Abstract
The C2-symmetrical “salen” ligand (+)-9 bearing two [5]ferrocenophane substituents has been prepared in five steps starting from readily available diacetylferrocene, p-hydroxybenzaldehyde and (R,R)-N,N’-diphenylethylenediamine. Reaction of (+)-9 with Mn(OAc)3, Co(OAc)2 ZnEt2 or UO2(OAc)2 gave the corresponding metal-complexes which were characterised by spectroscopic methods.
1. Introduction
Chiral N,N’-bis(salicylidene)ethylendiamine (salen) compounds are very popular ligands because of their easy formation and rich coordination chemistry with a large variety of metal ions, that has allowed their use as catalysts in different asymmetric reactions [1,2]. The Mn(salen) complexes have been mainly employed in the enantioselective epoxidation of cis-olefins [3,4] and their efficiency in the asymmetric induction has been related with their structural features [5,6]. The asymmetric ring opening of epoxides and the hydrolytic kinetic resolution of epoxides are actively promoted by Cr- or Co(salen) complexes [7,8,9], and the latter have been recently found interesting application in the stereoselective recognition of aminoalcohols and aminoacids [10,11].
Asymmetric alkylation of α-aminoacid enolates can be carried out in the presence of Cu(II)- or Co(II)salen catalysts [12], both possessing square-planar coordination, whereas the alkynylation of ketones has been performed with moderate enantioselectivity using Zn(salen) complexes [13,14].
More recently the salen framework has been used as building block in the creation of sophisticated homogeneous, multimetal catalysts and some salen complexes have been specifically designed for their application as functional materials [15]. The incorporation of “salen” moieties into macrocyclic structures able to give rise to supramolecular interactions [16] and the synthesis of salen compounds bearing Lewis acid or Lewis base activating groups are currently investigated for the development of more active catalysts [17,18].
Ferrocenyl compounds display unique electronic, steric and chemical properties which have been exploited in the synthesis of a great variety of derivatives and their useful application in different field of chemistry is well documented [19,20]. Our interest in the preparation of chiral ferrocenes [21,22] as well as in the development of new salen catalysts [23,24] prompted us to plan the synthesis of novel compounds bearing both these structural elements.
Some achiral ferrocene-containing salen ligands have been reported and their coordination behaviour [25,26] or catalytic activity [27] investigated. Enantiopure derivatives, potentially useful in asymmetric homogeneous catalysis, have been prepared by Bildstein and coworkers [28] incorporating planar chiral ferrocenes into the salen backbone.
The synthesis of two salen-type ligands having the ferrocenyl moiety on the chiral diimine bridge have been also described and one of them showed an unusual selectivity in the epoxidation of cis-olefin affording trans-epoxide as main isomer, but the asymmetric induction was not satisfactory [29]. Here we describe the synthesis of a chiral C2-symmetrical ligand, bearing a ferrocene system as substituent on the 5,5’-positions of the salicylidene unit, together with the spectroscopic characterization of its complexes with some metals.
2. Results and Discussion
2.1. Synthesis of the ligand and its metal complexes
Since the steric and electronic features of the 3,3’- or 5,5’-substituents of a salen catalyst can exert some influence on its efficiency in the asymmetric induction process [5,6,30,31], the introduction of a ferrocenyl substituent, with its peculiar properties, on the salicylaldeyde moiety was considered in the design of novel salen ligands. Among the possible ferrocenyl substituents, the ferrocenophane system, in which the two cyclopentadienyl rings are joined by an alkyl chain, appears attractive due to its rigidity and the presence of a cavity that could play a role in determining the direction of approach of the reagents to the catalytic site.
The synthesis of 1,5-dioxo-3-substituted [5]ferrocenophanes has been recently reported to occur in high yield by simple mixing of 1,1’-diacetylferrocene 1 and a suitable aldehyde in NaOH/ethanol solution under microwave irradiation [32]. The reaction can be also applied to salicylaldehydes, but better results were obtained using the corresponding O-benzylderivatives, so 2-hydroxybenzaldehyde or 4-hydroxybenzaldeyde were reacted with p-methylbenzylbromide in acetone in the presence of CsCO3 at room temperature for 12 h to afford compounds 2 and 3 in almost quantitative yields. The treatment of 1 with 2 in the reported conditions gave the [5]ferrocenophane 4 as a yellow solid, practically insoluble in all the common organic solvents, even after its O-deprotection. Deoxygenation of 4 by treatment with excess of borane in CH2Cl2 and removal of the benzyl group with hydrogen over Pd/C gave the alkylderivative 6 in 85% overall yield, this compound being soluble and more suitable for the following step of formylation on the aryl ring. The same reaction sequence was applied to prepare the isomeric [5]ferrocenophane 7 starting from 1 and 3 (Scheme 1).
Firstly a Gross-Rieche formylation was attempted on derivative 6, but it was quickly converted into a complex mixture of products and no selectivity for the formation of monoformyl derivative 8 was achieved either varying the reagent ratios or the reaction temperature. In the Riemar-Tiemann conditions, a selective 25% conversion of 6 into 8 was evidenced after 24 h, so the reaction was quenched at this stage and purified to recover the unreacted substrate that could be recycled and after four reaction cycles, the target aldehyde 8 was obtained in 55% overall yield.
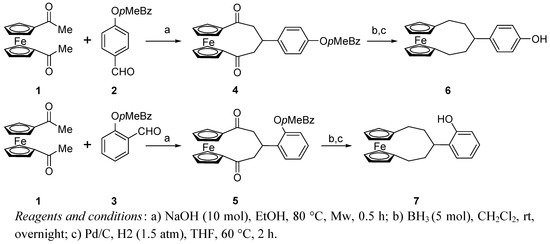
Scheme 1.
Synthesis of [5]ferrocenophane compounds.
