The Nrf2 System as a Potential Target for the Development of Indirect Antioxidants

Abstract
:1. Introduction: Reactive Oxygen Species and Oxidative Stress
2. Cellular Antioxidant System
2.1. Directly Acting Antioxidant Proteins
2.2. Phase 2 Detoxifying Enzymes as Antioxidant Proteins
3. Regulation of Antioxidant Genes by Nrf2
3.1. Antioxidant Response Element (ARE)
3.2. Nrf2 Signaling for the Regulation of ARE-Driven Genes
3.3. Keap1 as a Protein Inhibitor of Nrf2
4. Functional Role of the Nrf2 System: From Comparative Animal Studies
5. Indirect Antioxidants Activating the Nrf2 System
6. Pleiotropic Effects of Small Molecule Nrf2 Activators
6.1. Anti-Inflammatory Function of Small Molecule Nrf2 Activators
6.2. Modulation of Proteasome Function by Nrf2 Activators: Implication in Protein Toxicity-Associated Diseases
6.3. Lipid Metabolism and Nrf2 Activators.
6.4. Liver Regeneration and Nrf2
7. Conclusions
Acknowledgements
References
- Fridovich, I. Superoxide radical and superoxide dismutases. Annu. Rev. Biochem. 1995, 64, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Gutteridge, J. Free Radicals in Biology and Medicine, 3rd ed.; Oxford University Press: New York, NY, USA, 2001; pp. 82–95. [Google Scholar]
- Steinbrenner, H.; Sies, H. Protection against reactive oxygen species by selenoproteins. Biochim. Biophys. Acta 2009, 1790, 1478–1485. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Gutteridge, J. Free Radicals in Biology and Medicine, 3rd ed.; Oxford University Press: New York, NY, USA, 2001; pp. 262–316. [Google Scholar]
- Szatrowski, T.P.; Nathan, C.F. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991, 51, 794–798. [Google Scholar] [PubMed]
- Fita, I.; Rossmann, M.G. The active center of catalase. J. Mol. Biol. 1985, 185, 21–37. [Google Scholar] [CrossRef]
- Schneider, Y.; Vincent, F.; Duranton, B.; Badolo, L.; Gosse, F.; Bergmann, C.; Seiler, N.; Raul, F. Anti-proliferative effect of resveratrol, a natural component of grapes and wine, on human colonic cancer cells. Cancer Lett. 2000, 158, 85–91. [Google Scholar] [CrossRef]
- Brigelius-Flohe, R. Tissue-specific functions of individual glutathione peroxidases. Free Radic. Biol. Med. 1999, 27, 951–965. [Google Scholar] [CrossRef]
- Forgione, M.A.; Cap, A.; Liao, R.; Moldovan, N.I.; Eberhardt, R.T.; Lim, C.C.; Jones, J.; Goldschmidt-Clermont, P.J.; Loscalzo, J. Heterozygous cellular glutathione peroxidase deficiency in the mouse: abnormalities in vascular and cardiac function and structure. Circulation 2002, 106, 1154–1158. [Google Scholar] [CrossRef] [PubMed]
- Meister, A.; Anderson, M.E. Glutathione. Annu. Rev. Biochem. 1983, 52, 711–760. [Google Scholar] [CrossRef] [PubMed]
- Rahman, I.; Biswas, S.K.; Jimenez, L.A.; Torres, M.; Forman, H.J. Glutathione, stress responses, and redox signaling in lung inflammation. Antioxid. Redox. Signal 2005, 7, 42–59. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.S.; Chang, L.S.; Anderson, M.E.; Meister, A. Catalytic and regulatory properties of the heavy subunit of rat kidney gamma-glutamylcysteine synthetase. J. Biol. Chem. 1993, 268, 19675–19680. [Google Scholar] [PubMed]
- Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol. Lung. Cell Mol. Physiol. 2000, 279, L1005–1028. [Google Scholar] [CrossRef] [PubMed]
- Conney, A.H. Induction of drug-metabolizing enzymes: a path to the discovery of multiple cytochromes P450. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Talalay, P. Direct and indirect antioxidant properties of inducers of cytoprotective proteins. Mol. Nutr. Food Res. 2008, 52 (Suppl. 1), S128–S138. [Google Scholar] [CrossRef]
- Hayes, J.D.; McMahon, M.; Chowdhry, S.; Dinkova-Kostova, A.T. Cancer Chemoprevention Mechanisms Mediated Through the Keap1-Nrf2 Pathway. Antioxid. Redox. Signal 2010, in press. [Google Scholar] [CrossRef] [PubMed]
- Jernstrom, B.; Dock, L.; Martinez, M. Metabolic activation of benzoa.pyrene-7,8-dihydrodiol and benzoa.pyrene-7,8-dihydrodiol-9,10-epoxide to protein-binding products and the inhibitory effect of glutathione and cysteine. Carcinogenesis 1984, 5, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Flanagan, J.U.; Jowsey, I.R. Glutathione transferases. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 51–88. [Google Scholar] [CrossRef] [PubMed]
- Henderson, C.J.; Smith, A.G.; Ure, J.; Brown, K.; Bacon, E.J.; Wolf, C.R. Increased skin tumorigenesis in mice lacking pi class glutathione S-transferases. Proc. Natl. Acad. Sci. USA 1998, 95, 5275–5280. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, K.J.; Henderson, C.J.; Wang, X.J.; Vassieva, O.; Carrie, D.; Farmer, P.B.; Gaskell, M.; Park, K.; Wolf, C.R. Glutathione transferase pi plays a critical role in the development of lung carcinogenesis following exposure to tobacco-related carcinogens and urethane. Cancer Res. 2007, 67, 9248–9257. [Google Scholar] [CrossRef] [PubMed]
- Nazar-Stewart, V.; Vaughan, T.L.; Burt, R.D.; Chen, C.; Berwick, M.; Swanson, G.M. Glutathione S-transferase M1 and susceptibility to nasopharyngeal carcinoma. Cancer Epidemiol. Biomarkers Prev. 1999, 8, 547–551. [Google Scholar]
- Tukey, R.H.; Strassburg, C.P. Human UDP-glucuronosyltransferases: metabolism, expression, and disease. Annu. Rev. Pharmacol. Toxicol. 2000, 40, 581–616. [Google Scholar] [CrossRef] [PubMed]
- Vienneau, D.S.; DeBoni, U.; Wells, P.G. Potential genoprotective role for UDP-glucuronosyltransferases in chemical carcinogenesis: initiation of micronuclei by benzo(a)pyrene and benzo(e)pyrene in UDP-glucuronosyltransferase-deficient cultured rat skin fibroblasts. Cancer Res. 1995, 55, 1045–1051. [Google Scholar] [PubMed]
