Investigation of the Epimerization by Experimental and Computational Techniques. unpublished work.
- International Pharmacopoeia, Monographs for Antimalarial Drugs, 4th ed.; WHO Press: Geneva, Switzerland, 2006; pp. 215–218.
- Li, Y.; Wu, Y.-L. An Over Four Millennium Story Behind Qinghaosu (Artemisinin) – A Fantastic Antimalarial Drug from a Traditional Chinese Herb. Curr. Med. Chem. 2003, 47, 2945–2964. [Google Scholar]
- Lee, S. Artemisinin, promising lead natural product for various drug developments. Mini Rev. Med. Chem. 2007, 7, 411–422. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
D’Acquarica, I.; Gasparrini, F.; Kotoni, D.; Pierini, M.; Villani, C.; Cabri, W.; Di Mattia, M.; Giorgi, F. Stereodynamic Investigation of Labile Stereogenic Centres in Dihydroartemisinin. Molecules 2010, 15, 1309-1323. https://doi.org/10.3390/molecules15031309
D’Acquarica I, Gasparrini F, Kotoni D, Pierini M, Villani C, Cabri W, Di Mattia M, Giorgi F. Stereodynamic Investigation of Labile Stereogenic Centres in Dihydroartemisinin. Molecules. 2010; 15(3):1309-1323. https://doi.org/10.3390/molecules15031309
Chicago/Turabian StyleD’Acquarica, Ilaria, Francesco Gasparrini, Dorina Kotoni, Marco Pierini, Claudio Villani, Walter Cabri, Michela Di Mattia, and Fabrizio Giorgi. 2010. "Stereodynamic Investigation of Labile Stereogenic Centres in Dihydroartemisinin" Molecules 15, no. 3: 1309-1323. https://doi.org/10.3390/molecules15031309
APA StyleD’Acquarica, I., Gasparrini, F., Kotoni, D., Pierini, M., Villani, C., Cabri, W., Di Mattia, M., & Giorgi, F. (2010). Stereodynamic Investigation of Labile Stereogenic Centres in Dihydroartemisinin. Molecules, 15(3), 1309-1323. https://doi.org/10.3390/molecules15031309