Synthesis of Chiral 1,4-Disubstituted-1,2,3-Triazole Derivatives from Amino Acids

Abstract
1. Introduction
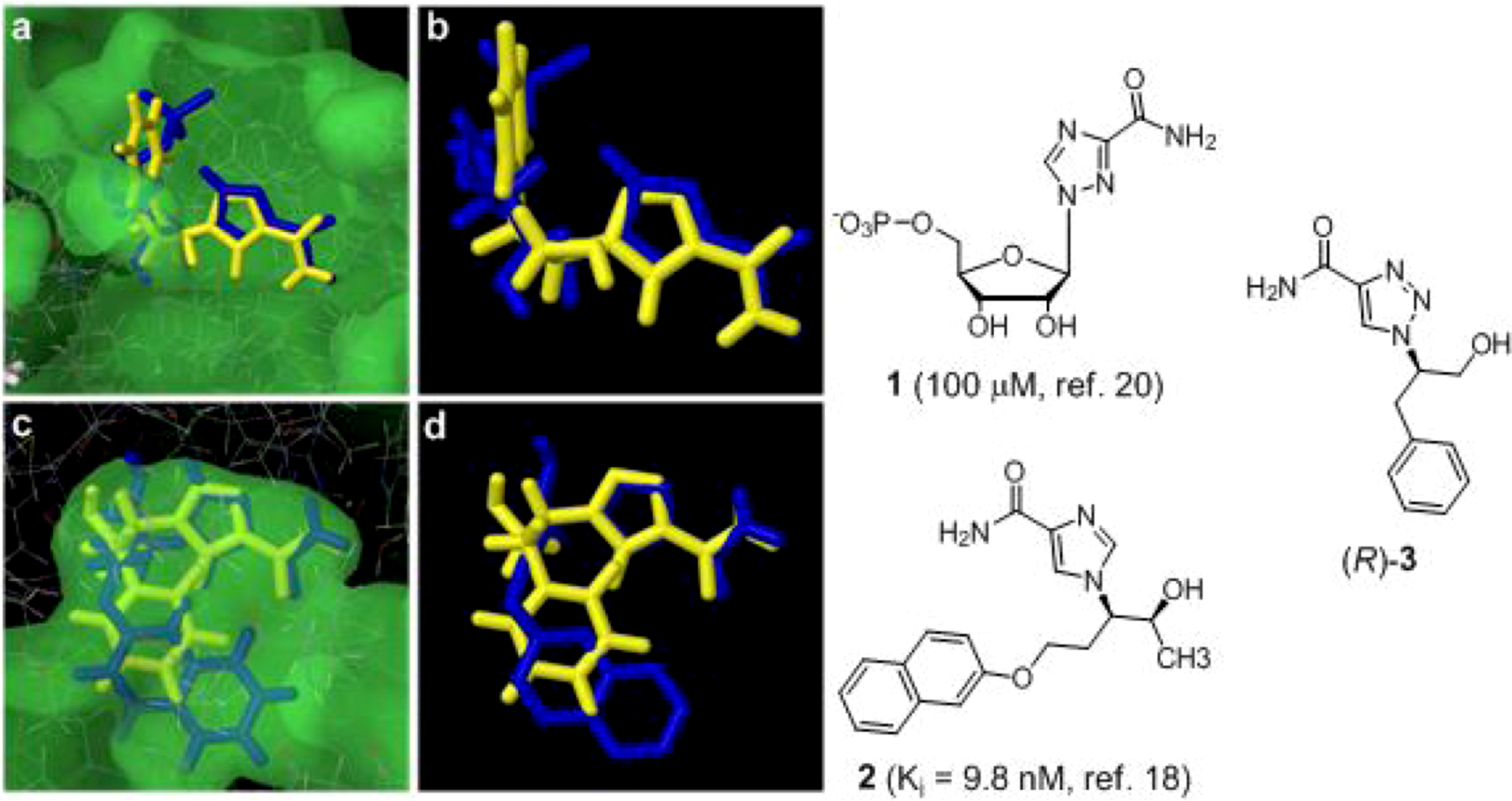
2. Results and Discussion
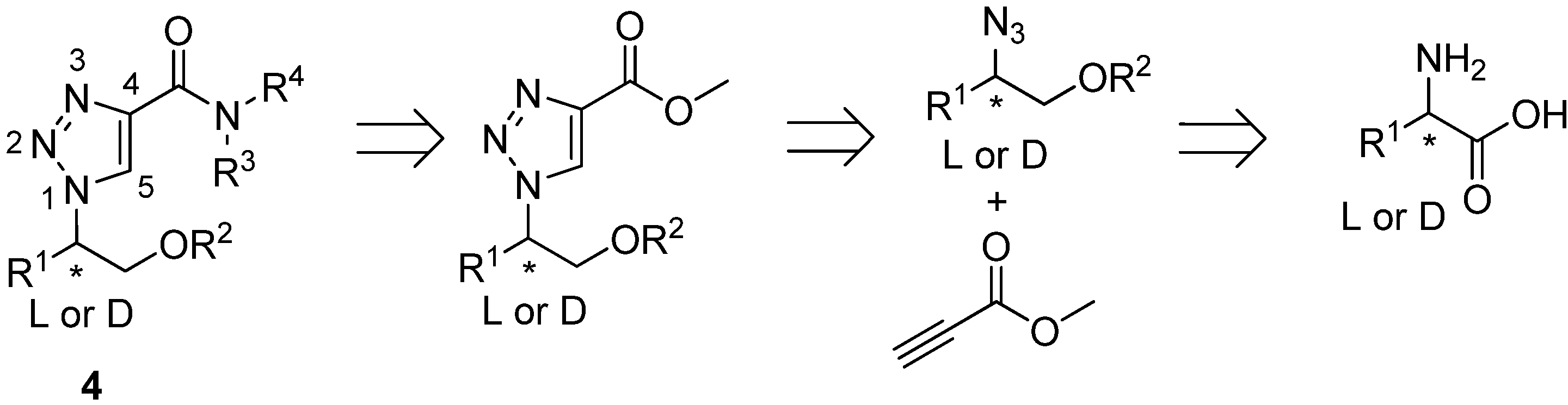

| Compound | R1 | R2 | Yield (%) |
|---|---|---|---|
| 5 | H | CH2Ph | - |
| 6 | CH2Ph | H | - |
| 7 | H | CH2CH2Ph | - |
| 8 | H | CH(CH3)2 | - |
| 9 | H | CH2OCH2Ph | - |
| 10 | H | CH2Ph | 79 |
| 11 | CH2Ph | H | 91 |
| 12 | H | CH2CH2Ph | 92 |
| 13 | H | CH(CH3)2 | 96 |
| 14 | H | CH2OCH2Ph | 97 |
| 15 | H | CH2Ph | 76 |
| 16 | CH2Ph | H | 72 |
| 17 | H | CH2CH2Ph | 74 |
| 18 | H | CH(CH3)2 | 70 |
| 19 | H | CH2OCH2Ph | 73 |
{kind=link}
{kind=link}
{kind=link}
| Compound | R1 | R2 | R3 | R4 | Yield (%) |
|---|---|---|---|---|---|
| 20 | H | CH2Ph | CH2Ph | - | 87 |
| 21 | CH2Ph | H | CH2Ph | - | 91 |
| 22 | H | CH2CH2Ph | CH2Ph | - | 92 |
| 23 | H | CH2OCH2Ph | Si(CH3)2C(CH3)3 | - | 67 |
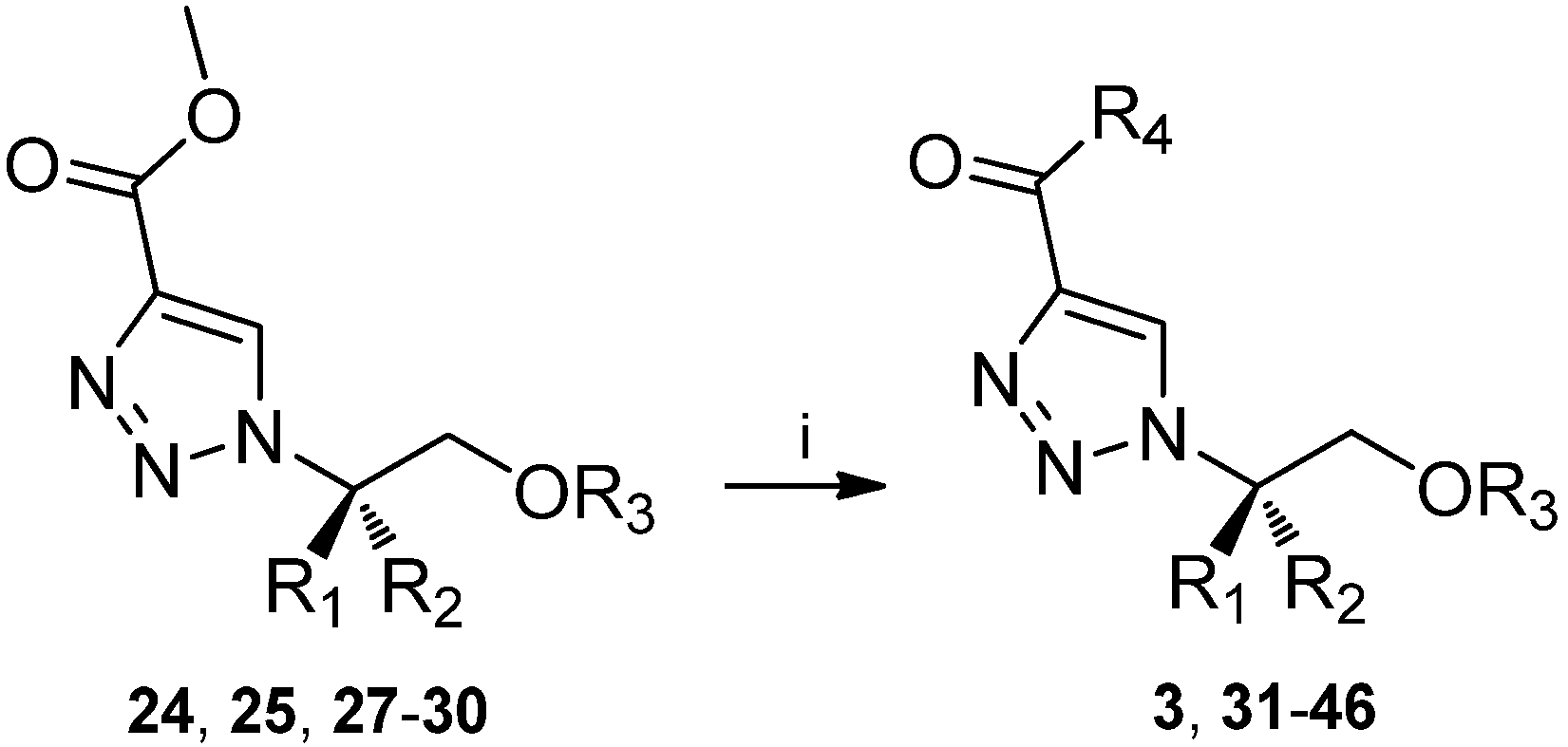
| 24 | H | CH2Ph | H | CO2CH3 | 71 |
| 25 | H | CH(CH3)2 | H | CO2CH3 | 74 |
| 26 | H | CH2Ph | H | Ph | 97 |
| 27 | H | CH2Ph | CH2Ph | CO2CH3 | 83 |
| 28 | CH2Ph | H | CH2Ph | CO2CH3 | 90 |
| 29 | H | CH2CH2Ph | CH2Ph | CO2CH3 | 82 |
| Compound | R1 | R2 | R3 | R4 | Yield (%) |
|---|---|---|---|---|---|
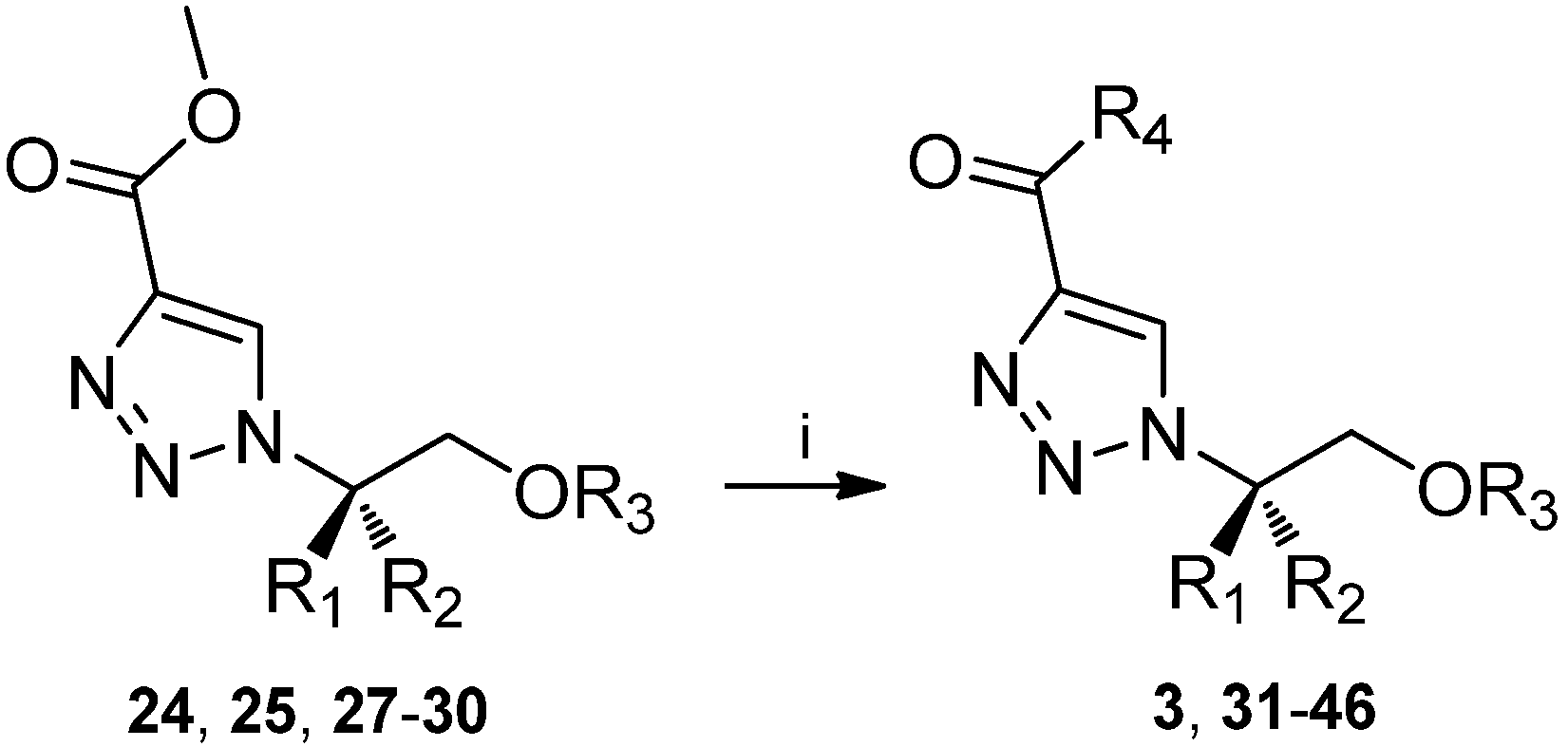
| (S)-3 | H | CH2Ph | H | NH2 | 97 |
| 31 | H | CH2Ph | H | NH(CH2)3CH3 | 58 |
| 32 | H | CH2Ph | H | NH(CH2)2OH | 4 |
| 33 | H | CH2Ph | H | NHCH2Ph | 35 |
| 34 | H | CH2Ph | H | N(CH2CH3)2 | 12 |
| 35 | H | CH2Ph | H | N(CH2CH2)2O | 30 |
| 36 | H | CH(CH3)2 | H | NH2 | 35 |
| 37 | H | CH2Ph | CH2Ph | NH2 | 92 |
| 38 | H | CH2Ph | CH2Ph | NH(CH2)3CH3 | 93 |