- Leung, H.Y.; Wang, Y.; Leung, L.K. Differential effect of over-expressing UGT1A1 and CYP1A1 on xenobiotic assault in MCF-7 cells. Toxicology 2007, 242, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Nioi, P.; Hayes, J.D. Contribution of NAD(P)H:quinone oxidoreductase 1 to protection against carcinogenesis, and regulation of its gene by the Nrf2 basic-region leucine zipper and the arylhydrocarbon receptor basic helix-loop-helix transcription factors. Mutat. Res. 2004, 555, 149–171. [Google Scholar] [CrossRef] [PubMed]
- Beyer, R.E.; Segura-Aguilar, J.; Di Bernardo, S.; Cavazzoni, M.; Fato, R.; Fiorentini, D.; Galli, M.C.; Setti, M.; Landi, L.; Lenaz, G. The role of DT-diaphorase in the maintenance of the reduced antioxidant form of coenzyme Q in membrane systems. Proc. Natl. Acad. Sci. USA 1996, 93, 2528–2532. [Google Scholar] [CrossRef] [PubMed]
- Landi, L.; Fiorentini, D.; Galli, M.C.; Segura-Aguilar, J.; Beyer, R.E. DT-Diaphorase maintains the reduced state of ubiquinones in lipid vesicles thereby promoting their antioxidant function. Free Radic. Biol. Med. 1997, 22, 329–335. [Google Scholar] [CrossRef]
- Siegel, D.; Bolton, E.M.; Burr, J.A.; Liebler, D.C.; Ross, D. The reduction of alpha-tocopherolquinone by human NAD(P)H: quinone oxidoreductase: the role of alpha-tocopherolhydroquinone as a cellular antioxidant. Mol. Pharmacol. 1997, 52, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Long, D.J., 2nd; Waikel, R.L.; Wang, X.J.; Perlaky, L.; Roop, D.R.; Jaiswal, A.K. NAD(P)H:quinone oxidoreductase 1 deficiency increases susceptibility to benzo(a)pyrene-induced mouse skin carcinogenesis. Cancer Res. 2000, 60, 5913–5915. [Google Scholar] [PubMed]
- Traver, R.D.; Siegel, D.; Beall, H.D.; Phillips, R.M.; Gibson, N.W.; Franklin, W.A.; Ross, D. Characterization of a polymorphism in NAD(P)H: quinone oxidoreductase (DT-diaphorase). Br. J. Cancer 1997, 75, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Wiemels, J.L.; Pagnamenta, A.; Taylor, G.M.; Eden, O.B.; Alexander, F.E.; Greaves, M.F. A lack of a functional NAD(P)H:quinone oxidoreductase allele is selectively associated with pediatric leukemias that have MLL fusions. United Kingdom Childhood Cancer Study Investigators. Cancer Res. 1999, 59, 4095–4099. [Google Scholar] [PubMed]
- Lafuente, M.J.; Casterad, X.; Trias, M.; Ascaso, C.; Molina, R.; Ballesta, A.; Zheng, S.; Wiencke, J.K.; Lafuente, A. NAD(P)H:quinone oxidoreductase-dependent risk for colorectal cancer and its association with the presence of K-ras mutations in tumors. Carcinogenesis 2000, 21, 1813–1819. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.H.; Li, Y.; Wang, R.; Sarbia, M.; Guo, W.; Wen, D.G.; Wei, L.Z.; Chen, Z.F.; Kuang, G.; Zhang, L.W.; He, M.; Wu, M.L.; Wang, S.J. The NAD(P)H: quinone oxidoreductase 1 C609T polymorphism and susceptibility to esophageal cancer. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2003, 20, 544–546. [Google Scholar] [PubMed]
- Loboda, A.; Jazwa, A.; Grochot-Przeczek, A.; Rutkowski, A.J.; Cisowski, J.; Agarwal, A.; Jozkowicz, A.; Dulak, J. Heme oxygenase-1 and the vascular bed: from molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal 2008, 10, 1767–1812. [Google Scholar] [CrossRef] [PubMed]
- Prestera, T.; Talalay, P.; Alam, J.; Ahn, Y.I.; Lee, P.J.; Choi, A.M. Parallel induction of heme oxygenase-1 and chemoprotective phase 2 enzymes by electrophiles and antioxidants: regulation by upstream antioxidant-responsive elements (ARE). Mol. Med. 1995, 1, 827–837. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.M.; Arosio, P. The ferritins: molecular properties, iron storage function and cellular regulation. Biochim. Biophys. Acta 1996, 1275, 161–203. [Google Scholar] [CrossRef]
- Tsuji, Y.; Ayaki, H.; Whitman, S.P.; Morrow, C.S.; Torti, S.V.; Torti, F.M. Coordinate transcriptional and translational regulation of ferritin in response to oxidative stress. Mol. Cell Biol. 2000, 20, 5818–5827. [Google Scholar] [CrossRef] [PubMed]
- Rushmore, T.H.; Pickett, C.B. Transcriptional regulation of the rat glutathione S-transferase Ya subunit gene. Characterization of a xenobiotic-responsive element controlling inducible expression by phenolic antioxidants. J. Biol. Chem. 1990, 265, 14648–14653. [Google Scholar] [PubMed]
- Rushmore, T.H.; Morton, M.R.; Pickett, C.B. The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J. Biol. Chem. 1991, 266, 11632–11639. [Google Scholar] [PubMed]
- Favreau, L.V.; Pickett, C.B. Transcriptional regulation of the rat NAD(P)H:quinone reductase gene. Identification of regulatory elements controlling basal level expression and inducible expression by planar aromatic compounds and phenolic antioxidants. J. Biol. Chem. 1991, 266, 4556–4561. [Google Scholar] [PubMed]
- Nioi, P.; McMahon, M.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Identification of a novel Nrf2-regulated antioxidant response element (ARE) in the mouse NAD(P)H:quinone oxidoreductase 1 gene: reassessment of the ARE consensus sequence. Biochem. J. 2003, 374, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, A.K. Human NAD(P)H:quinone oxidoreductase (NQO1) gene structure and induction by dioxin. Biochemistry 1991, 30, 10647–10653. [Google Scholar] [CrossRef] [PubMed]
- Erickson, A.M.; Nevarea, Z.; Gipp, J.J.; Mulcahy, R.T. Identification of a variant antioxidant response element in the promoter of the human glutamate-cysteine ligase modifier subunit gene. Revision of the ARE consensus sequence. J. Biol. Chem. 2002, 277, 30730–30737. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy, R.T.; Gipp, J.J. Identification of a putative antioxidant response element in the 5’-flanking region of the human gamma-glutamylcysteine synthetase heavy subunit gene. Biochem. Biophys. Res. Commun. 1995, 209, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy, R.T.; Wartman, M.A.; Bailey, H.H.; Gipp, J.J. Constitutive and beta-naphthoflavone-induced expression of the human gamma-glutamylcysteine synthetase heavy subunit gene is regulated by a distal antioxidant response element/TRE sequence. J. Biol. Chem. 1997, 272, 7445–7454. [Google Scholar] [CrossRef] [PubMed]
