Theoretical Studies of [2,3]-Sigmatropic Rearrangements of Allylic Selenoxides and Selenimides

Abstract
:1. Introduction
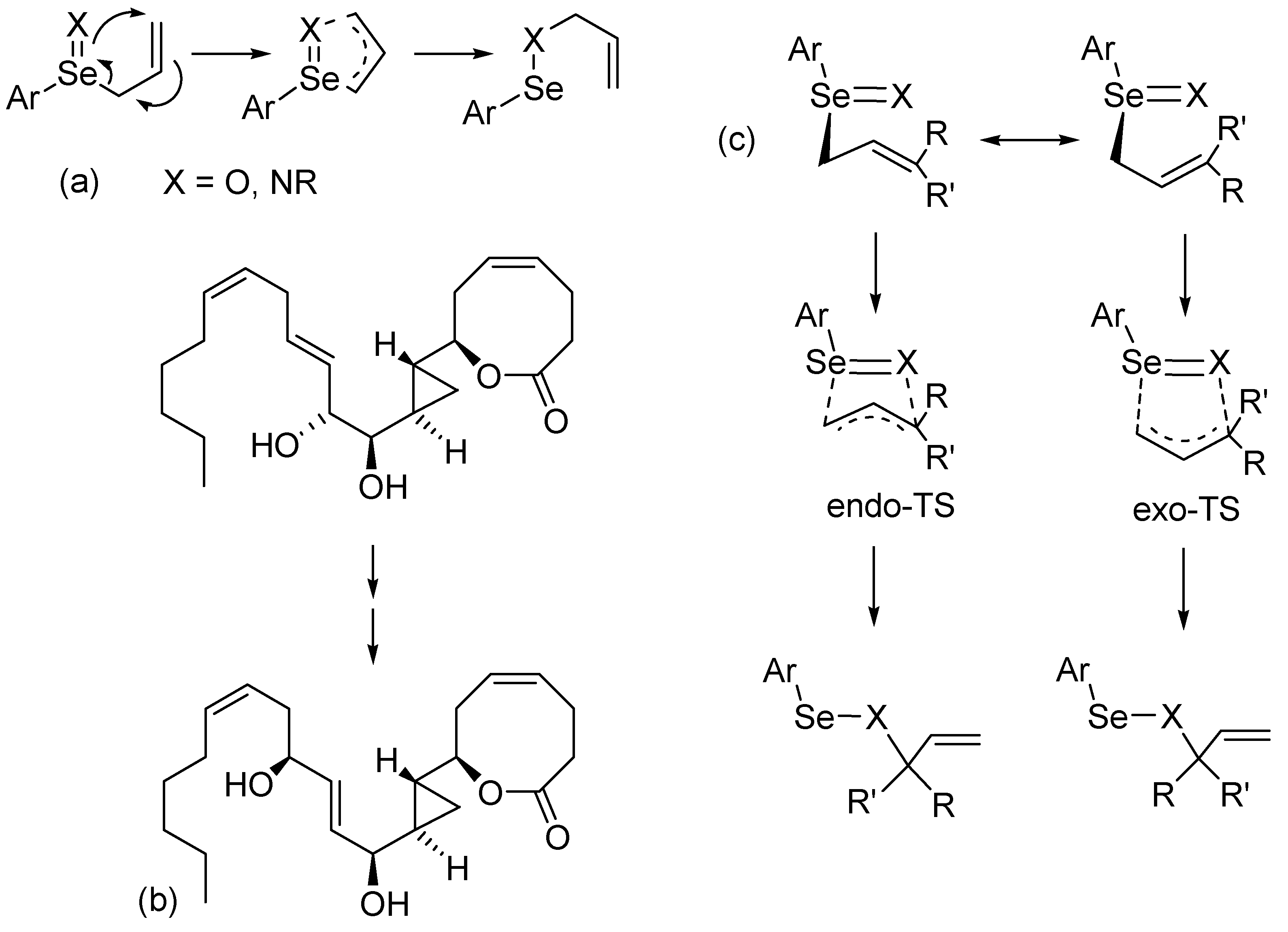
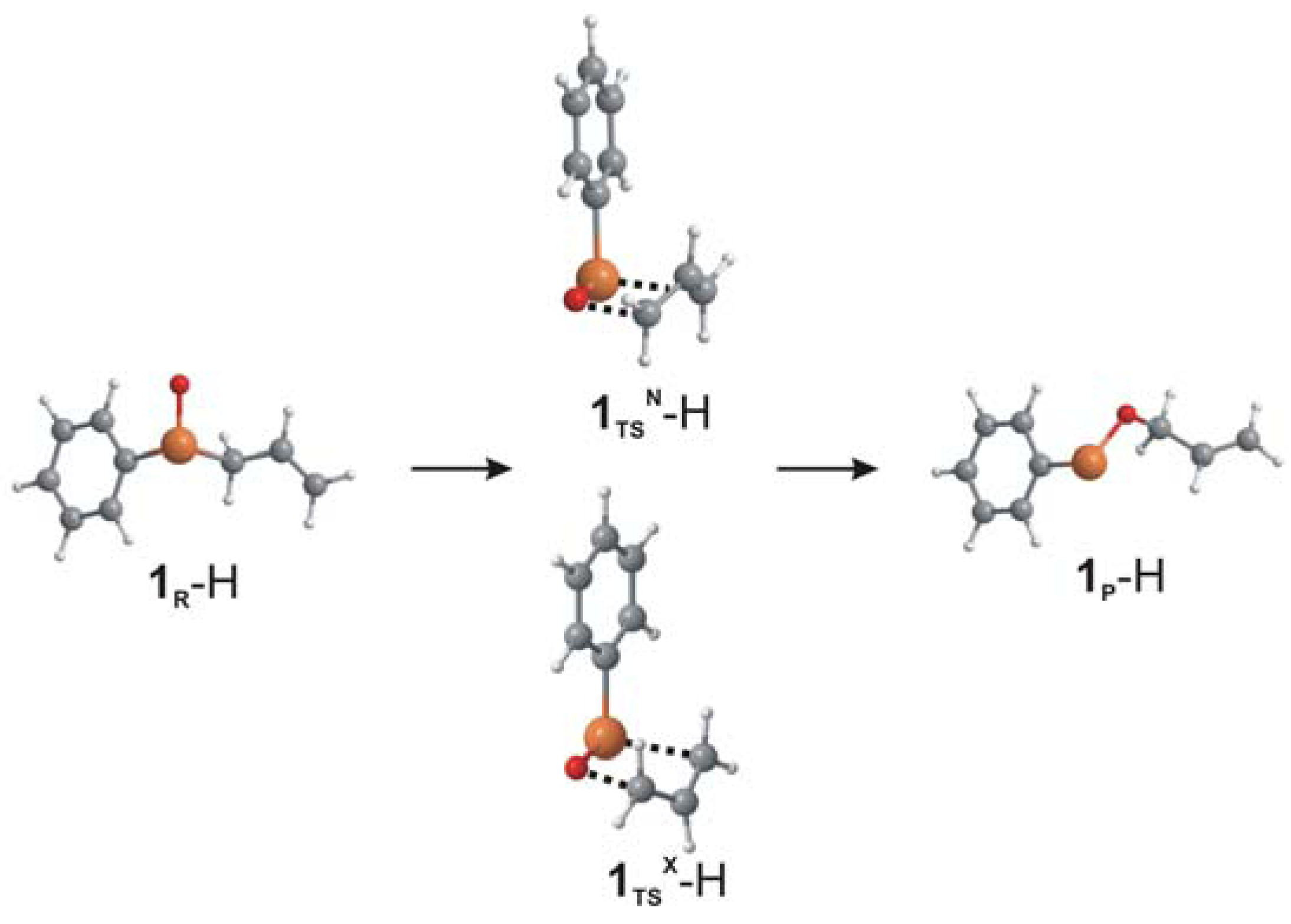
2. Results and Discussion
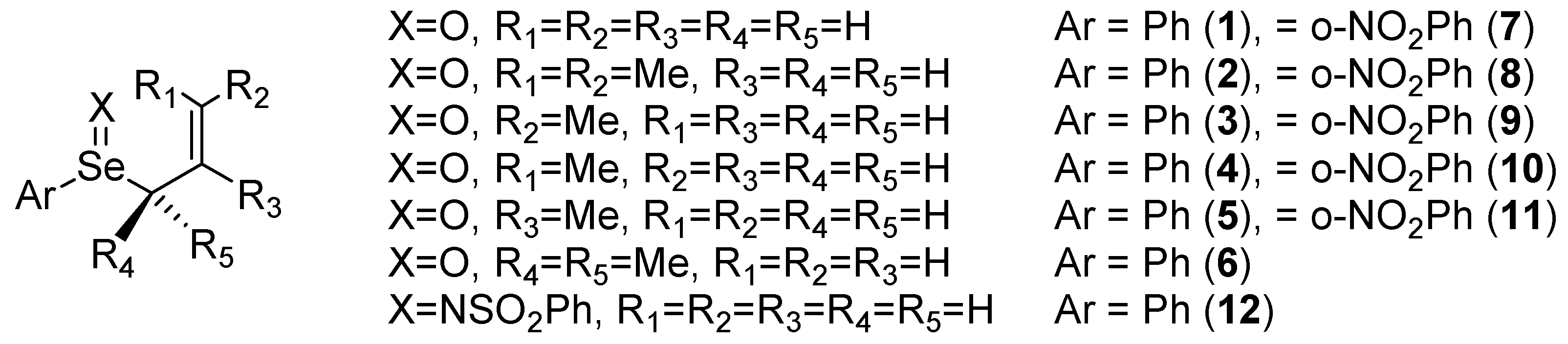