| 39 | H | CH2Ph | CH2Ph | NH(CH2)2OH | 81 |
| 40 | H | CH2Ph | CH2Ph | NHCH2Ph | 87 |
| 41 | H | CH2Ph | CH2Ph | N(CH2CH3)2 | 8 |
| 42 | H | CH2Ph | CH2Ph | N(CH2CH2)2O | 68 |
| 43 | CH2Ph | H | CH2Ph | NH(CH2)3CH3 | 98 |
| 44 | H | CH2OCH2Ph | CH2Ph | NH2 | 88 |
| 45 | H | CH2OCH2Ph | CH2Ph | NH(CH2)3CH3 | 93 |
| 46 | H | CH2OCH2Ph | Si(CH3)2C(CH3)3 | NH2 | 92 |
3. Experimental
3.1. General
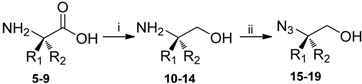
3.2. General procedure for preparation of amino alcohols
3.3. General procedure for preparation of azido alcohols by diazo transfer
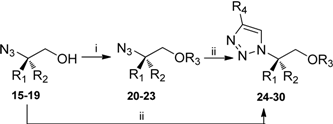
3.4. General procedure for benzylation of the hydroxyl functionality
3.5. General procedure for copper catalyzed cycloaddition reactions
3.6. General procedure for ester-amide conversion
3.7. Docking
4. Conclusions
Acknowledgements
- Sample Availability: Samples of the compounds are available from the authors.
References and Notes
- Fan, W.Q.; Katritzky, A.R. 1,2,3 Triazoles. In Comprehensive Heterocycle Chemistry II; Katritzky, A.R., Rees, C.W., Scriven, E.F.V., Eds.; Pergamon Press: New York, NY, USA, 1996; Volume 4, pp. 1–126. [Google Scholar]
- Katritzky, A.R.; Zhang, Y.; Singh, S.K. 1,2,3-Triazole formation under mild conditions via 1,3-dipolar cycloaddition of acetylenes with azides. Heterocycles 2003, 60, 1225–1239. [Google Scholar] [CrossRef]
- Alvarez, R.; Velazquez, S.; San, R.; Aquaro, S.; De, C.E.; Perno, C.F.; Karlsson, A.; Balzarini, J.; Camarasa, M.J. 1,2,3-Triazole-[2',5'-bis-O-(tert-butyldimethylsilyl)-beta-D- ribofuranosyl]-3'-spiro-5"-(4"-amino-1",2"-oxathiole 2",2"-dioxide) (TSAO) analogues: Synthesis and anti-HIV-1 activity. J. Med. Chem. 1994, 37, 4185–4194. [Google Scholar] [CrossRef]