- Wasserman, W.W.; Fahl, W.E. Functional antioxidant responsive elements. Proc. Natl. Acad. Sci. USA 1997, 94, 5361–5366. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; Yamamoto, M.; Nabeshima, Y. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Yamamoto, M. Molecular mechanisms activating the Nrf2-Keap1 pathway of antioxidant gene regulation. Antioxid. Redox Signal 2005, 7, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Kwak, M.K.; Egner, P.A.; Dolan, P.M.; Ramos-Gomez, M.; Groopman, J.D.; Itoh, K.; Yamamoto, M.; Kensler, T.W. Role of phase 2 enzyme induction in chemoprotection by dithiolethiones. Mutat. Res. 2001, 480-481, 305–315. [Google Scholar] [CrossRef]
- Venugopal, R.; Jaiswal, A.K. Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H:quinone oxidoreductase1 gene. Proc. Natl. Acad. Sci. USA 1996, 93, 14960–14965. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Gomez, M.; Kwak, M.K.; Dolan, P.M.; Itoh, K.; Yamamoto, M.; Talalay, P.; Kensler, T.W. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc. Natl. Acad. Sci. USA 2001, 98, 3410–3415. [Google Scholar] [CrossRef] [PubMed]
- Alam, J.; Stewart, D.; Touchard, C.; Boinapally, S.; Choi, A.M.; Cook, J.L. Nrf2, a Cap’n’Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J. Biol. Chem. 1999, 274, 26071–26078. [Google Scholar] [CrossRef] [PubMed]
- He, C.H.; Gong, P.; Hu, B.; Stewart, D.; Choi, M.E.; Choi, A.M.; Alam, J. Identification of activating transcription factor 4 (ATF4) as an Nrf2-interacting protein. Implication for heme oxygenase-1 gene regulation. J. Biol. Chem. 2001, 276, 20858–20865. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, K.; Kataoka, K.; Itoh, K.; Hayashi, N.; Nishizawa, M.; Yamamoto, M. Regulation of transcription by dimerization of erythroid factor NF-E2 p45 with small Maf proteins. Nature 1994, 367, 568–572. [Google Scholar] [CrossRef] [PubMed]
- Katsuoka, F.; Motohashi, H.; Ishii, T.; Aburatani, H.; Engel, J.D.; Yamamoto, M. Genetic evidence that small maf proteins are essential for the activation of antioxidant response element-dependent genes. Mol. Cell Biol. 2005, 25, 8044–8051. [Google Scholar] [CrossRef] [PubMed]
- Motohashi, H.; O’Connor, T.; Katsuoka, F.; Engel, J.D.; Yamamoto, M. Integration and diversity of the regulatory network composed of Maf and CNC families of transcription factors. Gene 2002, 294, 1–12. [Google Scholar] [CrossRef]
- Kwak, M.K.; Wakabayashi, N.; Itoh, K.; Motohashi, H.; Yamamoto, M.; Kensler, T.W. Modulation of gene expression by cancer chemopreventive dithiolethiones through the Keap1-Nrf2 pathway. Identification of novel gene clusters for cell survival. J. Biol. Chem. 2003, 278, 8135–8145. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Xu, C.; Shen, G.; Jain, M.R.; Khor, T.O.; Gopalkrishnan, A.; Lin, W.; Reddy, B.; Chan, J.Y.; Kong, A.N. Gene expression profiles induced by cancer chemopreventive isothiocyanate sulforaphane in the liver of C57BL/6J mice and C57BL/6J/Nrf2 (-/-) mice. Cancer Lett. 2006, 243, 170–192. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Xu, C.; Shen, G.; Jain, M.R.; Khor, T.O.; Gopalkrishnan, A.; Lin, W.; Reddy, B.; Chan, J.Y.; Kong, A.N. Identification of Nrf2-regulated genes induced by chemopreventive isothiocyanate PEITC by oligonucleotide microarray. Life Sci. 2006, 79, 1944–1955. [Google Scholar] [CrossRef] [PubMed]
- Kwak, M.K.; Kensler, T.W. Targeting NRF2 signaling for cancer chemoprevention. Toxicol. Appl. Pharmacol. 2010, 244, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Calkins, M.J.; Chan, K.; Kan, Y.W.; Johnson, J.A. Identification of the NF-E2-related factor-2-dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. J. Biol. Chem. 2003, 278, 12029–12038. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.; Xu, C.; Shen, G.; Hebbar, V.; Gopalakrishnan, A.; Hu, R.; Jain, M.R.; Lin, W.; Keum, Y.S.; Liew, C.; Chan, J.Y.; Kong, A.N. Pharmacogenomics of phenolic antioxidant butylated hydroxyanisole (BHA) in the small intestine and liver of Nrf2 knockout and C57BL/6J mice. Pharm. Res. 2006, 23, 2621–2637. [Google Scholar] [CrossRef] [PubMed]
- Thimmulappa, R.K.; Mai, K.H.; Srisuma, S.; Kensler, T.W.; Yamamoto, M.; Biswal, S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 2002, 62, 5196–5203. [Google Scholar] [PubMed]
- Yates, M.S.; Kwak, M.K.; Egner, P.A.; Groopman, J.D.; Bodreddigari, S.; Sutter, T.R.; Baumgartner, K.J.; Roebuck, B.D.; Liby, K.T.; Yore, M.M.; Honda, T.; Gribble, G.W.; Sporn, M.B.; Kensler, T.W. Potent protection against aflatoxin-induced tumorigenesis through induction of Nrf2-regulated pathways by the triterpenoid 1-2-cyano-3-,12-dioxooleana-1,9(11)-dien-28-oyl.imidazole. Cancer Res. 2006, 66, 2488–2494. [Google Scholar] [CrossRef] [PubMed]
- Yates, M.S.; Tran, Q.T.; Dolan, P.M.; Osburn, W.O.; Shin, S.; McCulloch, C.C.; Silkworth, J.B.; Taguchi, K.; Yamamoto, M.; Williams, C.R.; Liby, K.T.; Sporn, M.B.; Sutter, T.R.; Kensler, T.W. Genetic versus Chemoprotective Activation of Nrf2 Signaling: Overlapping yet Distinct Gene Expression Profiles between Keap1 Knockout and Triterpenoid Treated Mice. Carcinogenesis 2009, 2488–2494. [Google Scholar] [CrossRef] [PubMed]
- Rangasamy, T.; Cho, C.Y.; Thimmulappa, R.K.; Zhen, L.; Srisuma, S.S.; Kensler, T.W.; Yamamoto, M.; Petrache, I.; Tuder, R.M.; Biswal, S. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J. Clin. Invest. 2004, 114, 1248–1259. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, A.K.; McMahon, M.; Plummer, S.M.; Higgins, L.G.; Penning, T.M.; Igarashi, K.; Hayes, J.D. Characterization of the cancer chemopreventive NRF2-dependent gene battery in human keratinocytes: demonstration that the KEAP1-NRF2 pathway, and not the BACH1-NRF2 pathway, controls cytoprotection against electrophiles as well as redox-cycling compounds. Carcinogenesis 2009, 30, 1571–1580. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lee, J.M.; Johnson, J.A. Microarray analysis reveals an antioxidant responsive element-driven gene set involved in conferring protection from an oxidative stress-induced apoptosis in IMR-32 cells. J. Biol. Chem. 2002, 277, 388–394. [Google Scholar] [CrossRef] [PubMed]
