Synthesis and Characterization of Long-Chain Tartaric Acid Diamides as Novel Ceramide-Like Compounds

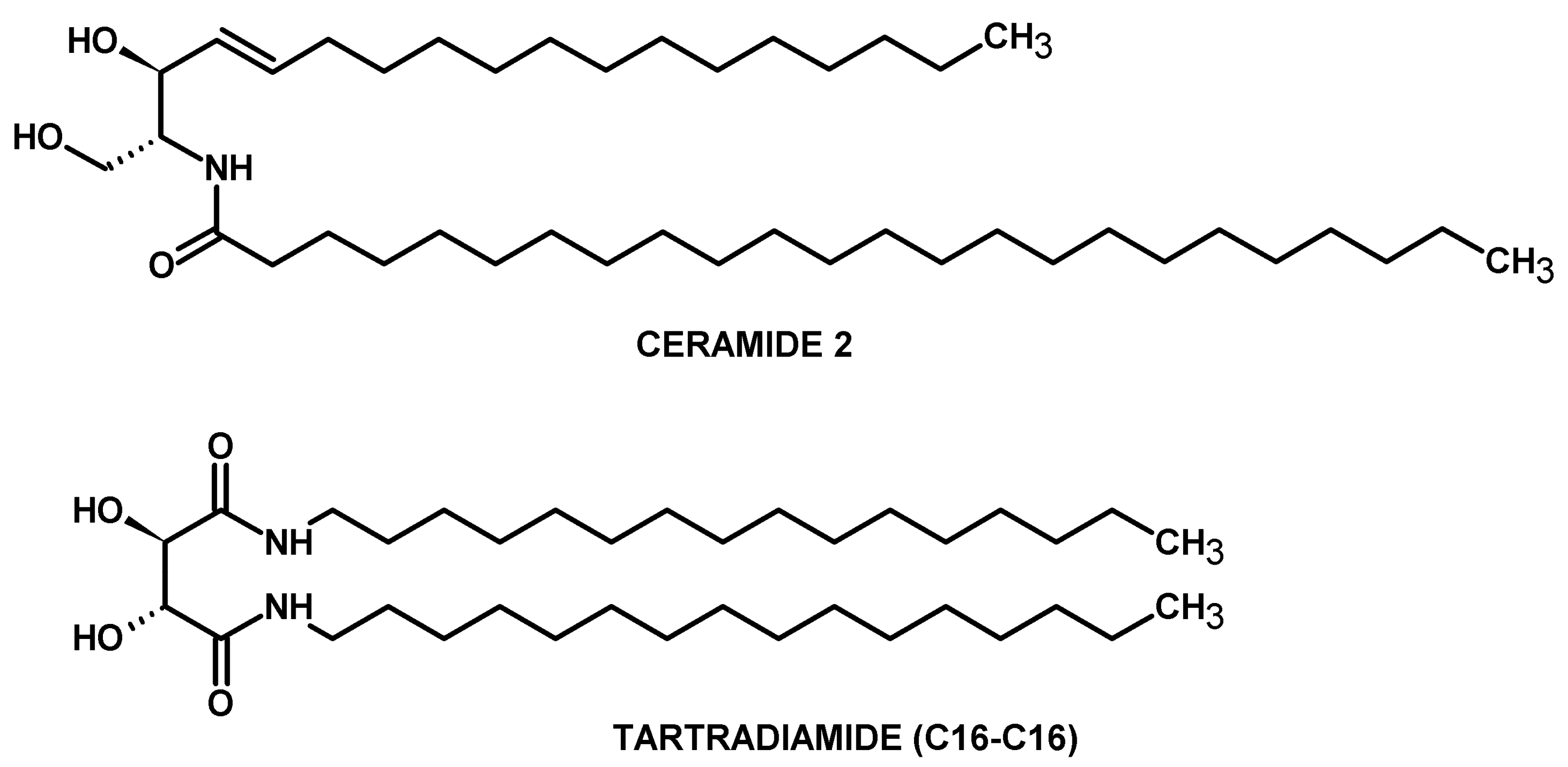{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
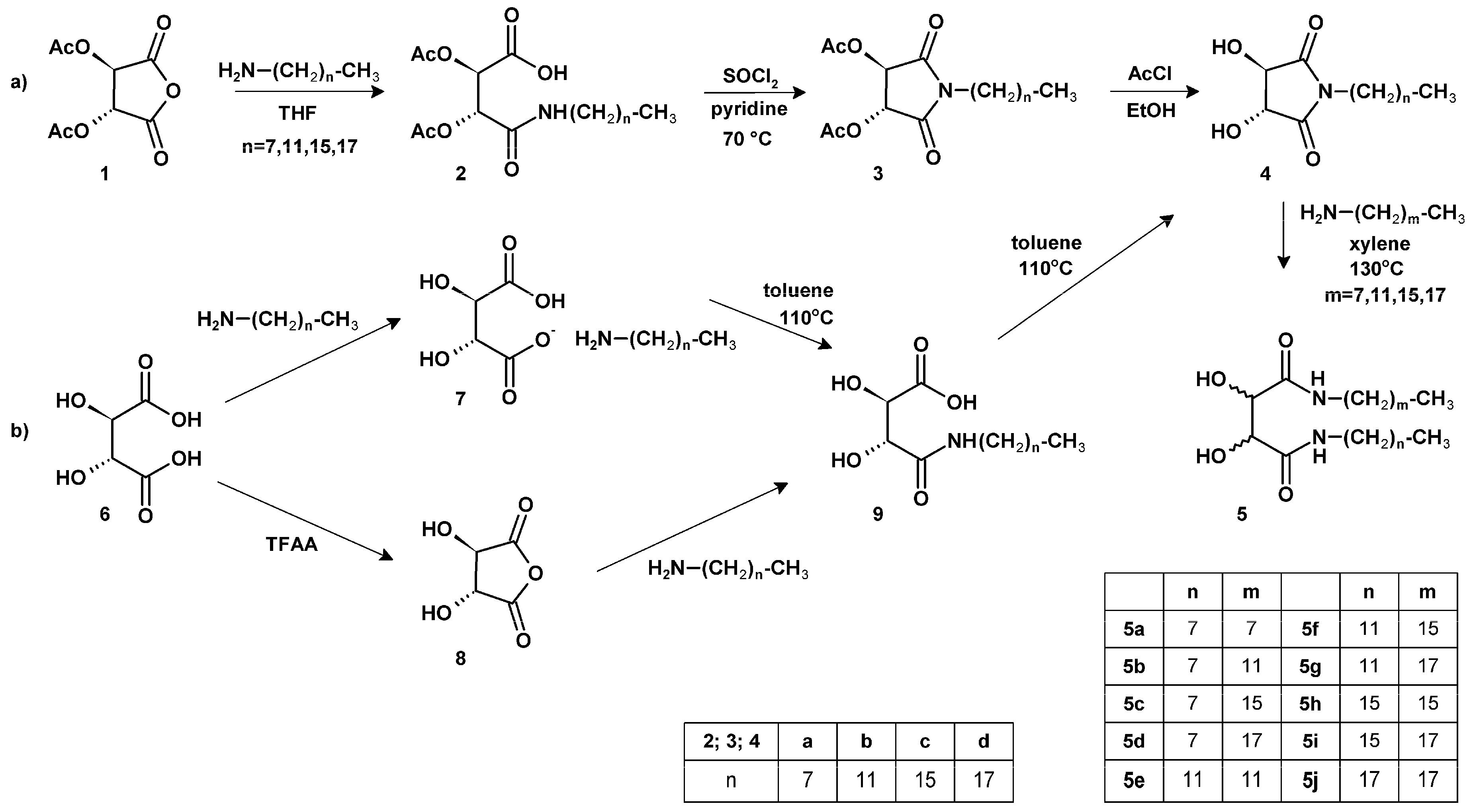
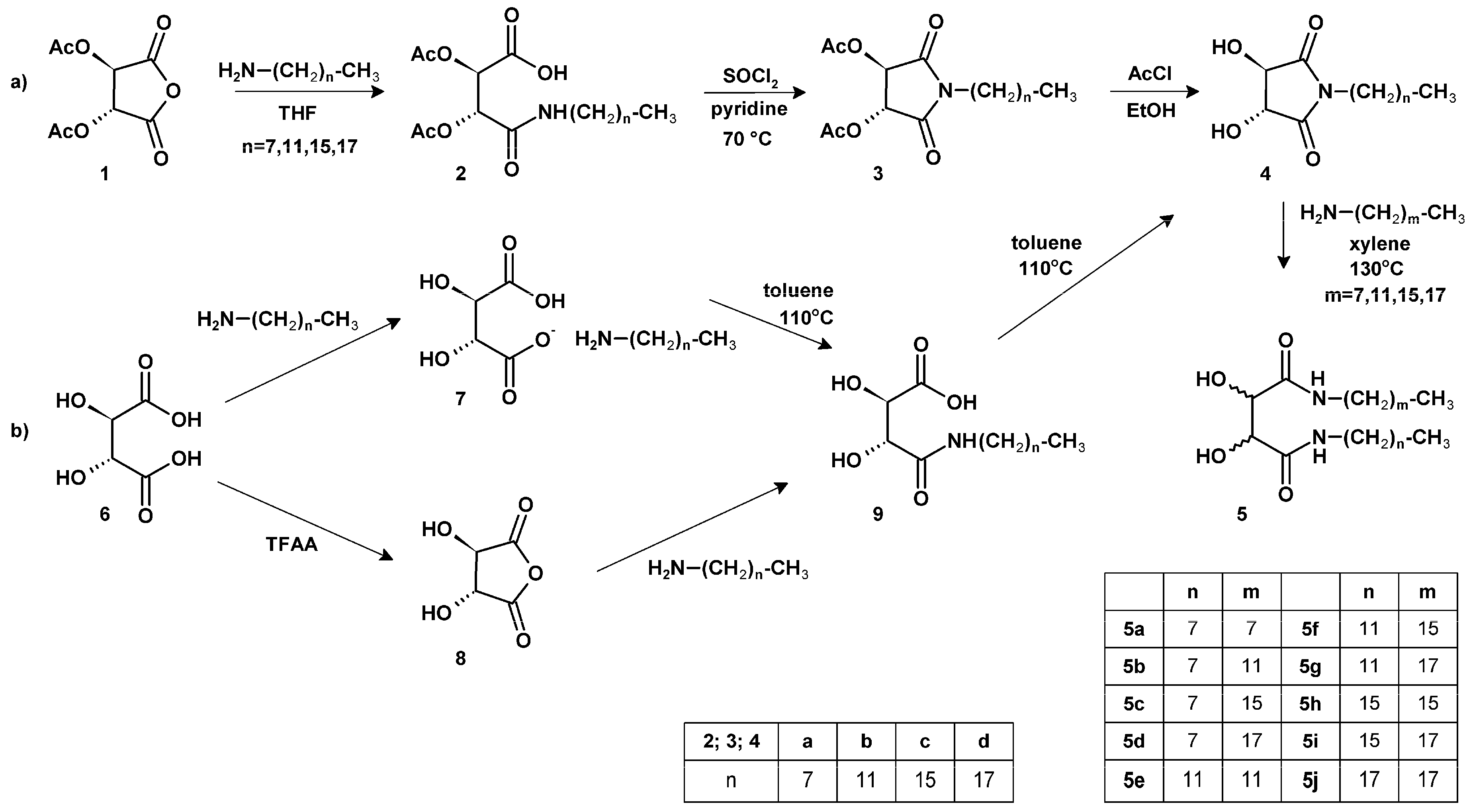
2.1. Synthesis of tartaric acid diamides
2.2. Characterization of tartaric acid diamides
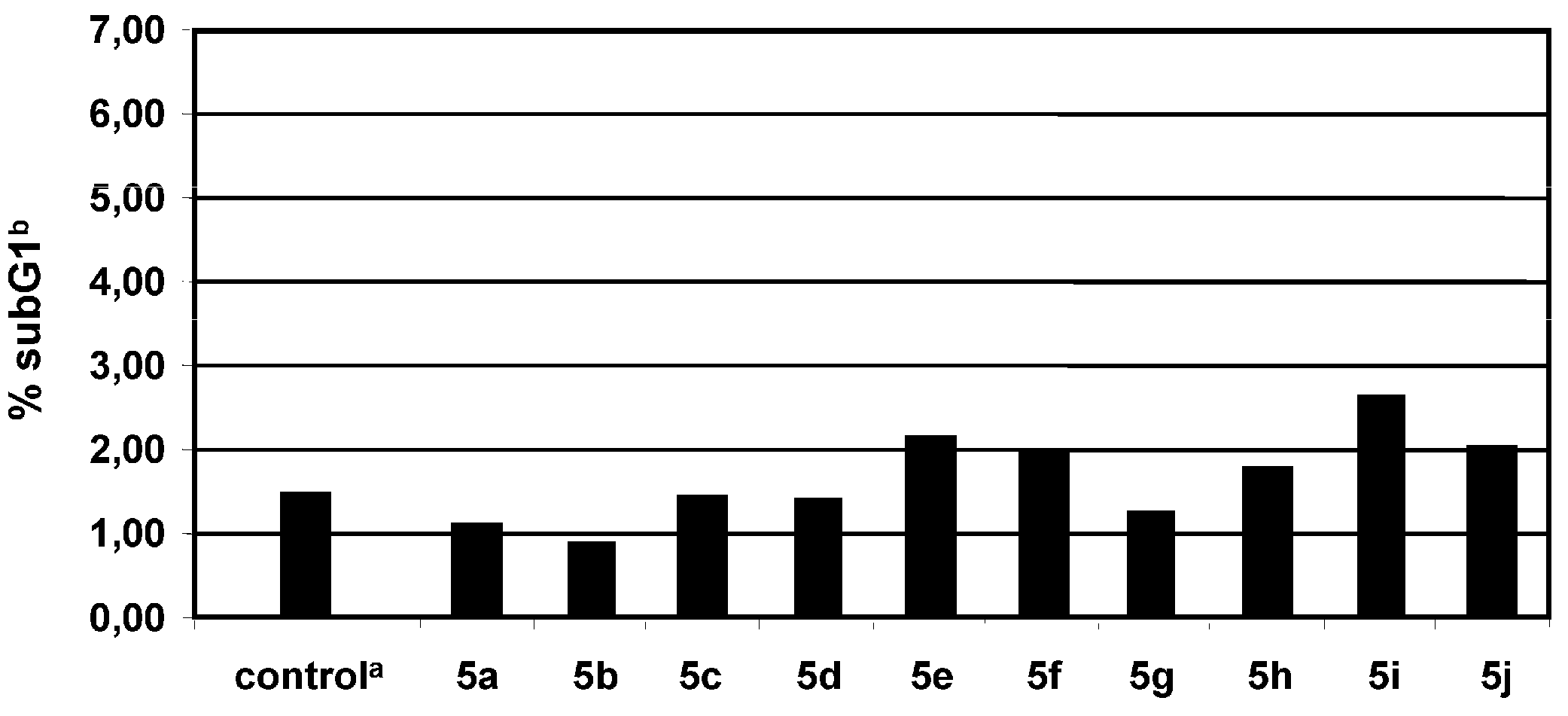
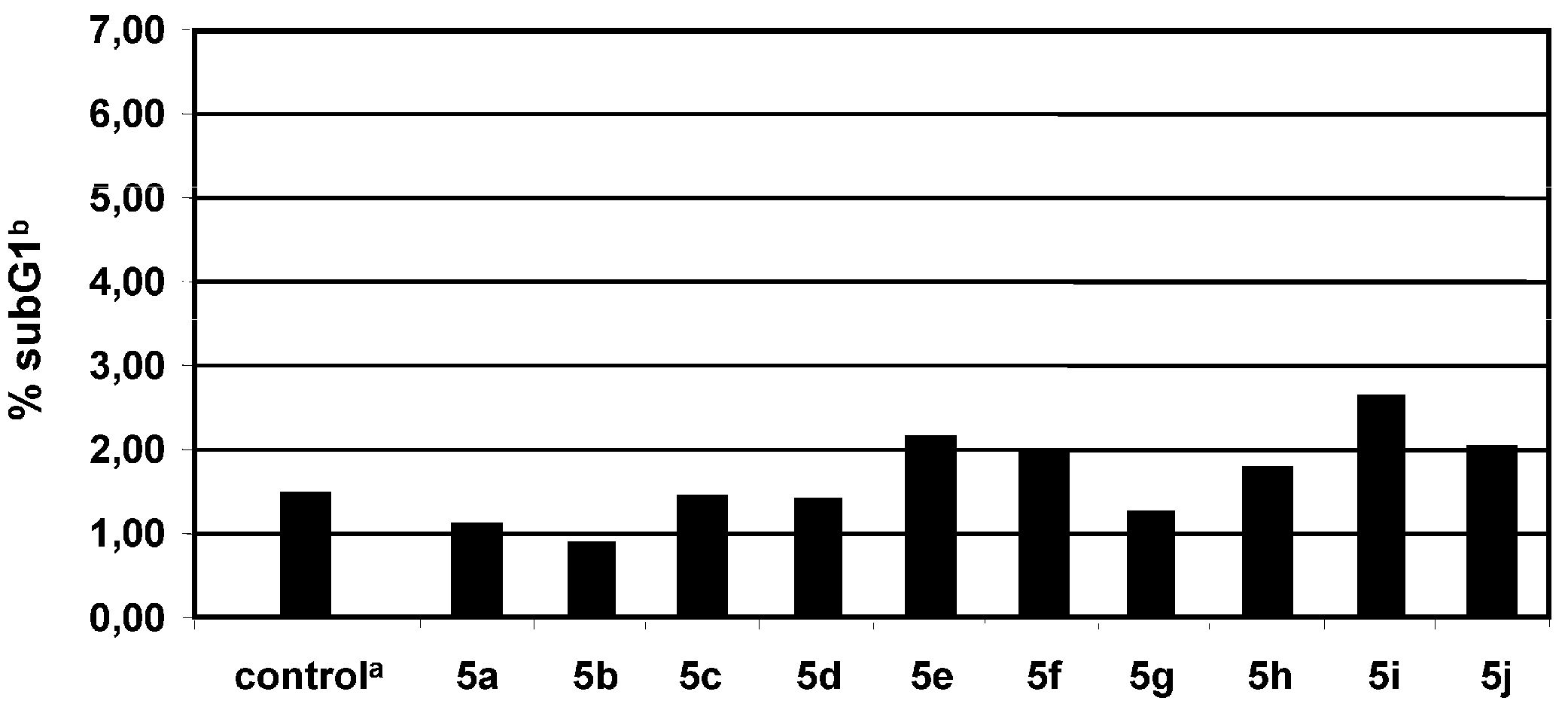
2.3. Proapoptotic effect of tartaric acid diamides
3. Experimental Section
3.1. General
3.2. General procedure for the synthesis of compounds 2a–d
3.3. General procedure for the synthesis of compounds 3a–d
3.4. General procedure for the synthesis of compounds 4a–d
3.5. General procedure for the synthesis of compounds 5a–j
4. Conclusions
Acknowledgments
References and Notes
- Hadgraft, J. Skin deep. Eur. J. Pharm. Biopharm. 2004, 58, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Frelichowska, J.; Bolzinger, M.A.; Pelletier, J.; Valour, J.P.; Chevalier, Y. Topical delivery of lipophilic drugs from o/w Pickering emulsions. Int. J. Pharm. 