- Velazquez, S.; Alvarez, R.; Perez, C.; Gago, F.; De, C.E.; Balzarini, J.; Camarasa, J.M. Regiospecific synthesis and anti-human immunodeficiency virus activity of novel 5-substituted N-alkylcarbamoyl and N,N-dialkylcarbamoyl 1,2,3-triazole-TSAO analogues. Antiviral Chem. Chemother. 1998, 9, 481–489. [Google Scholar]
- Whiting, M.; Muldoon, J.; Lin, Y.C.; Silverman, S.M.; Lindstrom, W.; Olson, A.J.; Kolb, H.C.; Finn, M.G.; Sharpless, B.K.; Elder, J.H.; Fokin, V.V. Inhibitors of HIV-1 protease by using in situ click chemistry. Angew. Chem. Int. Ed. 2006, 45, 1435–1439. [Google Scholar]
- Brik, A.; Muldoon, J.; Lin, Y.C.; Elder, J.C.; Goodsell, D.S.; Olson, A.J.; Fokin, V.V.; Sharpless, B.K.; Wong, C.H. Rapid diversity-oriented synthesis in microtiter plates for in situ screening of HIV protease inhibitors. Chem. Bio. Chem. 2003, 4, 1246–1248. [Google Scholar] [CrossRef]
- Genin, M.J.; Allwine, D.A.; Anderson, D.J.; Barbachyn, M.R.; Emmert, D.M.; Garmon, S.A.; Graber, D.R.; Grega, K.C.; Hester, J.B.; Hutchinson, D.K.; Morris, J.; Reischer, R.J.; Ford, C.W.; Zurenko, G.E.; Hamel, J.C.; Schaadt, R.D.; Stapert, D.; Yagi, B.H. Substituent effects on the antibacterial activity of nitrogen-carbon-linked (azolylphenyl)oxazolidinones with expanded activity against the fastidious gram-negative organisms Haemophilus influenzae and Moraxella catarrhalis. J. Med. Chem. 2000, 43, 953–970. [Google Scholar]