- Harvey, C.J.; Thimmulappa, R.K.; Singh, A.; Blake, D.J.; Ling, G.; Wakabayashi, N.; Fujii, J.; Myers, A.; Biswal, S. Nrf2-regulated glutathione recycling independent of biosynthesis is critical for cell survival during oxidative stress. Free Radic. Biol. Med. 2009, 46, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Reisman, S.A.; Yeager, R.L.; Yamamoto, M.; Klaassen, C.D. Increased Nrf2 activation in livers from Keap1-knockdown mice increases expression of cytoprotective genes that detoxify electrophiles more than those that detoxify reactive oxygen species. Toxicol. Sci. 2009, 108, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Rangasamy, T.; Thimmulappa, R.K.; Lee, H.; Osburn, W.O.; Brigelius-Flohe, R.; Kensler, T.W.; Yamamoto, M.; Biswal, S. Glutathione peroxidase 2, the major cigarette smoke-inducible isoform of GPX in lungs, is regulated by Nrf2. Am. J. Respir. Cell Mol. Biol. 2006, 35, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.Y.; Reddy, S.P.; Debiase, A.; Yamamoto, M.; Kleeberger, S.R. Gene expression profiling of NRF2-mediated protection against oxidative injury. Free Radic. Biol. Med. 2005, 38, 325–343. [Google Scholar] [CrossRef] [PubMed]
- Okawa, H.; Motohashi, H.; Kobayashi, A.; Aburatani, H.; Kensler, T.W.; Yamamoto, M. Hepatocyte-specific deletion of the keap1 gene activates Nrf2 and confers potent resistance against acute drug toxicity. Biochem. Biophys. Res. Commun. 2006, 339, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Banning, A.; Deubel, S.; Kluth, D.; Zhou, Z.; Brigelius-Flohe, R. The GI-GPx gene is a target for Nrf2. Mol. Cell Biol. 2005, 25, 4914–4923. [Google Scholar] [CrossRef] [PubMed]
- Habeos, I.G.; Ziros, P.G.; Chartoumpekis, D.; Psyrogiannis, A.; Kyriazopoulou, V.; Papavassiliou, A.G. Simvastatin activates Keap1/Nrf2 signaling in rat liver. J Mol. Med. 2008, 86, 1279–1285. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, I.; Mo, Y.; Gao, L.; Kazi, A.; Fisher, A.B.; Feinstein, S.I. Oxidant stress stimulates expression of the human peroxiredoxin 6 gene by a transcriptional mechanism involving an antioxidant response element. Free Radic. Biol. Med. 2009, 46, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.T.; Yen, G.C. Effect of sulforaphane on metallothionein expression and induction of apoptosis in human hepatoma HepG2 cells. Carcinogenesis 2005, 26, 2138–2148. [Google Scholar] [CrossRef] [PubMed]
- Mahaffey, C.M.; Zhang, H.; Rinna, A.; Holland, W.; Mack, P.C.; Forman, H.J. Multidrug-resistant protein-3 gene regulation by the transcription factor Nrf2 in human bronchial epithelial and non-small-cell lung carcinoma. Free Radic. Biol. Med. 2009, 46, 1650–1657. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Igarashi, K.; Hayashi, N.; Nishizawa, M.; Yamamoto, M. Cloning and characterization of a novel erythroid cell-derived CNC family transcription factor heterodimerizing with the small Maf family proteins. Mol. Cell Biol. 1995, 15, 4184–4193. [Google Scholar] [CrossRef] [PubMed]
- Katoh, Y.; Itoh, K.; Yoshida, E.; Miyagishi, M.; Fukamizu, A.; Yamamoto, M. Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells 2001, 6, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Nioi, P.; Nguyen, T.; Sherratt, P.J.; Pickett, C.B. The carboxy-terminal Neh3 domain of Nrf2 is required for transcriptional activation. Mol. Cell Biol. 2005, 25, 10895–10906. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Xue, F.; Cooley, L. kelch encodes a component of intercellular bridges in Drosophila egg chambers. Cell 1993, 72, 681–693. [Google Scholar] [CrossRef]
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.; Thomas, N.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Dimerization of substrate adaptors can facilitate cullin-mediated ubiquitylation of proteins by a "tethering" mechanism: a two-site interaction model for the Nrf2-Keap1 complex. J. Biol. Chem. 2006, 281, 24756–24768. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.D.; Lo, S.C.; Cross, J.V.; Templeton, D.J.; Hannink, M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol. Cell Biol. 2004, 24, 10941–10953. [Google Scholar] [CrossRef] [PubMed]
- Tong, K.I.; Kobayashi, A.; Katsuoka, F.; Yamamoto, M. Two-site substrate recognition model for the Keap1-Nrf2 system: a hinge and latch mechanism. Biol Chem 2006, 387, 1311–1320. [Google Scholar] [CrossRef] [PubMed]
- Tong, K.I.; Padmanabhan, B.; Kobayashi, A.; Shang, C.; Hirotsu, Y.; Yokoyama, S.; Yamamoto, M. Different electrostatic potentials define ETGE and DLG motifs as hinge and latch in oxidative stress response. Mol. Cell Biol. 2007, 27, 7511–7521. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, B.; Tong, K.I.; Ohta, T.; Nakamura, Y.; Scharlock, M.; Ohtsuji, M.; Kang, M.I.; Kobayashi, A.; Yokoyama, S.; Yamamoto, M. Structural basis for defects of Keap1 activity provoked by its point mutations in lung cancer. Mol. Cell 2006, 21, 689–700. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, N.; Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Kang, M.L.; Kobayashi, A.; Yamamoto, M.; Kensler, T.W.; Talalay, P. Protection against electrophile and oxidant stress by induction of the phase 2 response: Fate of cysteines of the Keap1 sensor modified by inducers. Proc. Natl. Acad. Sci. USA 2004, 101, 2040–2045. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Cole, R.N.; Itoh, K.; Wakabayashi, N.; Katoh, Y.; Yamamoto, M.; Talalay, P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. USA 2002, 99, 11908–11913. [Google Scholar] [CrossRef] [PubMed]