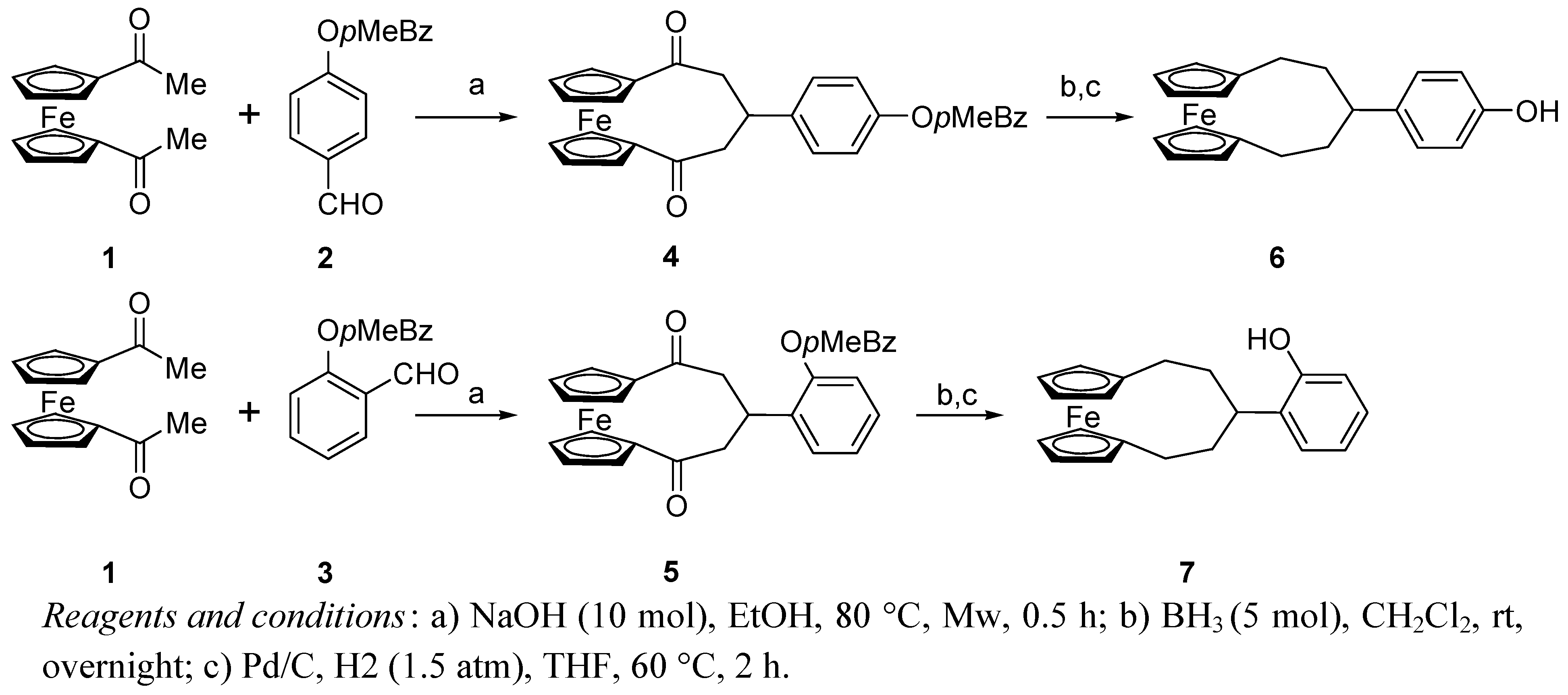
Unfortunately, all the attempts at ortho-formylation of 7 using different procedures failed and the para-substitution product was formed in CHCl3/KOH system, so that we were only able to prepare ligand (+)-9 by condensation of 8 with (+)-(1R,2R)-diphenylethylendiamine [(R,R)-dpen] (Scheme 2).
The symmetrical structure of (+)-9 was confirmed by its 1H-NMR spectrum and diagnostic resonances at δ 8.41 and 13.16 for the iminic and phenolic protons, respectively, together with a singlet at δ 4.76 for the protons on the diimine bridge were observed. The methylenic protons on the ferrocenophane bridge gave rise to four separate multiplets in the range 1.8-2.4 ppm whereas the methinic signal was overlapped by the cycplopentadienyl resonances. Complete assignment of the carbon resonances by 2D-NMR spectra and molecular peaks at m/z 924.2 in the ESI-MS spectrum also supported the structure of ligand (+)-9.
The treatment of (+)-9 with Mn(OAc)3 or Co(OAc)2 in EtOH gave the corresponding complexes (−)-10 and (+)-11 in high yield; due to their paramagnetic nature they were characterised by their ESI-MS and circular dichroism spectra (vide infra).
The structure of salen-Zn complex (–)-12, prepared by reacting the free ligand (+)-9 with Et2Zn in toluene, was mainly established by NMR analysis. The 1H-NMR spectrum of (–)-12 in CDCl3 was fully unresolved whereas in (CD3)2SO all the resonances could be assigned and the symmetric nature of the complex was evidenced; the main features of the spectrum are the absence of the phenolic signal and a slight upfield shift for both the resonances of azomethine and N-methinic protons compared with those of the free ligand. Complexes 10-12 (Figure 1) are quite stable and have the potential to be used as catalysts in asymmetric synthesis.
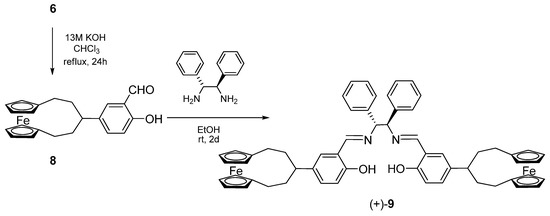
Scheme 2.
Synthesis of the salen ligand bearing ferrocenophane substituents.
Figure 1.
Metal complexes of salen ligand (+)-9.
Salen-uranyl(VI) complexes have been prepared as models for the pseudo-octahedral salen Mn=O species responsible for the selective oxygen transfer in the asymmetric epoxidation of olefins [23] and recently some activity in molecular recognition has been reported for these complexes [33,34].
The reaction of (+)-9 with (UO2)(OAc)2 gave the uranyl complex (+)-13 as a brown solid, whose structure was fully assigned from its 1H- and 13C-NMR spectra registered in (CD3)2SO. The lack of phenolic resonance in the protonic spectrum of 13 and the marked deshielding of the azomethinic and aminic protons (Δδ = +1.05 and Δδ = +1.47 ppm, respectively, compared with the free ligand 9) were taken as an evidence of the nitrogen and oxygen coordination with the metal, which acts as an electron withdrawing group. In the 13C-NMR spectrum, the diagnostic resonances for the C-OH and C=N carbons were also shifted downfield upon complexation showing large Δδ values (Δδ = +9.13 and +5.53 ppm, respectively).
The 1H-NMR spectrum of 13 registered in CDCl3 showed broad resonances for all the protons, but two broad singlets in 1:1 ratio at 298 K were distinguishable for the azomethinic group and assigned to two diastereomeric conformers which slowly interconverted on the NMR time scale. By warming, the above resonances tended to collapse and at 338K a 80:20 ratio of the two conformers was measured. In the case of uranyl complex of 14, with the same aminic moiety of (+)-13 and taken as a reference, two conformers were also observed in CDCl3, but not in (CD3)2SO, with a coalescence temperature of 300K. The slower interconversion rate of (+)-13 compared with the (UO2)-14 complex could be ascribed to the presence of bulky ferrocenophane substituents.
The additional coordination of a molecule of ethanol to the uranium atom has been reported for some salen-uranyl complexes on the basis of elemental analyses data [35] and it can be easily detected by 1H-NMR but no evidence of such coordination was obtained for (+)-13.