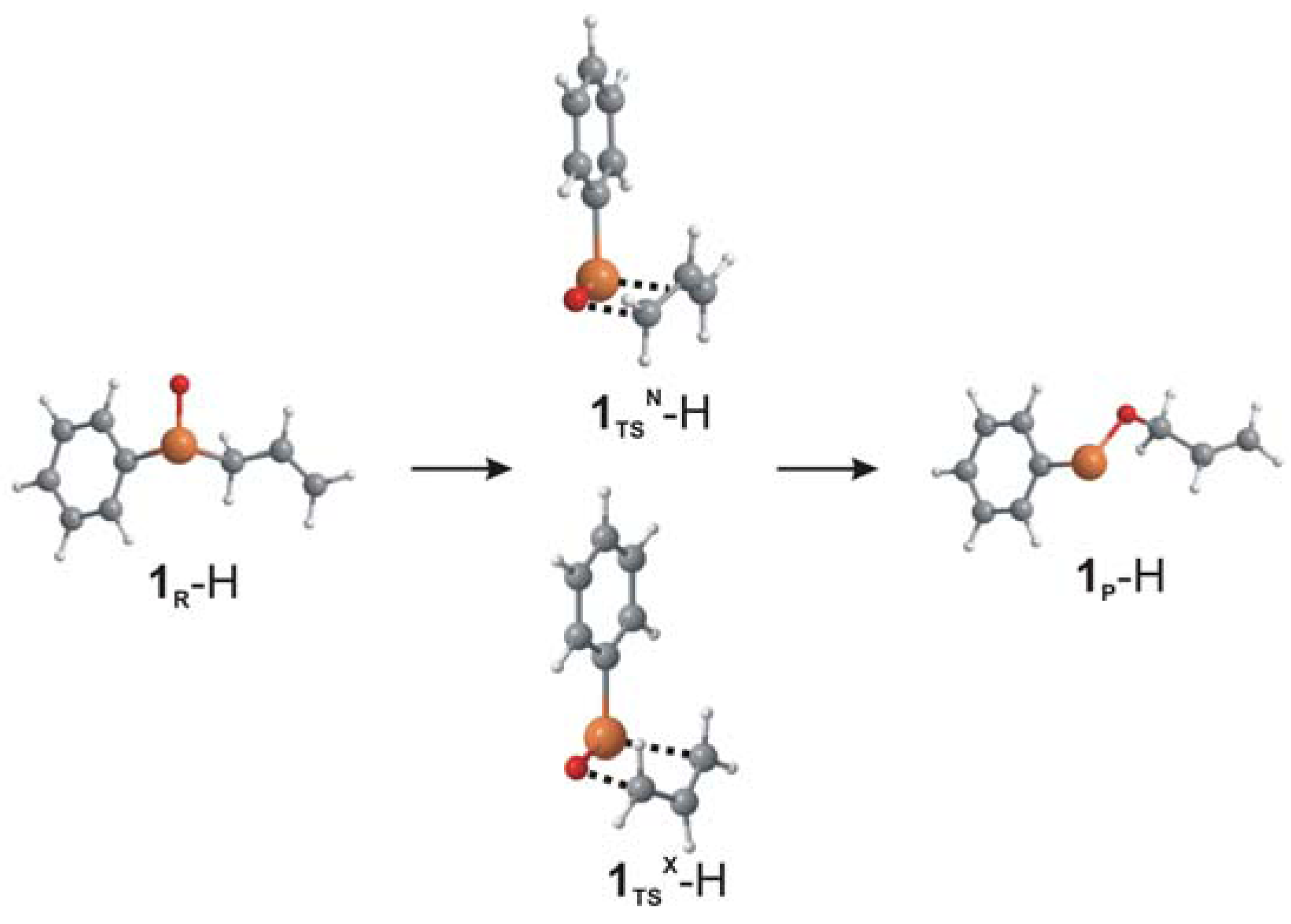
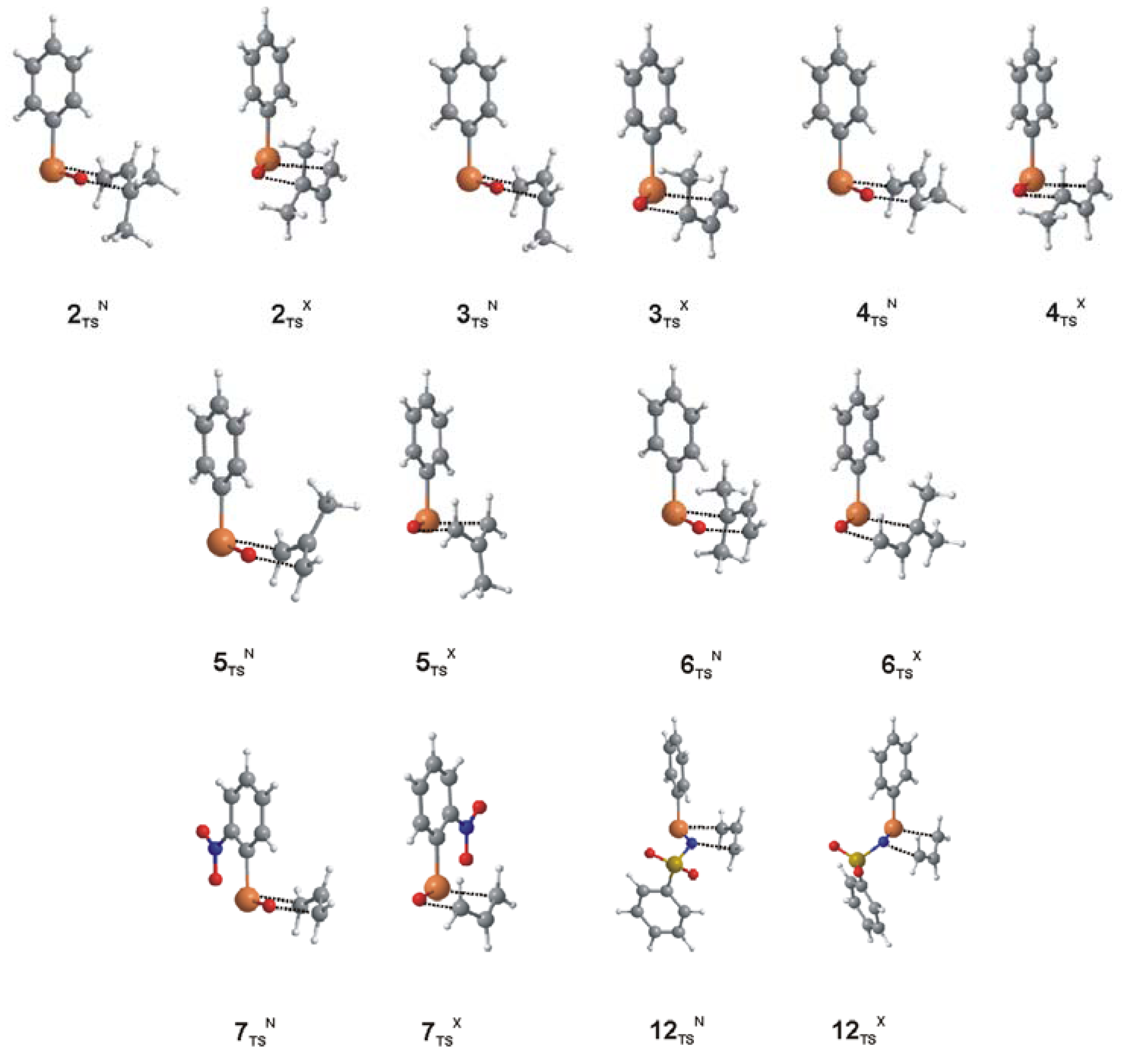

{kind=link}
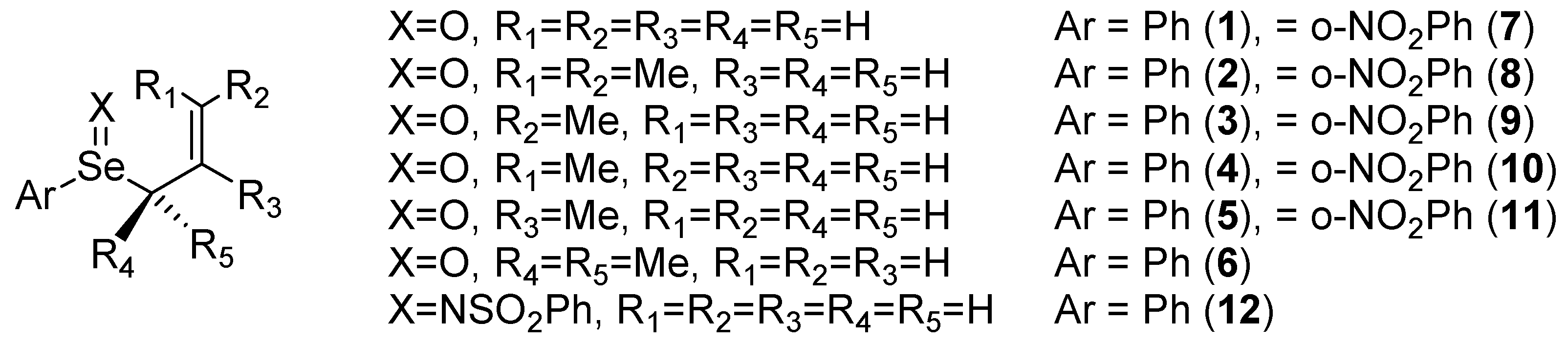{kind=link}
{kind=link}
{kind=link}
| Type a | d(Se/S-O,N), Å | d(Se/S-C), Å | d(C-O,N), Å | ΔG‡, kcal/mol | ΔΔG‡N-X, kcal/mol | ΔG, kcal/mol | |
|---|---|---|---|---|---|---|---|