- Brockunier, L.L.; Parmee, E.R.; Ok, H.O.; Candelore, M.R.; Cascieri, M.A.; Colwell, L.F.; Deng, L.; Feeney, W.P.; Forrest, M.J.; Hom, G.J.; MacIntyre, D.E.; Tota, L.; Wyvratt, M.J.; Fisher, M. H.; Weber, A.E. Human beta3-adrenergic receptor agonists containing 1,2,3-triazole-substituted benzenesulfonamides. Bioorg. Med. Chem. Lett. 2000, 10, 2111–2114. [Google Scholar] [CrossRef]
- Pande, V.; Ramos, M.J. Structural basis for the GSK-3beta binding affinity and selectivity against CDK-2 of 1-(4-aminofurazan-3yl)-5-dialkylaminomethyl-1H-[1,2,3]triazole-4-carboxylic acid derivatives. Bioorg. Med. Chem. Lett. 2005, 15, 5129–5135. [Google Scholar] [CrossRef]
- Olesen, P.H.; Sørensen, A.R.; Ursö, B.; Kurtzhals, P.; Bowler, A.N.; Ehrbar, U.; Hansen, B.F. Synthesis and in vitro characterization of 1-(4-Aminofurazan-3-yl)-5-dialkylaminomethyl-1H-[1,2,3]triazole-4-carboxylic acid derivatives. A new class of selective GSK-3 Inhibitors. J. Med. Chem. 2003, 46, 3333–3341. [Google Scholar] [CrossRef]
- Krasinski, A.; Radic, Z.; Manetsch, R.; Raushel, J.; Taylor, P.; Sharpless, B.K.; Kolb, H.C. In situ selection of lead compounds by click chemistry: Target-guided optimization of aceylcholinesterase inhibitors. J. Am. Chem. Soc. 2005, 127, 6686–6692. [Google Scholar]
- Mocharla, V.P.; Colasson, B.; Lee, L.V.; Roeper, S.; Sharpless, B.K.; Wong, C.H.; Kolb, H.C. In situ click chemistry: Enzyme-generated inhibitors of carbonic anhydrase II. Angew. Chem. Int. Ed. 2005, 44, 116–120. [Google Scholar]
- Micetich, R.G.; Maiti, S.N.; Spevak, P.; Hall, T.W.; Yamabe, S.; Ishida, N.; Tanaka, M.; Yamazaki, T.; Nakai, A.; Ogawa, K. Synthesis and .beta.-lactamase inhibitory properties of 2.beta.-[(1,2,3-triazol-1-yl)methyl]-2.alpha.-methylpenam-3.alpha.-carboxylic acid 1,1-dioxide and related triazolyl derivatives. J. Med. Chem. 1987, 30, 594–601. [Google Scholar]