2.2. Circular dichroism spectra
Circular dichroism (CD) spectroscopy has been applied as a useful tool in the assignment of absolute configuration as well as conformational analysis of chiral molecules and biological polymers [36,37,38]. The development of quantum chemical CD calculations to be compared with experimental CD investigations has increased the reliability of the method and its extension to several classes of compounds [39,40]. The electronic absorption and the associated circular dichroism spectra have been reported for several salicylaldimine complexes with different cations and related with the coordination geometry of the central metal and the conformation of the diamine chelate ring [41,42,43,44].
The electronic and CD spectra of the free ligand (+)-9 and the corresponding metal complexes were registered in CH3CN, except for (–)-12 that was unsoluble in this solvent, and no substantial differences were observed in CH3Cl.
In the UV-vis spectrum of (+)-9 the azomethine π-π* transition band was observed at 338 nm and the benzene π-π* transitions gave rise to overlapping bands below 250 nm. Although two bands assigned to metal-ligand and intraligand charge transfer transitions could be expected for the ferrocene chromophore in the range 300-600 nm [45], they were not discernible, perhaps due to their low-intensity.
The CD spectrum of (+)-9 displayed a negative Cotton effect (CE) associated with the π-π* transition of the azomethine chromophore, in agreement with the R,R-configuration of the diamine moiety [41]. The region 210-270 nm appeared better resolved in the CD than the UV-vis spectrum, but is clearly complicated by multiple CEs arising from benzenoid transitions involving salicylaldimine rings, phenyl rings on the (R,R)-dpen moiety and cyclopentadienyl rings (Figure 2).
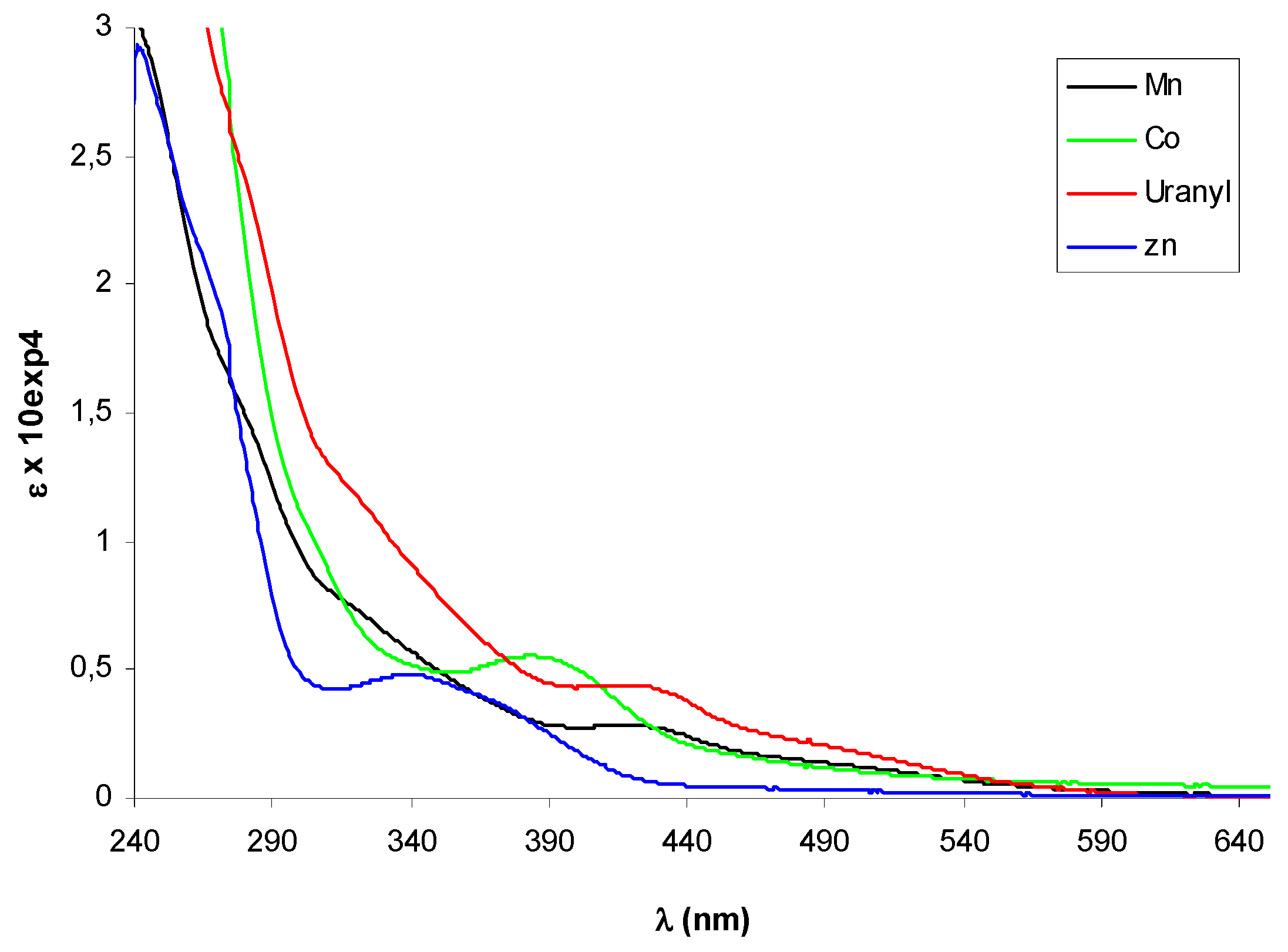
In the UV-vis spectra of complexes 10-13 additional absorption bands due to the presence of the metal could be expected in the 380-420 nm region, where ligand-metal charge transfer transitions (LMCT) as d-π∗ (azomethine) are usually observed, and in the 500-600 nm range, the typical absorption region for d-d transitions. The diagnostic azomethine π-π* transition was easily detected for the Zn-complex (–)-12 appearing slight shifted with a maximum at 348 nm, but it seemed hidden by other bands in the spectra of the other complexes, that displayed a weaks band (ε < 6000) in the LMCT region (Figure 3).
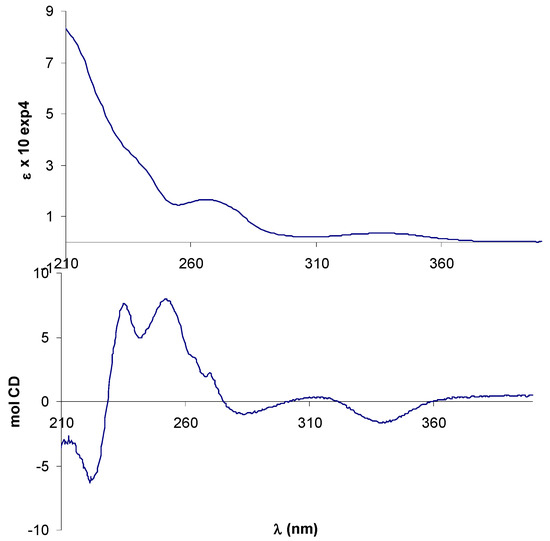
Figure 2.