| 1 | N | 1.699 | 2.481 | 2.006 | 13.2 | 0.3 | -7.7 |
| 1(S) | N | 1.566 | 2.463 | 1.994 | 16.4 | 0.4 | -1.8 |
| 2 | Nb | 1.697 | 2.567 | 2.077 | 12.7 | 1.3 | -6.2 |
| 3 | Nb | 1.696 | 2.513 | 2.060 | 13.5 | 1.4 | -7.4 |
| 4 | N | 1.699 | 2.521 | 2.026 | 12.0 | 0.1 | -7.8 |
| 5 | X | 1.696 | 2.513 | 2.049 | 14.4 | -1.0 | -7.7 |
| 6 | N | 1.693 | 2.616 | 2.122 | 11.1 | 1.3 | -7.4 |
| 7 | N | 1.695 | 2.477 | 2.075 | 11.2 | 0.7 | -15.1 |
| 7(S) | N | 1.562 | 2.462 | 2.050 | 14.2 | 0.9 | -7.3 |
| 8 | Nb | 1.693 | 2.561 | 2.157 | 10.4 | 1.7 | -12.9 |
| 9 | Nb | 1.692 | 2.510 | 2.134 | 11.4 | 2.1 | -14.1 |
| 10 | N | 1.696 | 2.517 | 2.099 | 9.9 | 0.4 | -14.9 |
| 11 | X | 1.694 | 2.508 | 2.102 | 12.4 | -0.9 | -15.9 |
| 12 | X | 1.775 | 2.441 | 2.276 | 13.4 | -0.6 | -19.0 |
3. Theoretical Section
4. Conclusions
Acknowledgements
- Sample Availability: Not available.