- Eggler, A.L.; Liu, G.; Pezzuto, J.M.; van Breemen, R.B.; Mesecar, A.D. Modifying specific cysteines of the electrophile-sensing human Keap1 protein is insufficient to disrupt binding to the Nrf2 domain Neh2. Proc. Natl. Acad. Sci. USA 2005, 102, 10070–10075. [Google Scholar] [CrossRef] [PubMed]
- Eggler, A.L.; Luo, Y.; van Breemen, R.B.; Mesecar, A.D. Identification of the highly reactive cysteine 151 in the chemopreventive agent-sensor Keap1 protein is method-dependent. Chem. Res. Toxicol. 2007, 20, 1878–1884. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.D.; Hannink, M. Distinct cysteine residues in keap1 are required for keap1-dependent ubiquitination of nrf2 and for stabilization of nrf2 by chemopreventive agents and oxidative stress. Mol. Cell Biol. 2003, 23, 8137–8151. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.; Kan, Y.W. Nrf2 is essential for protection against acute pulmonary injury in mice. Proc. Natl. Acad. Sci. USA 1999, 96, 12731–12736. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.Y.; Jedlicka, A.E.; Reddy, S.P.; Kensler, T.W.; Yamamoto, M.; Zhang, L.Y.; Kleeberger, S.R. Role of NRF2 in protection against hyperoxic lung injury in mice. Am. J. Respir. Cell Mol. Biol. 2002, 26, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.Y.; Jedlicka, A.E.; Reddy, S.P.; Zhang, L.Y.; Kensler, T.W.; Kleeberger, S.R. Linkage analysis of susceptibility to hyperoxia. Nrf2 is a candidate gene. Am. J. Respir. Cell Mol. Biol. 2002, 26, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.M.; Manandhar, S.; Lee, H.R.; Park, H.M.; Kwak, M.K. Role of the Nrf2-antioxidant system in cytotoxicity mediated by anticancer cisplatin: implication to cancer cell resistance. Cancer Lett. 2008, 260, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Kwak, M.K.; Ramos-Gomez, M.; Wakabayashi, N.; Kensler, T.W. Chemoprevention by 1,2-dithiole-3-thiones through induction of NQO1 and other phase 2 enzymes. Methods Enzymol. 2004, 382, 414–423. [Google Scholar] [PubMed]
- Osburn, W.O.; Wakabayashi, N.; Misra, V.; Nilles, T.; Biswal, S.; Trush, M.A.; Kensler, T.W. Nrf2 regulates an adaptive response protecting against oxidative damage following diquat-mediated formation of superoxide anion. Arch. Biochem. Biophys. 2006, 454, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Gomez, M.; Dolan, P.M.; Itoh, K.; Yamamoto, M.; Kensler, T.W. Interactive effects of nrf2 genotype and oltipraz on benzoa.pyrene-DNA adducts and tumor yield in mice. Carcinogenesis 2003, 24, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Aoki, Y.; Sato, H.; Nishimura, N.; Takahashi, S.; Itoh, K.; Yamamoto, M. Accelerated DNA adduct formation in the lung of the Nrf2 knockout mouse exposed to diesel exhaust. Toxicol. Appl. Pharmacol. 2001, 173, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Iida, K.; Itoh, K.; Kumagai, Y.; Oyasu, R.; Hattori, K.; Kawai, K.; Shimazui, T.; Akaza, H.; Yamamoto, M. Nrf2 is essential for the chemopreventive efficacy of oltipraz against urinary bladder carcinogenesis. Cancer Res. 2004, 64, 6424–6431. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Huang, Z.; Chan, J.Y.; Zhang, D.D. Nrf2 protects against As(III)-induced damage in mouse liver and bladder. Toxicol. Appl. Pharmacol. 2009, 240, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Thimmulappa, R.K.; Scollick, C.; Traore, K.; Yates, M.; Trush, M.A.; Liby, K.T.; Sporn, M.B.; Yamamoto, M.; Kensler, T.W.; Biswal, S. Nrf2-dependent protection from LPS induced inflammatory response and mortality by CDDO-Imidazolide. Biochem. Biophys. Res. Commun. 2006, 351, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Jia, Z.; Zhang, L.; Yamamoto, M.; Misra, H.P.; Trush, M.A.; Li, Y. Antioxidants and phase 2 enzymes in macrophages: regulation by Nrf2 signaling and protection against oxidative and electrophilic stress. Exp. Biol. Med. (Maywood) 2008, 233, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.Y.; Reddy, S.P.; Yamamoto, M.; Kleeberger, S.R. The transcription factor NRF2 protects against pulmonary fibrosis. FASEB J. 2004, 18, 1258–1260. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.H.; Park, H.M.; Jung, K.A.; Choi, H.G.; Kim, J.A.; Kim, D.D.; Kim, S.G.; Kang, K.W.; Ku, S.K.; Kensler, T.W.; Kwak, M.K. The NRF2-heme oxygenase-1 system modulates cyclosporin A-induced epithelial-mesenchymal transition and renal fibrosis. Free Radic. Biol. Med. 2010, 48, 1051–1063. [Google Scholar] [CrossRef] [PubMed]
- Surh, Y.J.; Kundu, J.K.; Na, H.K. Nrf2 as a master redox switch in turning on the cellular signaling involved in the induction of cytoprotective genes by some chemopreventive phytochemicals. Planta Med. 2008, 74, 1526–1539. [Google Scholar] [CrossRef] [PubMed]
- Chung, F.L.; Conaway, C.C.; Rao, C.V.; Reddy, B.S. Chemoprevention of colonic aberrant crypt foci in Fischer rats by sulforaphane and phenethyl isothiocyanate. Carcinogenesis 2000, 21, 2287–2291. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.D.; Dashwood, R.H.; Ho, E. Multi-targeted prevention of cancer by sulforaphane. Cancer Lett. 2008, 269, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Fahey, J.W.; Haristoy, X.; Dolan, P.M.; Kensler, T.W.; Scholtus, I.; Stephenson, K.K.; Talalay, P.; Lozniewski, A. Sulforaphane inhibits extracellular, intracellular, and antibiotic-resistant strains of Helicobacter pylori and prevents benzoa.pyrene-induced stomach tumors. Proc. Natl. Acad. Sci. USA 2002, 99, 7610–7615. [Google Scholar] [CrossRef] [PubMed]