- Actor, P.; Uri, J.V.; Phillips, L.; Sachs, C.S.; Zajac, J.R.G.; Berges, D.A.; Dunn, G.L.; Hoover, J.R.E.; Weisbach, J.A. Laboratory studies with cefatrizine (SK + F 60771), a new broad-spectrum orally-active cephalosporin. J. Antibiot. 1975, 28, 594–601. [Google Scholar] [CrossRef] [Green Version]
- Sidwell, R.W.; Huffman, J.H.; Khare, G.P.; Allen, L.B.; Witkowski, J.T.; Robins, R.K. Broad-spectrum antiviral activity of Virazole: 1-Beta-D-ribofuranosyl-1,2,4-triazole-3-carboxamide. Science 1972, 177, 705–706. [Google Scholar]
- Terasaka, T.; Kinoshita, T.; Kuno, M.; Nakanishi, I. A highly potent non-nucleoside adenosine deaminase inhibitor: Efficient drug discovery by intentional lead hybridization. J. Am. Chem. Soc. 2004, 126, 34–35. [Google Scholar] [CrossRef]
- Terasaka, T.; Kinoshita, T.; Kuno, M.; Seki, N.; Tanaka, K.; Nakanishi, I. Structure-based design, synthesis, and structure-activity relationship studies of novel non-nucleoside adenosine deaminase inhibitors. J. Med. Chem. 2004, 47, 3730–3743. [Google Scholar] [CrossRef]
- Terasaka, T.; Okumura, H.; Tsuji, K.; Kato, T.; Nakanishi, I.; Kinoshita, T.; Kato, Y.; Kuno, M.; Seki, N.; Naoe, Y.; Inoue, T.; Tanaka, K.; Nakamura, K. Structure-based design and synthesis of non-nucleoside, potent, and orally bioavailable adenosine deaminase inhibitors. J. Med. Chem. 2004, 47, 2728–2731. [Google Scholar] [CrossRef]
- Terasaka, T.; Tsuji, K.; Kato, T.; Nakanishi, I.; Kinoshita, T.; Kato, Y.; Kuno, M.; Inoue, T.; Tanaka, K.; Nakamura, K. Rational design of non-nucleoside, potent, and orally bioavailable adenosine deaminase inhibitors: predicting enzyme conformational change and metabolism. J. Med. Chem. 2005, 48, 4750–4753. [Google Scholar] [CrossRef]