Electronic (up) and CD (bottom) spectra of free ligand (+)-9 in CH3CN.
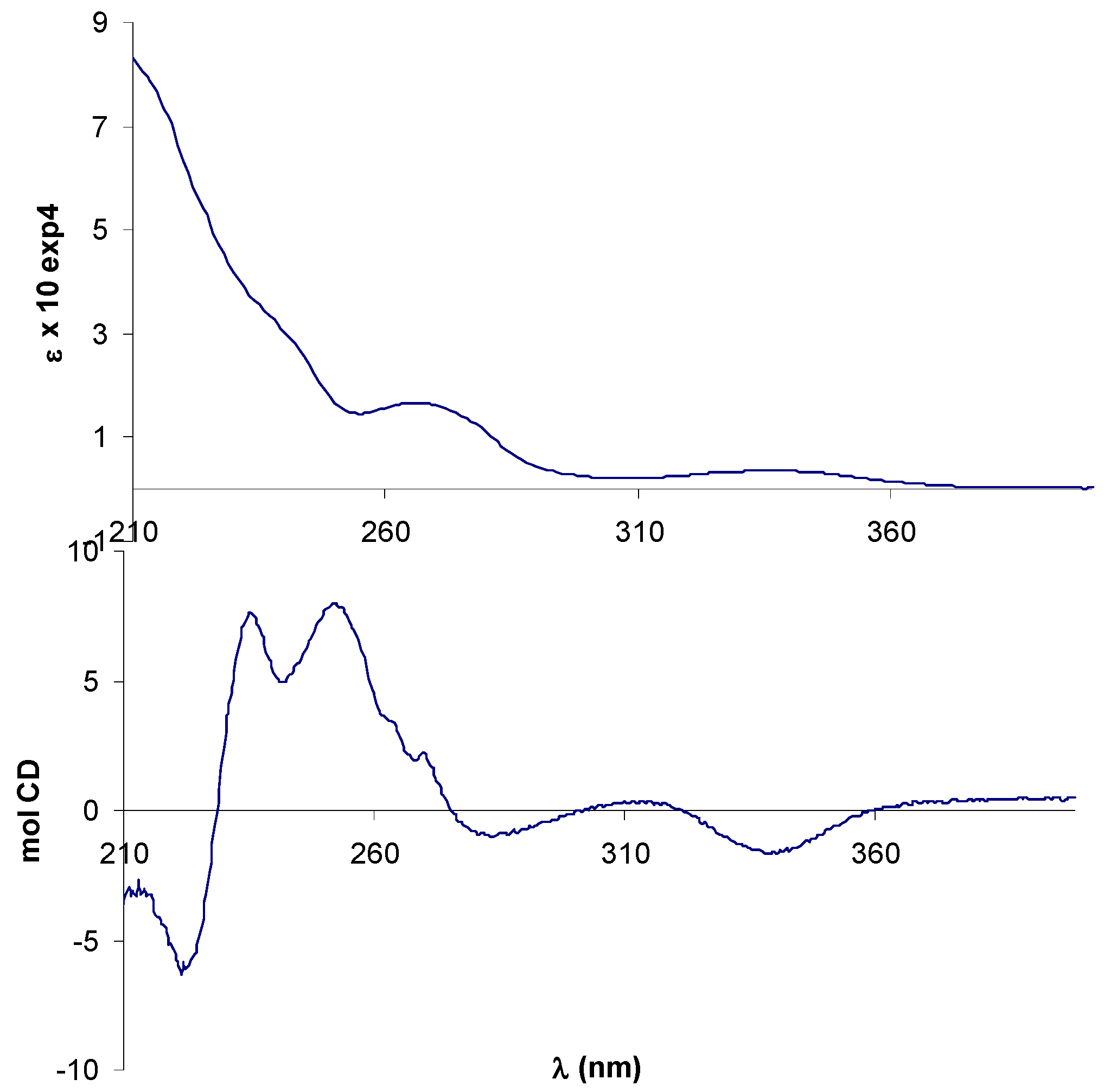
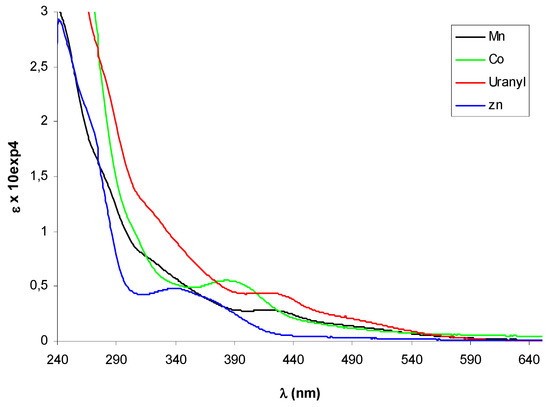
Figure 3.
UV-vis spectra of metal complexes of (+)-9. The spectrum of Zn(II)-complex was registered in CHCl3, the others in CH3CN.
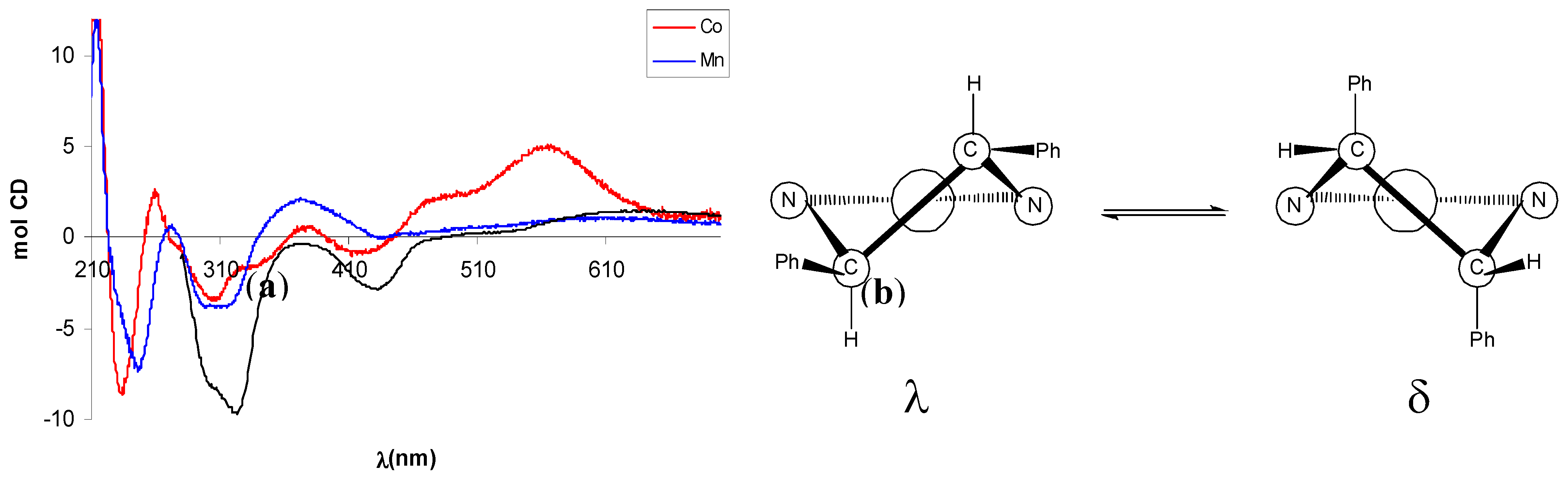
In the CD spectrum of Mn(III)-complex (–)-10 the negative CE around 320 nm, better resolved in CHCl3, can be related to a λ configuration of the five-membered ethylendiamine chelate ring on the basis of literature data [42]. Since the absolute configuration of the amine moiety is (R,R), a λ conformation of the chelate ring implies an equatorial disposition of the phenyl rings, in agreement with a repulsion between the axial phenyl groups and the apical acetate ligand that makes the other possible conformation δ less favoured (Figure 4).