- Juge, N.; Mithen, R.F.; Traka, M. Molecular basis for chemoprevention by sulforaphane: a comprehensive review. Cell Mol Life Sci. 2007, 64, 1105–1127. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kensler, T.W.; Cho, C.G.; Posner, G.H.; Talalay, P. Anticarcinogenic activities of sulforaphane and structurally related synthetic norbornyl isothiocyanates. Proc. Natl. Acad. Sci. USA 1994, 91, 3147–3150. [Google Scholar] [CrossRef] [PubMed]
- Kensler, T.W.; Egner, P.A.; Dolan, P.M.; Groopman, J.D.; Roebuck, B.D. Mechanism of protection against aflatoxin tumorigenicity in rats fed 5-(2-pyrazinyl)-4-methyl-1,2-dithiol-3-thione (oltipraz) and related 1,2-dithiol-3-thiones and 1,2-dithiol-3-ones. Cancer Res. 1987, 47, 4271–4277. [Google Scholar] [PubMed]
- Kensler, T.W.; Groopman, J.D.; Eaton, D.L.; Curphey, T.J.; Roebuck, B.D. Potent inhibition of aflatoxin-induced hepatic tumorigenesis by the monofunctional enzyme inducer 1,2-dithiole-3-thione. Carcinogenesis 1992, 13, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Kensler, T.W.; Groopman, J.D.; Sutter, T.R.; Curphey, T.J.; Roebuck, B.D. Development of cancer chemopreventive agents: oltipraz as a paradigm. Chem. Res. Toxicol. 1999, 12, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Roebuck, B.D.; Curphey, T.J.; Li, Y.; Baumgartner, K.J.; Bodreddigari, S.; Yan, J.; Gange, S.J.; Kensler, T.W.; Sutter, T.R. Evaluation of the cancer chemopreventive potency of dithiolethione analogs of oltipraz. Carcinogenesis 2003, 24, 1919–1928. [Google Scholar] [CrossRef] [PubMed]
- Aziz, M.H.; Kumar, R.; Ahmad, N. Cancer chemoprevention by resveratrol: in vitro and in vivo studies and the underlying mechanisms (review). Int. J. Oncol. 2003, 23, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Juan, M.E.; Vinardell, M.P.; Planas, J.M. The daily oral administration of high doses of trans-resveratrol to rats for 28 days is not harmful. J. Nutr. 2002, 132, 257–260. [Google Scholar] [CrossRef] [PubMed]
- Surh, Y.J. Cancer chemoprevention with dietary phytochemicals. Nat. Rev. Cancer 2003, 3, 768–780. [Google Scholar] [CrossRef] [PubMed]
- Athar, M.; Back, J.H.; Tang, X.; Kim, K.H.; Kopelovich, L.; Bickers, D.R.; Kim, A.L. Resveratrol: a review of preclinical studies for human cancer prevention. Toxicol. Appl. Pharmacol. 2007, 224, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Khar, A. Biological effects of curcumin and its role in cancer chemoprevention and therapy. Anticancer Agents Med. Chem. 2006, 6, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, N.; Aggarwal, B.B.; Newman, R.A.; Wolff, R.A.; Kunnumakkara, A.B.; Abbruzzese, J.L.; Ng, C.S.; Badmaev, V.; Kurzrock, R. Phase II trial of curcumin in patients with advanced pancreatic cancer. Clin. Cancer Res. 2008, 14, 4491–4499. [Google Scholar] [CrossRef] [PubMed]
- Hatcher, H.; Planalp, R.; Cho, J.; Torti, F.M.; Torti, S.V. Curcumin: from ancient medicine to current clinical trials. Cell Mol. Life Sci. 2008, 65, 1631–1652. [Google Scholar] [CrossRef] [PubMed]
- Nagaoka, T.; Banskota, A.H.; Tezuka, Y.; Saiki, I.; Kadota, S. Selective antiproliferative activity of caffeic acid phenethyl ester analogues on highly liver-metastatic murine colon 26-L5 carcinoma cell line. Bioorg. Med. Chem. 2002, 10, 3351–3359. [Google Scholar] [CrossRef]
- Zhang, Y.; Talalay, P.; Cho, C.G.; Posner, G.H. A major inducer of anticarcinogenic protective enzymes from broccoli: isolation and elucidation of structure. Proc. Natl. Acad. Sci. USA 1992, 89, 2399–2403. [Google Scholar] [CrossRef] [PubMed]
- Conaway, C.C.; Wang, C.X.; Pittman, B.; Yang, Y.M.; Schwartz, J.E.; Tian, D.; McIntee, E.J.; Hecht, S.S.; Chung, F.L. Phenethyl isothiocyanate and sulforaphane and their N-acetylcysteine conjugates inhibit malignant progression of lung adenomas induced by tobacco carcinogens in A/J mice. Cancer Res. 2005, 65, 8548–8557. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Munday, R. Dithiolethiones for cancer chemoprevention: where do we stand? Mol. Cancer Ther. 2008, 7, 3470–3479. [Google Scholar] [CrossRef] [PubMed]
- O’Dwyer, P.J.; Szarka, C.E.; Yao, K.S.; Halbherr, T.C.; Pfeiffer, G.R.; Green, F.; Gallo, J.M.; Brennan, J.; Frucht, H.; Goosenberg, E.B.; Hamilton, T.C.; Litwin, S.; Balshem, A.M.; Engstrom, P.F.; Clapper, M.L. Modulation of gene expression in subjects at risk for colorectal cancer by the chemopreventive dithiolethione oltipraz. J. Clin. Invest. 1996, 98, 1210–1217. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.S.; Shen, X.; He, X.; Zhu, Y.R.; Zhang, B.C.; Wang, J.B.; Qian, G.S.; Kuang, S.Y.; Zarba, A.; Egner, P.A.; Jacobson, L.P.; Munoz, A.; Helzlsouer, K.J.; Groopman, J.D.; Kensler, T.W. Protective alterations in phase 1 and 2 metabolism of aflatoxin B1 by oltipraz in residents of Qidong, People’s Republic of China. J. Natl. Cancer Inst. 1999, 91, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Leiro, J.; Alvarez, E.; Arranz, J.A.; Laguna, R.; Uriarte, E.; Orallo, F. Effects of cis-resveratrol on inflammatory murine macrophages: antioxidant activity and down-regulation of inflammatory genes. J. Leukoc. Biol. 2004, 75, 1156–1165. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Velez, M.; Martinez-Martinez, F.; Del Valle-Ribes, C. The study of phenolic compounds as natural antioxidants in wine. Crit. Rev. Food Sci. Nutr. 2003, 43, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Miller, N.J.; Rice-Evans, C.A. Antioxidant activity of resveratrol in red wine. Clin. Chem. 1995, 41, 1789. [Google Scholar] [PubMed]
- Chen, C.Y.; Jang, J.H.; Li, M.H.; Surh, Y.J. Resveratrol upregulates heme oxygenase-1 expression via activation of NF-E2-related factor 2 in PC12 cells. Biochem. Biophys. Res. Commun. 2005, 331, 993–1000. [Google Scholar] [CrossRef] [PubMed]
- Kode, A.; Rajendrasozhan, S.; Caito, S.; Yang, S.R.; Megson, I.L.; Rahman, I. Resveratrol induces glutathione synthesis by activation of Nrf2 and protects against cigarette smoke-mediated oxidative stress in human lung epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 294, L478–488. [Google Scholar] [CrossRef] [PubMed]