2009, 371, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Cross, S.E.; Innes, B.; Roberts, M.S.; Tsuzuki, T.; Robertson, T.A.; McCormick, P. Human skin penetration of sunscreen nanoparticles: In-vitro assessment of a novel micronized zinc oxide formulation. Skin Pharm. Physiol. 2007, 20, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Kansy, M.; Senner, F.; Gubernator, K. Physicochemical High Throughput Screening: Parallel Artificial Membrane Permeability Assay in the Description of Passive Absorption Processes. J. Med. Chem. 1998, 41, 1007–1010. [Google Scholar] [CrossRef] [PubMed]
- Bouwstra, J.A.; Honeywell-Nguyen, P.L.; Gooris, G.S.; Ponec, M. Structure of the skin barrier and its modulation by vesicular formulations. Prog. Lipid Res. 2003, 42, 1–36. [Google Scholar] [CrossRef]
- Nussbaumer, P. Medicinal chemistry aspects of drug targets in sphingolipid metabolism. ChemMedChem 2008, 3, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Delgado, A.; Casas, J.; Llibaria, A.; Abad, J.L.; Fabrias, G. Chemical tools to investigate sphingolipid metabolism and functions. ChemMedChem 2007, 2, 580–606. [Google Scholar] [CrossRef] [PubMed]
- Pettus, B.J.; Chalfant, C.E.; Hannun, Y.A. Ceramide in apoptosis: An overview and current perspectives. Biochim. Biophys. Acta 2002, 1585, 114–125. [Google Scholar] [CrossRef]
- Giroux, S.; Rubini, P.; Gerardin, C.; Selve, C.; Henry, B. Hydrophobic tartaric acid monoamides as complexing and tensioactive agents. New J. Chem. 2000, 24, 173–178. [Google Scholar] [CrossRef]
- Schitter, R.M.E.; Jocham, D.; Saf, R.; Mirtl, C.; Stelzer, F.; Hummel, K. Synthesis and characterization of a new chiral functional polymer. J. Mol. Catal. A-Chem. 1998, 133, 75–82. [Google Scholar] [CrossRef]
- Ilmarinen, K.; Kriis, K.; Paju, A.; Pehk, T.; Lopp, M. Synthesis of new N-tetrasubstituted derivatives of R,R – tartaric acid and their use as chiral ligands in oxidation catalysts. Proc. Estonian Acad. Sci. Chem. 2001, 50, 147–155. [Google Scholar]