References
- Nishibatashi, Y.; Uemura, S. Sigmatropic rearrangements of organoselenium compounds. In Organoselenium Chemistry. A Practical Approach; Back, T.G, Ed.; Oxford University Press: Oxford, UK, 1999; pp. 207–221. [Google Scholar]
- Zanoni, G.; Castronovo, F.; Perani, E.; Vidari, G. A → J prostaglandin swap: A new tactic for cyclopentenone prostaglandin synthesis. J. Org. Chem. 2003, 68, 6803–6805. [Google Scholar]
- Davoren, J.E.; Harcken, C.; Martin, S.F. Enantioselective synthesis and structure revision of solandelactone E. J. Org. Chem. 2008, 73, 391–402. [Google Scholar] [CrossRef]
- Back, T.G.; McPhee, D.J. A caveat concerning the use of N-(phenylseleno)phthalimide and tri-n-butylphosphine for the conversion of alcohols to selenides. Formation of 3-phthalimido derivatives from an allylic 3-sterol. J. Org. Chem. 1984, 49, 3842–3843. [Google Scholar] [CrossRef]
- Shea, R.G.; Fitzner, J.N.; Fankhauser, J.E.; Spaltenstein, A.; Carpino, P.A.; Peevey, R.M.; Pratt, D.V.; Tenge, B.J.; Hopkins, P.B. Allylic selenides in organic synthesis: New methods for the synthesis of allylic amines. J. Org. Chem. 1986, 51, 5243–5252. [Google Scholar]
- Peng, X.; Hong, I.S.; Li, H.; Seidman, M.M.; Greenberg, M.M. Intrastrand cross-link formation in duplex and triplex DNA by modified pyrimidines. J. Am. Chem. Soc. 2008, 130, 10299–10306. [Google Scholar]
- Davis, F.A.; Reddy, R.T. Asymmetric oxidation of simple selenides to selenoxides in high enantiopurity. Stereochemical aspects of the allylic selenoxide/allyl selenenate rearrangement. J. Org. Chem. 1992, 57, 2599–2606. [Google Scholar] [CrossRef]
- Nishibayashi, Y.; Singh, J.D.; Fukuzawa, S.; Uemura, S. Synthesis of [R,S;R,S]- and [S,R,S,R]-bis[2-[1-(dimethylamino)ethyl]ferrocenyl] diselenides and their application to asymmetric selenoxide elimination and [2,3]sigmatropic rearrangement. J. Org. Chem. 1995, 60, 4114–4120. [Google Scholar] [CrossRef]
- Reich, H.J.; Yelm, K.E. Asymmetric induction in the oxidation of [2.2]paracyclophane-substituted selenides. Application of chirality transfer in the selenoxide [2,3] sigmatropic rearrangement. J. Org. Chem. 1991, 56, 5672–5679. [Google Scholar] [CrossRef]
- Soma, T.; Shimizu, T.; Hirabayashi, K.; Kamigata, N. Stabilizing effect of intramolecular Lewis base toward racemization of optically active selenoxides. Heteroatom Chem. 2007, 18, 301–311. [Google Scholar] [CrossRef]
- Fankhauser, J.E.; Peevey, R.M.; Hopkins, P.B. Synthesis of protected allylic amines from allylic phenyl selenides: Improved conditions for the chloramine T oxidation of allylic phenyl selenides. Tetrahedron Lett. 1984, 25, 15–18. [Google Scholar]
- Shea, R.G.; Fitzner, J.N.; Fankhauser, J.E.; Hopkins, P.B. Direct conversion of allylic selenides to protected allylic amines. J. Org. Chem. 1984, 49, 3647–3650. [Google Scholar] [CrossRef]
- Shea, R.G.; Fitzner, J.N.; Fankhauser, J.E.; Spaltenstein, A.; Carpino, P.A.; Peevey, R.M.; Pratt, D.V.; Tenge, B.J.; Hopkins, P.B. Allylic selenides in organic synthesis: New methods for the synthesis of allylic amines. J. Org. Chem. 1986, 51, 5243–5252. [Google Scholar] [CrossRef]