- Rubiolo, J.A.; Mithieux, G.; Vega, F.V. Resveratrol protects primary rat hepatocytes against oxidative stress damage: activation of the Nrf2 transcription factor and augmented activities of antioxidant enzymes. Eur. J. Pharmacol. 2008, 591, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Ammon, H.P.; Wahl, M.A. Pharmacology of Curcuma longa. Planta Med. 1991, 57, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Balogun, E.; Hoque, M.; Gong, P.; Killeen, E.; Green, C.J.; Foresti, R.; Alam, J.; Motterlini, R. Curcumin activates the haem oxygenase-1 gene via regulation of Nrf2 and the antioxidant-responsive element. Biochem. J. 2003, 371, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.; Gupta, S.; Maru, G.B. Dietary curcumin modulates transcriptional regulators of phase I and phase II enzymes in benzoa.pyrene-treated mice: mechanism of its anti-initiating action. Carcinogenesis 2008, 29, 1022–1032. [Google Scholar] [CrossRef] [PubMed]
- Calabro, P.; Yeh, E.T. The pleiotropic effects of statins. Curr Opin Cardiol 2005, 20, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Shaw, S.M.; Fildes, J.E.; Yonan, N.; Williams, S.G. Pleiotropic effects and cholesterol-lowering therapy. Cardiology 2009, 112, 4–12. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, G. Statins and renal diseases: from primary prevention to renal replacement therapy. J. Am. Soc. Nephrol. 2006, 17, S148–S152. [Google Scholar] [CrossRef] [PubMed]
- Dulak, J.; Jozkowicz, A. Anti-angiogenic and anti-inflammatory effects of statins: relevance to anti-cancer therapy. Curr. Cancer Drug Targets 2005, 5, 579–594. [Google Scholar] [CrossRef] [PubMed]
- Immenschuh, S.; Baumgart-Vogt, E.; Mueller, S. Heme Oxygenase-1 and Iron in Liver Inflammation: a Complex Alliance. Curr. Drug Targets 2010. [Google Scholar] [CrossRef]
- Paine, A.; Eiz-Vesper, B.; Blasczyk, R.; Immenschuh, S. Signaling to heme oxygenase-1 and its anti-inflammatory therapeutic potential. Biochem. Pharmacol. 2010. [Google Scholar] [CrossRef] [PubMed]
- Khor, T.O.; Yu, S.; Kong, A.N. Dietary cancer chemopreventive agents - targeting inflammation and Nrf2 signaling pathway. Planta Med. 2008, 74, 1540–1547. [Google Scholar] [CrossRef] [PubMed]
- Kwak, M.K.; Wakabayashi, N.; Greenlaw, J.L.; Yamamoto, M.; Kensler, T.W. Antioxidants enhance mammalian proteasome expression through the Keap1-Nrf2 signaling pathway. Mol. Cell Biol. 2003, 23, 8786–8794. [Google Scholar] [CrossRef] [PubMed]
- Kwak, M.K.; Huang, B.; Chang, H.; Kim, J.A.; Kensler, T.W. Tissue specific increase of the catalytic subunits of the 26S proteasome by indirect antioxidant dithiolethione in mice: enhanced activity for degradation of abnormal protein. Life Sci. 2007, 80, 2411–2420. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Hypothesis: proteasomal dysfunction: a primary event in neurogeneration that leads to nitrative and oxidative stress and subsequent cell death. Ann. N Y Acad. Sci. 2002, 962, 182–194. [Google Scholar] [CrossRef] [PubMed]
- Poppek, D.; Grune, T. Proteasomal defense of oxidative protein modifications. Antioxid. Redox Signal 2006, 8, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Stefani, M.; Dobson, C.M. Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J. Mol. Med. 2003, 27, 27. [Google Scholar] [CrossRef] [PubMed]
- Kwak, M.K.; Cho, J.M.; Huang, B.; Shin, S.; Kensler, T.W. Role of increased expression of the proteasome in the protective effects of sulforaphane against hydrogen peroxide-mediated cytotoxicity in murine neuroblastoma cells. Free Radic. Biol. Med. 2007, 43, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.; Wakabayashi, N.; Misra, V.; Biswal, S.; Lee, G.H.; Agoston, E.S.; Yamamoto, M.; Kensler, T.W. NRF2 modulates aryl hydrocarbon receptor signaling: influence on adipogenesis. Mol. Cell Biol. 2007, 27, 7188–7197. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.; Wakabayashi, J.; Yates, M.S.; Wakabayashi, N.; Dolan, P.M.; Aja, S.; Liby, K.T.; Sporn, M.B.; Yamamoto, M.; Kensler, T.W. Role of Nrf2 in prevention of high-fat diet-induced obesity by synthetic triterpenoid CDDO-imidazolide. Eur. J. Pharmacol. 2009, 620, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Aleksunes, L.M.; Yeager, R.L.; Gyamfi, M.A.; Esterly, N.; Guo, G.L.; Klaassen, C.D. NF-E2-related factor 2 inhibits lipid accumulation and oxidative stress in mice fed a high-fat diet. J. Pharmacol. Exp. Ther. 2008, 325, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Beyer, T.A.; Xu, W.; Teupser, D.; auf dem Keller, U.; Bugnon, P.; Hildt, E.; Thiery, J.; Kan, Y.W.; Werner, S. Impaired liver regeneration in Nrf2 knockout mice: role of ROS-mediated insulin/IGF-1 resistance. EMBO J. 2008, 27, 212–223. [Google Scholar] [CrossRef] [PubMed]
- Beyer, T.A.; Werner, S. The cytoprotective Nrf2 transcription factor controls insulin receptor signaling in the regenerating liver. Cell Cycle 2008, 7, 874–878. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, N.; Shin, S.; Slocum, S.L.; Agoston, E.S.; Wakabayashi, J.; Kwak, M.K.; Misra, V.; Biswal, S.; Yamamoto, M.; Kensler, T.W. Regulation of notch1 signaling by nrf2: implications for tissue regeneration. Sci. Signal 2010, 3, ra52. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.W.; Choi, S.H.; Ha, J.R.; Kim, C.W.; Kim, S.G. Inhibition of dimethylnitrosamine-induced liver fibrosis by 5-(2-pyrazinyl)-4-methyl-1,2-dithiol-3-thione. (oltipraz) in rats: suppression of transforming growth factor-beta1 and tumor necrosis factor-alpha expression. Chem. Biol. Interact 2002, 139, 61–77. [Google Scholar] [CrossRef]