The CD spectrum of Co(II)-complex (+)-11 showed some degree of similarity with that of (–)-10, with a more intense CE related with d-d transitions at 560 nm (Figure 4). In analogy with tetradentate Schiff base Ni(II) or Cu(II) complexes [43,44], possessing the same square-planar geometry of Co(II) complexes, this band is diagnostic for the conformational assignment and a positive CE has been related with a shift of the conformational λ,δ-equilibrium toward the λ form.
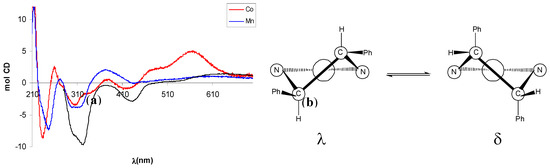
Figure 4.
(a) CD spectra of Mn(III)-complex (–)-10 and Co(II)-complex (+)-11. Black line refers to (–)-10 in CHCl3. (b) Conformations and phenyl orientation for (R,R)-dpen diamine chelate ring.
The CD spectra of (–)-12 and (+)-13, shown in Figure 5, are in good agreement with the reported data for the analogue complexes of 14 [35], but they not allow any conformational assignment since empirical rules establishing its relationship with the sign of a specific CE have not been developed for the salen complexes with these metals.
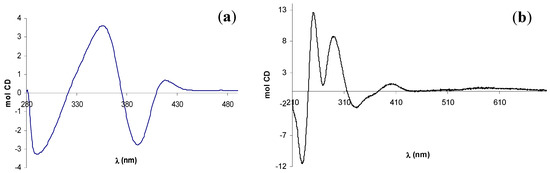
Figure 5.
CD spectra of (a) zinc(II) complex (–)-12 and (b) UO2-complex (+)-13.
3. Experimental
3.1. General methods
Reactions under microwave irradiation were performed in a 100 mL open vessel using a CEM Discover Benchmate equipped with temperature control device. Microprilled sodium hydroxide was purchased from Riedel-de-Haën. Diacetylferrocene and (1R,2R)-diphenylethylendiamine were available from Aldrich. Column chromatography was performed on Si 60 (230-400 mesh) silica gel using the specified eluants. 1H- and 13C-NMR spectra were registered in the specified deuterated solvents at 400 and 100 MHz, respectively. 2D-NMR were performed using standard Bruker programs. Chemical shifts (δ) are given as ppm relative to the residual solvent peak and coupling constants (J) are in Hz. In the NMR assignments, Cp and Ar refers to cyclopentadienyl and phenyl rings respectively. Melting points are uncorrected. Optical rotations were measured on a DIP 135 JASCO instrument. CD spectra were run at 20 °C on a JASCO J-810 spectropolarimeter using solutions in CH3CN ranging from 10-4 to 10-5 mol/L and 1 cm quartz cells. ESI mass spectra were obtained by employing an ES-MS Thermo-Finnigan LCQ-DECA spectrometer equipped with an ion trap analyzer and acquired in positive mode using 5V capillary voltage and 220 °C capillary temperature.
3.2. Synthesis of 1,5-dioxo-3-(p-methylbenzyloxyphenyl)[5]ferrocenophane (4)
To a solution of 4-hydroxybenzaldehyde in acetone (2.5 g, 20 mmol), 4-methylbenzylbromide (3.8 g, 20 mmol) and CsCO3 (9.1 g, 28 mmol) were added and the mixture left to react under stirring at rt. After 20 h the mixture was filtered and the solution was taken to dryness. The residue was purified on a Silica gel column and protected aldehyde 2 was recovered in 92% yield. In a 100 mL flask equipped with a reflux condenser, diacetylferrocene 1 (2 mmol), aldehyde 2 and NaOH were mixed in 1:1:10 mol ratio and dissolved in 4:1 v/v EtOH/H2O (20 mL) and the mixture was subjected to 50W microwave irradiation for 30 min with 80 °C temperature setting [32]. After dilution with CH2Cl2 the mixture was extracted with satd. NH4Cl and the organic layer taken to dryness. The residue was washed twice with n-hexane/EtOAc to remove the reagents, leaving compound 4 as a gold yellow solid in 90% yield which was used without further purification. 1H-NMR (CDCl3) δ: 2.38 (s, 3H, CH3), 2.50 (m, 2H, CH2), 2.91 (t, 2H, J = 11.9, CH2), 4.31 (br t, 1H, CH), 4.58 (s, 2H, Cp), 4.62 (2H, Cp), 4.88 (s, 4H, Cp), 5.03 (s, 2H, CH2), 6.97 (d, 2H, J = 8.6, Ar), 7.21 (d, 2H, J = 7.9, Ar), 7.28 (d, 2H, J = 8.6, Ar), 7.34 (d, 2H, J = 7.9, Ar); Anal. Calcd. For C29H26FeO3: C, 72.81; H, 5.48. Found: C, 73.25; H, 5.54.