- Synoradzki, L.; Ruskowski, P.; Bernas, U. Tartaric acid and its O-acyl derivatives. Part 1. Synthesis of tartaric acid and O-acyl tartaric acids and anhydrides. Org. Prep. Proc. Int. 2005, 37, 37–63. [Google Scholar] [CrossRef]
- Gonzalez, S.V.; Carlsen, P. Tartradiamide formation by thermolysis of tartaric acid with alkylamines. Tetrahedron Lett. 2008, 49, 3925–3926. [Google Scholar] [CrossRef]
- Platier-Royon, R.; Massicot, F.; Sudha, A.V.R.L.; Portella, C.; Dupont, L.; Mohamadou, A.; Aplincourt, M. Synthesis of functionalized bis-amides of L-(+)-tartaric acid and application as copper (II) ligands. C.R. Chim. 2004, 7, 119–123. [Google Scholar] [CrossRef]
- Gonzalez, S.V.; Carlsen, P. Tartaric acid amides by the gabriel route. Eur. J. Org. Chem. 2007, 21, 3495–3502. [Google Scholar] [CrossRef]
- Karasavvas, N.; Erukulla, R.K.; Bittman, R.; Lockshin, R.; Zakeri, Z. Stereospecific induction of apoptosis in U937 cells by N-octanoyl-sphingosine stereoisomers and N-octyl-sphingosine: The ceramide amide group is not required for apoptosis. Eur. J. Biochem. 