- Kang, K.W.; Kim, Y.G.; Cho, M.K.; Bae, S.K.; Kim, C.W.; Lee, M.G.; Kim, S.G. Oltipraz regenerates cirrhotic liver through CCAAT/enhancer binding protein-mediated stellate cell inactivation. FASEB J. 2002, 16, 1988–1990. [Google Scholar] [CrossRef] [PubMed]
- Talalay, P.; De Long, M.J.; Prochaska, H.J. Identification of a common chemical signal regulating the induction of enzymes that protect against chemical carcinogenesis. Proc. Natl. Acad. Sci. USA 1988, 85, 8261–8265. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Li, L.; Iwamoto, N.; Nakajima-Takagi, Y.; Kaneko, H.; Nakayama, Y.; Eguchi, M.; Wada, Y.; Kumagai, Y.; Yamamoto, M. The antioxidant defense system Keap1-Nrf2 comprises a multiple sensing mechanism for responding to a wide range of chemical compounds. Mol. Cell Biol. 2009, 29, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Holland, R.; Hawkins, A.E.; Eggler, A.L.; Mesecar, A.D.; Fabris, D.; Fishbein, J.C. Prospective type 1 and type 2 disulfides of Keap1 protein. Chem. Res. Toxicol. 2008, 21, 2051–2060. [Google Scholar] [CrossRef] [PubMed]
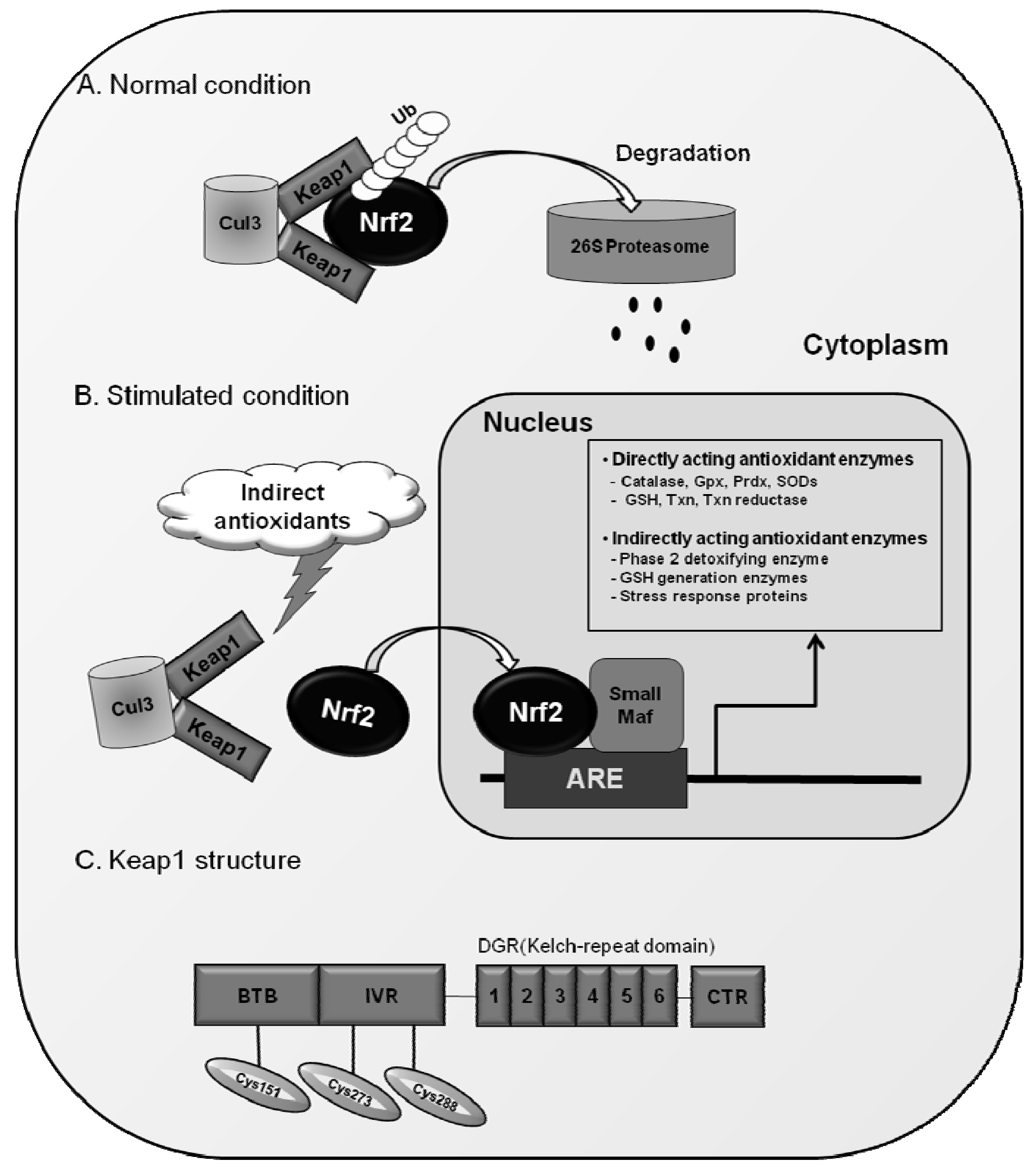

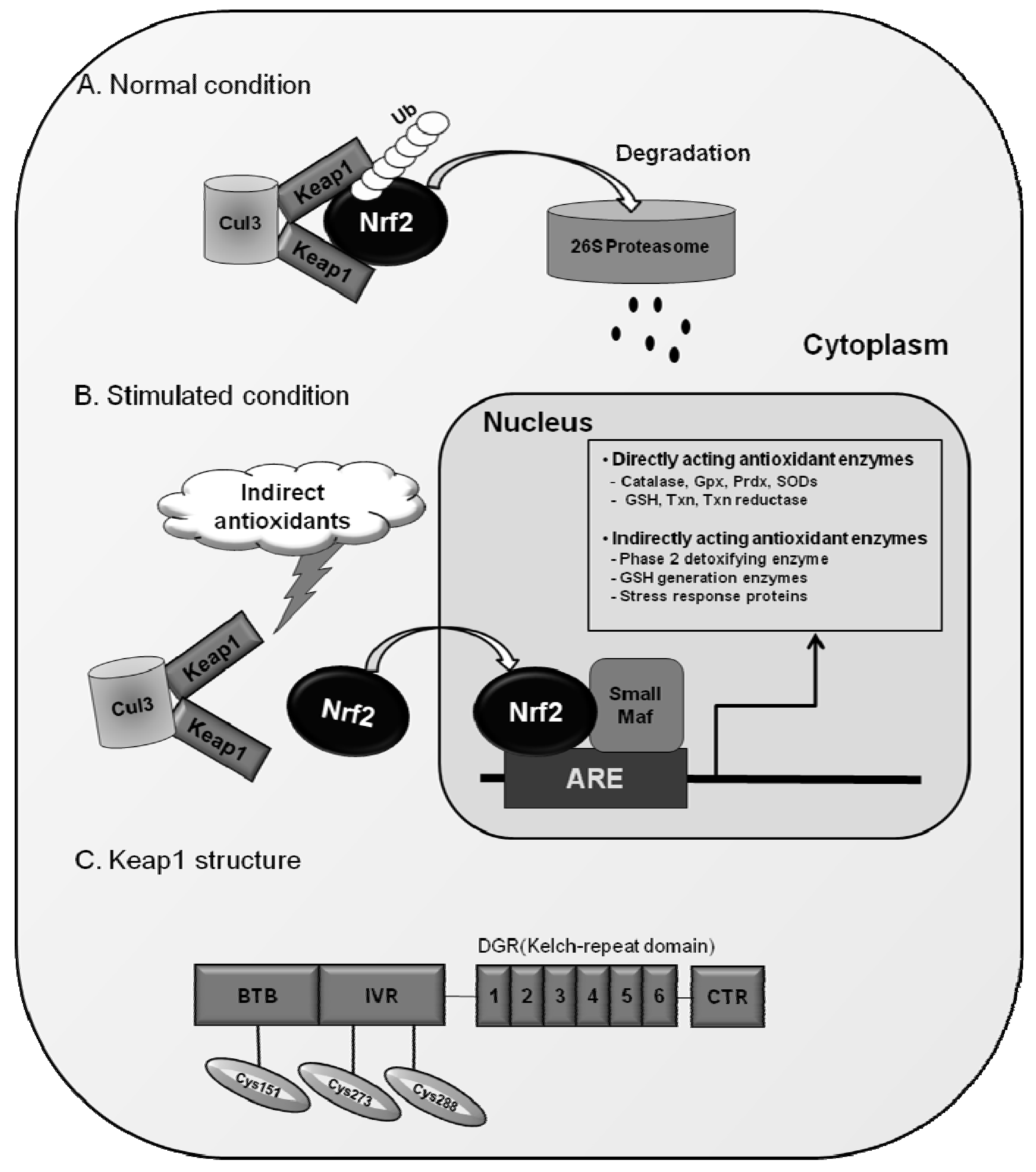{kind=link}
| Function | Gene | Species (organs) | Reference |
|---|---|---|---|
| GSH biosynthesis | GCLC | Mouse (liver, lung) Human (HaCaT;keratinocyte) | [58,67] [68] |
| GCLM | Mouse (liver, lung) Human (IMR-32;neuroblastoma cell) (HaCaT) | [58,67] [68,69] | |
| GSR | Mouse (liver, lung) Human (IMR-32, HaCaT) | [70,71] [68,69] | |
| Glutathione peroxidases | GPx1 | Mouse (cardiovascular, lung) | [72] |
| GPx2 | Mouse (liver, lung) Rat (liver) Human (Caco-2;colon Cell) Rat (liver) | [73,74] [75] [76] | |
| Thioredoxin reductase | TXNRD | Mouse (liver, lung) Human (IMR-32) | [65,67] [69] |
| Thioredoxin | TXN | Mouse (liver) | [58] |
| Peroxiredoxin | PRDX1 | Mouse (liver,lung) | [67,74] |
| PRDX6 | Human (A549; lung derived cell line) | [77] | |
| Superoxide dismutase | SOD1 | Mouse (liver) | [71] |
| SOD2 | Mouse (liver) | [71] | |
| SOD3 | Mouse (lung) | [67] | |
| Catalase | Mouse (liver, lung) | [71,73] | |
| Glutathione S-transferases | GSTA1 | Mouse (liver, lung, small intestine) | [64,67,71] |
| GSTA2 | Mouse (liver, lung, small intestine) | [58,64,67] | |
| GSTA3 | Mouse (liver, lung, small intestine) | [64,67,71] | |
| GSTA4 | Mouse (liver) | [65] | |
| GSTM1 | Mouse (liver, small intestine) | [58,64,65] | |
| GSTM2 | Mouse (liver, small intestine) | [58,64,65] | |
| GSTM3 | Mouse (liver, small intestine) Human (IMR-32) | [58,64,65] [69] | |
| GSTM4 | Mouse (liver) | [65] | |
| GSTM5 | Mouse (liver) | [59] | |
| GSTM6 | Mouse (liver) | [65] | |
| MGST2 | Mouse (small intestine) | [64] | |
| MGST3 | Mouse (liver, small intestine) | [58,64] | |
| UDP-glucuronosyl transferase | UGT1A6 | Mouse (liver) | [74] |
| UGT2B1 | Mouse (liver) | [71] | |
| UGT2B5 | Mouse (liver, small intestine) | [58,64] | |
| Reduction | NQO1 AKR1A AKR1B8 | Mouse (liver, lung, small intestine) Human (IMR-32) Mouse (liver, small intestine) Mouse (liver, small intestine) | [58,64,67] [69] [58,64] [64,67] |
| Heme oxygenase-1 | HO-1 | Mouse (liver) Rat (liver) Human (IMR-32, HaCaT) | [59,76] [68,69] |
| Hydrolysis | EPHX | Mouse (liver, small intestine) | [58,64] |
| Iron transport | Ferritin H | Mouse (lung) Human (HaCaT) | [73] [68] |
| Ferritin L | Mouse (liver) Human (HaCaT) | [65] [68] | |
| Detoxication of heavy metals Transport | MT І | Mouse (embryonic fibroblasts) Human (HepG2 cell;hepatoma) | [56] [78] |
| MT ІІ | Mouse (embryonic fibroblasts) Human (HepG2 Cell) | [56] [78] | |
| MRP2 | Mouse (liver) | [71] | |
| MRP3 | Mouse (liver) Human (NSCLC, HBE1) | [71] [79] | |
| 26S Proteasome | Psma1,4,5,6,7 Psmb1,2,3,4,5,6 | Mouse (liver) Mouse (liver) | [58] [58] |
| Psmc1,3 Psmd1,5,7,11,12,13 | Mouse (liver) Mouse (liver) | [58] [58] | |
| Indirect antioxidants | Effect on target organ toxicity |
|---|---|
Sulforaphane 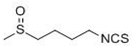 | Protection against tumor formation induced by many carcinogens: mammary, colon, lung, pancreatic, gastric, intestine, skin, and bladder (mouse, rat, hamster) [16,111,112,113,114,115] |
D3T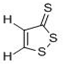 | Inhibition of aflatoxin B1 induced hepatic tumorigenesis (rat) [116,117]. |
Oltipraz | Inhibition of carcinogenesis induced by various carcinogens in bladder, colon, kidney, liver, lung, pancreas, and stomach (mouse, rat) [116,118,119]. |
Resveratrol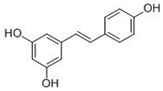 | Inhibition of growth of variety tumors: skin, breast, gastric, colon, small intestine, lung, esophageal, prostate, liver, and pancreatic cancers (mice, rat) [120,121,122]. Human clinical trials in breast cancer patients [123]. |
Curcumin 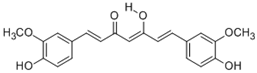 | Inhibition of tumor development in skin, liver, oral, esophageal, stomach, intestinal, colon, and bladder (mouse, rat) [124]. Human clinical trials in patients with advanced pancreatic cancer and other disease [125,126]. |
CAPE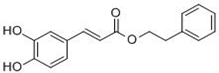 | Anti-proliferation property in cancer cells [127]. |
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jung, K.-A.; Kwak, M.-K. The Nrf2 System as a Potential Target for the Development of Indirect Antioxidants. Molecules 2010, 15, 7266-7291. https://doi.org/10.3390/molecules15107266
Jung K-A, Kwak M-K. The Nrf2 System as a Potential Target for the Development of Indirect Antioxidants. Molecules. 2010; 15(10):7266-7291. https://doi.org/10.3390/molecules15107266
Chicago/Turabian StyleJung, Kyeong-Ah, and Mi-Kyoung Kwak. 2010. "The Nrf2 System as a Potential Target for the Development of Indirect Antioxidants" Molecules 15, no. 10: 7266-7291. https://doi.org/10.3390/molecules15107266
APA StyleJung, K.-A., & Kwak, M.-K. (2010). The Nrf2 System as a Potential Target for the Development of Indirect Antioxidants. Molecules, 15(10), 7266-7291. https://doi.org/10.3390/molecules15107266