- Benarroch, D.; Egloff, M.P.; Mulard, L.; Guerreiro, C.; Romette, J.L.; Canard, B. A structural basis for the inhibition of the NS5 dengue virus mRNA 2′-O-methyltransferase domain by ribavirin 5′-triphosphate. J. Biol. Chem. 2004, 279, 35638–35643. [Google Scholar]
- Tornøe, C.; Christensen, C.; Meldal, M. Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, B.K. A stepwise huisgen cycloaddition process: Copper(I)-catalyzed regioselective ligation of azides and terminal alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Laufer, S.A.; Domeyer, D.M.; Scior, T.R.F.; Albrecht, W.; Hauser, D.R.J. Synthesis and biological testing of purine derivatives as potential ATP-competitive kinase inhibitors. J. Med. Chem. 2005, 48, 710–722. [Google Scholar] [CrossRef]
- Zaloom, J.; Roberts, D.C. Preparation of azido derivatives from amino acids and peptides by diazo transfer. J. Org. Chem. 1981, 46, 5173–5176. [Google Scholar] [CrossRef]
- Vasella, A.; Witzig, C.; Chiara, J.L.; Lomas, M.M. Convenient synthesis of 2-Azido-2-deoxy-aldoses by diazo transfer. Helv. Chim. Acta 1991, 74, 2073–2077. [Google Scholar] [CrossRef]
- Hedenström, M.; Holm, L.; Yuan, Z.Q.; Emtenas, H.; Kihlberg, J. Stereoselective Synthesis of Ψ[CH2O] Pseudodipeptides and Conformational Analysis of a PheΨ[CH2O]Ala Containing Analogue of the Drug Desmopressin. Bioorg. Med. Chem. Lett. 2002, 12, 841–844. [Google Scholar] [CrossRef]