- Reich, H.J.; Yelm, K.E.; Wollowitz, S. Kinetics, thermodynamics, and stereochemistry of the allyl sulfoxide-sulfenate and selenoxide-selenenate [2,3] sigmatropic rearrangements. J. Am. Chem. Soc. 1983, 105, 2503–2504. [Google Scholar] [CrossRef]
- Freeman, F.; Bathala, R.M.; Cavillo, J.E.; Huang, A.C.; Jackson, T.K.; Lopez-Mercado, A.Z.; Phung, S.; Suh, J.; Valencia, D.O. [2,3]-sigmatropic rearrangements of hydrogen and alkyl 3-propenyl sulfoxides: A computational study. Intern. J. Quant. Chem. 2006, 106, 2390–2397. [Google Scholar] [CrossRef]
- Bayse, C.A.; Allison, B.D. Activation energies of selenoxide elimination from Se-substituted selenocysteine. J. Mol. Model. 2007, 13, 47–53. [Google Scholar]
- Macdougall, P.E.; Smith, N.A.; Schiesser, C.H. Substituent effects in selenoxide elimination chemistry. Tetrahedron 2008, 64, 2824–2831. [Google Scholar] [CrossRef]
- Bayse, C.A.; Baker, R.A.; Ortwine, K.N. Relative strengths of Se···N,O interactions: Implications for glutathione peroxidase activity. Inorg. Chim. Acta 2005, 358, 3849–3854. [Google Scholar]
- PQS Version 3.3. Parallel Quantum Solutions: Fayetteville, AR, USA.
- Dunning, T.H. Gaussian basis functions for use in molecular calculations. III. Contraction of (10s6p) atomic basis sets for the first row atoms. J. Chem. Phys. 1971, 55, 716–723. [Google Scholar] [CrossRef]
- Hurley, M.M.; Pacios, L.F.; Christiansen, P.A.; Ross, R.B.; Ermler, W.C. Ab initio relativistic effective core potentials with spin-orbit operators. II. K through Kr. J. Chem. Phys. 1986, 84, 6840–6853. [Google Scholar]
- Wadt, W.R.; Hay, P.J. Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi. J. Chem. Phys. 1985, 82, 284–298. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian basis functions for use in molecular calculations. I. Contraction of (9s5p) atomic basis sets for the first-row atoms. J. Chem. Phys. 1970, 53, 2823–2833. [Google Scholar] [CrossRef]
- Bayse, C.A.; Antony, S. Molecular modeling of bioactive selenium compounds. Main Group Chem. 2007, 6, 185–200. [Google Scholar] [CrossRef]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bayse, C.A.; Antony, S. Theoretical Studies of [2,3]-Sigmatropic Rearrangements of Allylic Selenoxides and Selenimides. Molecules 2009, 14, 3229-3236. https://doi.org/10.3390/molecules14093229
Bayse CA, Antony S. Theoretical Studies of [2,3]-Sigmatropic Rearrangements of Allylic Selenoxides and Selenimides. Molecules. 2009; 14(9):3229-3236. https://doi.org/10.3390/molecules14093229
Chicago/Turabian StyleBayse, Craig A., and Sonia Antony. 2009. "Theoretical Studies of [2,3]-Sigmatropic Rearrangements of Allylic Selenoxides and Selenimides" Molecules 14, no. 9: 3229-3236. https://doi.org/10.3390/molecules14093229
APA StyleBayse, C. A., & Antony, S. (2009). Theoretical Studies of [2,3]-Sigmatropic Rearrangements of Allylic Selenoxides and Selenimides. Molecules, 14(9), 3229-3236. https://doi.org/10.3390/molecules14093229