3.3. Synthesis of 4-([5]ferrocenophane-3-yl)-phenol (6)
To a solution of 4 (0.5 g, 1.1 mmol) in CH2Cl2 (150 mL) was added BH3/Me2S (2M in THF, 1.2 mL) and the reaction stirred overnight at room temperature. After quenching with MeOH (caution: gas evolution) the reaction mixture was extracted with sat. NH4Cl and the organic layer taken to dryness. The residue was dissolved in THF (20 mL) and to this solution Pd/C (50 mg) was added and the suspension stirred under H2 atmosphere (1.2 bar) at 60 °C for 3 h. As soon as quantitative conversion of the substrate was detected by TLC, the solid was removed by filtration through a short plug of Celite and the solution taken to dryness. The residue was purified by silica gel chromatography (n-hexane:EtOAc 8:2) to afford pure 6 as a gold-colored solid (0.3 g, 85% yield), mp 138-139 °C, 1H-NMR (CDCl3) δ: 1.93 (m, 2H, CH2), 2.02 (m, 2H, CH2), 2.21 (dt, 2H, J = 15.5, 5.1 and 5.1, CH2), 2.47 (ddd, 2H, J = 15.5, 10.1 and 3.9, CH2), 4.04 (s, 2H, Cp), 4.07 (br s, 5H, Cp and CH), 4.20 (s, 2H, Cp), 4.74 (s, 1H, OH), 6.82 (d, 2H, J = 8.3, Ar), 7.22 (d, 2H, J = 8.3, Ar); 13C-NMR (CDCl3) δ: 23.93 (CH2), 32.69 (CH2), 39.42 (CH), 66.20 (CpH), 67.16 (CpH), 68.03 (CpH), 69.24 (CpH), 89.33 (Cp), 115.20 (Ar-H), 128.73 (Ar), 140.57 (Ar), 153.65 (Ar-OH); ESI-MS: m/z 346.4 [M+]; Anal. Calcd. For C21H22FeO: C, 72.85; H, 6.40. Found: C, 73.58; H, 6.47.
3.4. Synthesis of 2-([5]ferrocenophane-3-yl)-phenol (7)
Starting from salicylaldehyde and applying the same reaction sequence described for compound 6, the isomeric derivative 7 was obtained in 78% overall yield (referred to 1). 1H-NMR (CDCl3) δ: 2.00 (m, 4H, 2 x CH2), 2.28 (dt, 2H, J = 15.6, 4.9 and 4.9, CH2), 2.54 (ddd, 2H, J = 15.6, 10.3 and 4.6, CH2), 4.08 (s, 2H, Cp), 4.10 (s, 2H, Cp), 4.22 (s, 2H, Cp), 4.26 (s, 2H, Cp), 4.61 (m, 1H, CH), 4.88 (s, 1H, OH), 6.78 (d, 1H, J = 7.5, Ar), 6.95 (t, 1H, J = 7.5, Ar), 7.10 (t, 1H, J = 7.5, Ar), 7.31 (d, 1H, J = 7.5, Ar); 13C-NMR (CDCl3) δ: 24.08 (CH2), 31.71 (CH2), 32.00 (CH), 66.50 (CpH), 67.74 (CpH), 68.19 (CpH), 69.50 (CpH), 90.15 (Cp), 115.22 (Ar-H), 121.03 (Ar-H), 126.60 (Ar-H), 128.49 (Ar-H), 134.30 (Ar), 152.98 (Ar-OH); mp 134-135 °C, ESI-MS: m/z 346.6 [M+]; Anal. Calcd. For C21H22FeO: C, 72.85; H, 6.40. Found: C, 73.34; H, 6.46.
3.5. Synthesis of 4-([5]ferrocenophane-3-yl)-salicylaldehyde (8)
Compound 6 (300 mg, 0.77 mmol) was dissolved in CHCl3 (20 mL) and 13M KOH (2 mL) was added. The mixture was then refluxed for 24 h under vigorous stirring and the reaction monitored by TLC analysis (n-hexane:EtOAc 85:15). The reaction was then quenched at about 25% conversion of substrate, when 8 was detected as the sole product, by addition of H2O and CH3COOH to acidic pH. After extraction the organic layer was dried over MgSO4 and taken to dryness. Column chromatography on Si gel (n-hexane:EtOAc 85:15) gave pure 8 (60 mg, 21% yield) and the unreacted 6 (210 mg, 70% yield), which was then recycled. mp 139-140 °C; 1H-NMR (CDCl3) δ: 1.96 (m, 2H, CH2), 2.06 (m, 2H, CH2), 2.22 (ddd, 2H, J = 15.6, 6.2 and 4.3, CH2), 2.48 (ddd, 2H, J = 15.6, 10.1 and 6.4, CH2), 4.04 (s, 2H, Cp), 4.09 (s, 4H, Cp), 4.21 (br s, 3H, Cp and CH), 6.99 (d, 1H, J = 8.5, Ar), 7.49 (d, 1H, J = 2.2, Ar), 7.53 (dd, 1H, J = 8.5 and 2.2), 9.95 (s, 1H, CHO), 10.90 (s, 1H, OH); 13C-NMR (CDCl3) δ: 23.82 (CH2), 32.45 (CH2), 39.36 (CH), 66.21 (CpH), 67.11 (CpH), 68.11 (CpH), 69.06 (CpH), 89.99 (Cp), 117.55 (Ar-H), 120.50 (Ar), 131.97 (Ar-H), 136.31 (Ar-H), 139.73 (Ar), 160.25 (Ar-OH) 196.61 (CHO); ESI-MS: m/z 374.3 [M+]; Anal. Calcd. For C22H22FeO2: C, 70.60; H, 5.93. Found: C, 70.98; H, 6.01.
3.6. Synthesis of (+)-(R,R)-N,N’-bis[4-([5]ferrocenophane-3-yl)-salicylidene]-1,2-diphenylethyle-nediamine [(+)-9]
To a solution of aldehyde 8 (47 mg, 0.126 mmol) in absolute ethanol (100 mL) (1R,2R)-1,2-diphenylethylenediamine (13.3 mg, 0.063 mmol) was added and the reaction mixture maintained under stirring at room temperature for 2 days. When the TLC analysis indicated the disappearance of the aldehyde, the solvent was removed in vacuo and the residue applied to a silica gel column (n-hexane:EtOAc 9:1) to give pure (+)-9 (55 mg, 0.059 mmol, 94% yield) as a pale orange solid, mp 139-140 °C; [α]D = +20.2 (c 0.48, CHCl3); 1H-NMR (CDCl3) δ: 1.85 (m, 4H, -CH2), 1.96 (m, 4H, -CH2), 2.16 (m, 4H, -CH2), 2.41 (m, 4H, -CH2), 3.98 (br s, 2H, Cp), 4.00 (br s, 2H, Cp), 4.07 ( s, 10H, Cp and CH), 4.20 (m, 4H, Cp), 4.76 (s, 2H, CH-N), 6.95 (d, 2H, J = 8.3, Ar), 7.11 (m, 2H, Ar), 7.24 (m, 12H, Ar), 8.41 (s, 2H, CH=N), 13.15 (s, 2H, OH); 13C-NMR (CDCl3) δ: 23.88 (CH2), 32.50 (CH2), 32.56 (CH2), 39.35 (CH), 66.20 (CpH), 67.16 (CpH), 68.00 (CpH), 69.20 (CpH), 69.25 (CpH), 80.39 (CH-N), 89.20 (Cp), 116.77 (Ar-H), 118.40 (Ar), 127.58 (Ar-H), 127.94 (Ar-H), 128.36 (Ar-H), 130.64 (Ar-H), 132.00 (Ar-H), 138.45 (Ar), 139.48 (Ar), 159.08 (Ar-OH), 166.46 (C=N); ESI-MS: m/z 924.2 [M+], 462.4 [M++]; Anal. Calcd. For C58H56Fe2N2O2: C, 75.33; H, 6.10; N, 3.03. Found: C, 75.91; H, 6.16; N, 3.08.