- Hedenström, M.; Yuan, Z.; Brickmann, K.; Carlsson, J.; Ekholm, K.; Johansson, B.; Kreutz, E.; Nilsson, A.; Sethson, I.; Kihlberg, J. Conformations and receptor activity of desmopressin analogues, which contain gamma-turn mimetics or a psi[CH(2)O] isostere. J. Med. Chem. 2002, 45, 2501–2511. [Google Scholar] [CrossRef]
- Ramanathan, S.K.; Keeler, J.; Lee, H.L.; Reddy, D.S.; Lushington, G.; Aube, J. Modular synthesis of cyclic peptidomimetics inspired by γ-turns. Org. Lett. 2005, 7, 1059–1062. [Google Scholar] [CrossRef]
- Gerard, B.; Ryan, J.; Beeler, A.B.; Porco, J.A., Jr. Synthesis of 1,4,5-trisubstituted-1,2,3-triazoles by copper-catalyzed cycloaddition-coupling of azides and terminal. Tetrahedron 2006, 62, 6405–6411. [Google Scholar] [CrossRef]
- Wang, Q.; Chan, T.R.; Hilgraf, R.; Fokin, V.V.; Sharpless, B.K.; Finn, M.G. Bioconjugation by copper(I)-catalyzed azide-alkyne [3+2] cycloaddition. J. Am. Chem. Soc. 2003, 125, 3192–3193. [Google Scholar]
- Himo, F.; Lovell, T.; Hilgraf, R.; Rostovtsev, V.V.; Noodleman, L.; Sharpless, B.K.; Fokin, V.V. Copper(I)-catalyzed synthesis of azoles. DFT study predicts unprecedented reactivity and intermediates. J. Am. Chem. Soc. 2005, 127, 210–216. [Google Scholar]
- Chan, T.R.; Hilgraf, R.; Sharpless, B.K.; Fokin, V.V. Polytriazoles as copper(I)-stabilizing ligands in catalysis. Org. Lett. 2004, 6, 2853–2855. [Google Scholar] [CrossRef]
- Merlo, A.A.; Fernandes, M.S. An efficient strategy for the synthesis of chiral liquid crystals using Evans' methodology. Synth. Commun. 2003, 33, 1167–1178. [Google Scholar] [CrossRef]
- Park, C.S.; Kim, M.S.; Sim, T.B.; Pyun, D.K.; Lee, C.H.; Choi, D.; Lee, W.K.; Chang, J.W.; Ha, H.J. Novel stereoselective synthesis of functionalized oxazolidinones from chiral aziridines. J. Org. Chem. 2003, 68, 43–49. [Google Scholar]