1996, 236, 729–737. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.P.; Traganos, F.; Darzynkiewicz, Z. A selective procedure for DNA extraction from apoptotic cells applicable for gel electrophoresis and flow cytometry. Anal. Biochem. 1994, 218, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Imre, G.; Dunai, Zs.; Petak, I.; Mihalik, R. Cystein cathepsin and Hsp90 activities determine the balance between apoptotic and necrotic cell death pathways in caspase-compromised U937 cells. Biochim. Biophys. Acta 2007, 1173, 1546–1557. [Google Scholar] [CrossRef] [PubMed]
- Traganos, F. Mechanism of antitumor drug action assessed by cytometry. Methods Cell Biol. 2004, 75, 257–305. [Google Scholar] [PubMed]
- Sinkó, B.; Kökösi, J.; Avdeef, A.; Takács-Novák, K. A PAMPA study of the permeability-enhancing effect of new ceramide analogues. Chem. Biodiver. 2009, 11, 1867–1874. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 5a–j are available from the authors. |
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sinkó, B.; Pálfi, M.; Béni, S.; Kökösi, J.; Takács-Novák, K. Synthesis and Characterization of Long-Chain Tartaric Acid Diamides as Novel Ceramide-Like Compounds. Molecules 2010, 15, 824-833. https://doi.org/10.3390/molecules15020824
Sinkó B, Pálfi M, Béni S, Kökösi J, Takács-Novák K. Synthesis and Characterization of Long-Chain Tartaric Acid Diamides as Novel Ceramide-Like Compounds. Molecules. 2010; 15(2):824-833. https://doi.org/10.3390/molecules15020824
Chicago/Turabian StyleSinkó, Bálint, Melinda Pálfi, Szabolcs Béni, József Kökösi, and Krisztina Takács-Novák. 2010. "Synthesis and Characterization of Long-Chain Tartaric Acid Diamides as Novel Ceramide-Like Compounds" Molecules 15, no. 2: 824-833. https://doi.org/10.3390/molecules15020824
APA StyleSinkó, B., Pálfi, M., Béni, S., Kökösi, J., & Takács-Novák, K. (2010). Synthesis and Characterization of Long-Chain Tartaric Acid Diamides as Novel Ceramide-Like Compounds. Molecules, 15(2), 824-833. https://doi.org/10.3390/molecules15020824