3.7. General procedure for the synthesis of metal complexes
To a solution of (+)-9 (50 mg, 54 mmol) in absolute EtOH (10 mL), the appropriate M(OAc)n·xH2O (54 mmol, M = Mn(III), Co(II) or [UO2]2+) was added and the mixture left to react at rt under stirring until TLC analysis showed the complete conversion of substrate. The complex was separated from the solution by filtration and air-dried (80–90% yield)
Mn(III)-complex (–)-10: [α]D = –642.1 (c 0.05, CHCl3); ESI-MS: m/z 1023.3 [M + EtOH]+; CD: λ (Δε) 625 (+1.08), 370 (+2.08), 306 (–3.82), 268 (+0.38), 243 (–7.16); Anal. Calcd. for C60H57Fe2MnN2O4: C, 69.51; H, 5.54; N, 2.70. Found: C, 69.48; H, 5.50; N, 2.67.
Co(II)-complex (+)-11: [α]D = +610.2 (c 0.05, CHCl3); ESI-MS: m/z 981.2 [M] +, 490.8 [M] ++; CD: λ (Δε) 562 (+4.90), 470 (+1.83), 416 (–0.77), 372 (+0.42), 300 (–3.78), 257 (+2.07), 232 ( –8.31); Anal. Calcd. For C58H54CoFe2N2O2 : C, 70.69; H, 5.54; N, 2.85. Found: C, 70.66; H, 5.49; N, 2.82.
UO2 complex (+)-13: [α]D = +142.7 (c 0.15, C6H6); 1H-NMR (d6-DMSO) δ: 1.91 (m, 4H, -CH2), 2.03 (m, 4H, -CH2), 2.12 (m, 4H, -CH2), 2.42 (m, 4H, -CH2), 4.07 (br s, 10H, Cp and -CH), 4.10 (s, 4H, Cp), 4.19 (s, 4H, Cp), 6.22 (s, 2H, CH-N), 6.97 (d, 2H, J = 8.6, Ar), 7.20 (m, 6H, Ar), 7.49 (s, 2H, Ar), 7.62 (dd, 2H, J = 2.0 and 8.6, Ar), 7.66 (d, 4H, J = 7.5, Ar), 9.44 (s, 2H, CH=N); 13C-NMR (d6-DMSO) δ: 23.94 (CH2), 32.98 (CH2), 38.81 (CH), 66.35 (CpH), 67.34 (CpH), 68.17 (CpH), 69.22 (CpH), 80.61 (CH-N), 89.00 (Cp), 120.70 (Ar-H), 123.22 (Ar), 127.60 (Ar-H), 127.79 (Ar-H), 128.68 (Ar-H), 133.84 (Ar-H), 135.08 (Ar-H), 136.06 (Ar), 142.2 (Ar), 168.21 (Ar-O), 171.99 (C=N); ESI-MS: m/z 1192.3 [M]+; CD: λ (Δε) 575 (+0.68), 398 (+1.16), 331 (–2.52), 287 (+8.60), 250 (+12.45), 228 (–11.48); Anal. Calcd. For C58H54Fe2N2O4U: C, 58.40; H, 4.56; N, 2.35. Found: C, 58.38; H, 4.49; N, 2.31.
3.8. Synthesis of Zn(II) complex (-)-12
A 2M solution of Et2Zn in toluene (0.030 mL, 60 mmol) was added to a solution of (+)-9 (50 mg, 54 mmol) in toluene (5 mL) and the mixture stirred at rt overnight. The solvent was removed under vacuum, the residue suspended in CHCl3 and the solid removed by filtration. The final solution was taken to dryness to give pure complex in quantitative yield: [α]D = – 21.3 (c 0.3, CHCl3); 1H-NMR (d6-DMSO) δ: 1.77 (m, 4H, -CH2), 1.92 (m, 4H, -CH2), 2.05 (m, 4H, -CH2), 2.37 (m, 4H, -CH2), 3.86 (m, 2H, CH), 4.00 (s, 4H, Cp), 4.03 (s, 4H, Cp), 4.06 (s, 4H, Cp), 4.14 (s, 4H, Cp), 5.05 (s, 2H, CH-N), 6.64 (d, 2H, J = 8.7, Ar), 7.00 (d, 2H, J = 2.2, Ar), 7.17 (dd, 2H, J = 8.7 and 2.2, Ar), 7.26 (br t, 2H, Ar), 7.36 (t, 4H, J = 7.5, Ar), 7.44 (d, 4H, J = 7.5, Ar), 8.36 (s, 2H, CH=N); 13C-NMR (d6-DMSO) δ: 23.87 (CH2), 32.56 (CH2), 39.23 (CH), 66.20 (CpH), 67.22 (CpH), 68.02 (CpH), 69.33 (CpH), 73.13 (CH-N), 89.40 (Cp), 118.9 (Ar), 123.09 (Ar-H), 127.85 (Ar-H), 127.92 (Ar-H), 128.87 (Ar-H), 131.4 (Ar), 133.44 (Ar-H), 133.56 (Ar-H), 142.37 (Ar), 170.32 (Ar-O), 170.65 (C=N); ESI-MS: m/z 987.3 [M]+, CD: λ (Δε) 416 (+0.63), 389 (–2.74), 353 (3.58), 289 (–3.25); Anal. Calcd. For C58H54Fe2N2O2Zn : C, 70.50; H, 5.51; N, 2.83. Found: C, 70.46; H, 5.48; N, 2.80.
4. Conclusion
A novel salen ligand (+)-9 bearing two [5]ferrocenophane para-substituents on the salicylideneamine rings has been synthesized starting from readily available diacetylferrocene, p-hydroxybenzaldehyde and (R,R)-dpen and its complexes with four different metals have been also prepared and characterised. The evaluation of the use of these complexes as ligands in asymmetric synthesis is currently under investigation.
Acknowledgements
The authors thank University of Catania, MIUR and CNR for the financial support.
- Sample Availability: Samples of compounds 4, 5 and 9 are available from the authors.