- Welch, J.T.; Seper, K.W. Synthesis, regioselective deprotonation, and stereoselective alkylation of fluoro ketimines. J. Org. Chem. 1988, 53, 2991–2999. [Google Scholar] [CrossRef]
- Danklmaier, J.; Hoenig, H. Synthese diastereomerenreiner 2,5-disubstituierter 3-Oxoperhydro-1,4-oxazine. Liebigs Ann. Chem. 1988, 851–854. [Google Scholar] [CrossRef]
- Macromodel [9.1]; Schrödinger, LLC: New York, NY, USA, 2007.
- Glide [4.0]; Schrödinger, LLC: New York, NY, USA, 2005.
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein−ligand complexes. J. Med. Chem. 2006, 49, 6196. [Google Scholar]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Klein, M.; Krainz, K.; Redwan, I.N.; Dinér, P.; Grøtli, M. Synthesis of Chiral 1,4-Disubstituted-1,2,3-Triazole Derivatives from Amino Acids. Molecules 2009, 14, 5124-5143. https://doi.org/10.3390/molecules14125124
Klein M, Krainz K, Redwan IN, Dinér P, Grøtli M. Synthesis of Chiral 1,4-Disubstituted-1,2,3-Triazole Derivatives from Amino Acids. Molecules. 2009; 14(12):5124-5143. https://doi.org/10.3390/molecules14125124
Chicago/Turabian StyleKlein, Michael, Karin Krainz, Itedale Namro Redwan, Peter Dinér, and Morten Grøtli. 2009. "Synthesis of Chiral 1,4-Disubstituted-1,2,3-Triazole Derivatives from Amino Acids" Molecules 14, no. 12: 5124-5143. https://doi.org/10.3390/molecules14125124
APA StyleKlein, M., Krainz, K., Redwan, I. N., Dinér, P., & Grøtli, M. (2009). Synthesis of Chiral 1,4-Disubstituted-1,2,3-Triazole Derivatives from Amino Acids. Molecules, 14(12), 5124-5143. https://doi.org/10.3390/molecules14125124