References
- Cozzi, P.G. Metal-salen Schiff base complexes in catalysis: Practical aspects. Chem. Soc. Rev. 2004, 33, 410–421. [Google Scholar] [CrossRef]
- Bailezao, C.; Garcia, H. Chiral salen complexes: An overview to recoverable and reusable homogeneous and heterogeneous catalysts. Chem. Rev. 2006, 106, 3987–4043. [Google Scholar] [CrossRef]
- Zhang, W.; Loebach, J.L.; Wilson, S.R.; Jacobsen, E.N. Enantioselective epoxidation of unfunctionalized olefins catalyzed by (salen) manganese complexes. J. Am. Chem. Soc. 1990, 112, 2801–2803. [Google Scholar] [CrossRef]
- Jacobsen, E.N.; Zhang, W.; Muci, A.R.; Ecker, J.R.; Deng, L. Highly enantioselective epoxidation catalysts derived from 1,2-diaminocyclohexane. J. Am. Chem. Soc. 1991, 113, 7063–7064. [Google Scholar] [CrossRef]
- Srinivasan, K.; Michaud, P.; Kochi, J.K. Epoxidation of olefins with cationic (salen) manganese(III) complexes. The modulation of catalytic activity by substituents. J. Am. Chem. Soc. 1986, 108, 2309–2320. [Google Scholar] [CrossRef]
- Cavallo, L.; Jacobsen, H. Electronic effects in (salen)Mn-based epoxidation catalysts. J. Org. Chem. 2003, 68, 6202–6207. [Google Scholar] [CrossRef]
- Schaus, S.E.; Brandes, B.D.; Larrow, J.F.; Togunaga, M.; Hansen, K.B.; Gould, A.E.; Furrow, M.E.; Jacobsen, E.N. Highly selective hydrolytic kinetic resolution of terminal epoxides catalyzed by chiral (salen) CoIII complexes. Practical synthesis of enantioenriched terminal epoxides and 1,2-Diols. J. Am. Chem. Soc. 2002, 124, 1307–1315. [Google Scholar]
- Oh, C.R.; Choo, D.J.; Shim, W.H.; Lee, D.H.; Roh, E.J.; Lee, S.; Song, C.E. Chiral Co(III) (salen)-catalysed hydrolytic kinetic resolution of racemic epoxides in ionic liquids. Chem. Commun. 2003, 1100–1101. [Google Scholar]
- Dioos, B.M.L.; Jacobs, P.A. Microwave-assisted Cr(salen)-catalysed asymmetric ring opening of epoxides. J. Catal. 2005, 235, 428–430. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, W.; Lough, A.J.; Kim, B.M.; Chin, J. A cobalt(III)-salen complex with an axial substituent in the diamine backbone: Stereoselective recognition of amino alcohols. J. Am. Chem. Soc. 2005, 127, 16776–16777. [Google Scholar] [CrossRef]
- Dzygiel, P.; Reeve, T.B.; Piarulli, U.; Krupicka, M.; Tvaroska, I.; Gennari, C. Resolution of racemic N-benzyl α-amino acids by liquid-liquid extraction. Eur. J. Org. Chem. 2008, 1253–1264. [Google Scholar]
- Belokon, Y.N.; Fuentes, J.; North, M.; Steed, J.W. Influence of the metal and chiral diamine on metal(II)salen catalysed, asymmetric synthesis of α-methyl α-amino acids. Tetrahedron 2004, 60, 3191–3204. [Google Scholar] [CrossRef]
- Cozzi, P.G. Enantioselective alkynylation of ketones catalyzed by Zn(salen) complexes. Angew. Chem. Int. Ed. 2003, 42, 2895–2898. [Google Scholar] [CrossRef]
- Pathak, K.; Bhatt, A.P.; Abdi, S.H.R.; Kureshy, R.I.; Khan, N.H.; Ahmad, I.; Jasra, R. Enantioselective phenylacetylene addition to aldehydes and ketones catalyzed by recyclable polymeric Zn (salen) complex. Chirality 2007, 19, 82–88. [Google Scholar] [CrossRef]
- Wezemberg, S.J.; Kleij, A.W. Material application for salen frameworks. Angew. Chem. Int. Ed. 2008, 47, 2354–2364. [Google Scholar] [CrossRef]
- Kleij, A.W. New templating strategies with salen scaffolds (salen = N,N’-Bis (salicylidene) ethylenediamine dianion). Chem. Eur. J. 2008, 14, 10520–10529. [Google Scholar] [CrossRef]
- Di Mauro, E.F.; Kozlowski, M.C. Salen-derived catalysts containing secondary basic groups in the addition of diethylzinc to aldehydes. Org. Lett. 2001, 3, 3053–3056. [Google Scholar] [CrossRef]
- Jiang, C.J.; Chen, Z.R. Chiral cobalt salen complexes containing Lewis acid: A highly reactive and enantioselective catalyst for the hydrolytic kinetic resolution of terminal epoxides. Kinet. Catal. 2008, 49, 474–478. [Google Scholar] [CrossRef]
- Ferrocenes; Hayashi, T.; Togni, A. (Eds.) Wiley-VCH Verlag: Weinheim, Germany, 2008.
- Arrayas, R.G.; Adrio, J.; Carretero, J.C. Recent applications of chiral ferrocene ligands in asymmetric catalysis. Angew. Chem. In. Ed. 2006, 45, 7674–7715. [Google Scholar] [CrossRef]
- Patti, A.; Pedotti, S. Synthesis of chiral 1-ferrocenylaldols and 1-ferrocenyl-1,3-diols via asymmetric reduction. Tetrahedron Asymmetry 2006, 17, 1824–1830. [Google Scholar] [CrossRef]
- Patti, A.; Pedotti, S. Synthesis of chiral alcohols containing the 1,3-diferrocenylpropane structural motif. Tetrahedron Asymmetry 2008, 19, 1891–1897. [Google Scholar] [CrossRef]
- Amato, M.E.; Ballistreri, F.P.; Pappalardo, A.; Tomaselli, G.A.; Toscano, R.M.; Williams, D.J. Novel chiral (salen)MnIII complexes containing a calix[4]arene unit as catalysts for enantioselective epoxidation reactions of (Z)-aryl alkenes. Eur. J. Org. Chem. 2005, 3562–3570. [Google Scholar]
- Amato, M.E.; Ballistreri, F.P.; Pappalardo, A.; Sciotto, D.; Tomaselli, G.A.; Toscano, R.M. Synthesis and conformational aspects of 20- and 40-membered macrocyclic mono and dinuclear uranyl complexes incorporating salen and (R)-Binol units. Tetrahedron 2007, 63, 9751–9757. [Google Scholar] [CrossRef]
- Gibson, V.C.; Long, N.J.; Marshall, E.L.; Oxford, P.J.; White, A.J.P.; Williams, D.J. The synthesis and metal coordination chemistry of new 1,1’-N-substituted ferrocenediyl ligands derived from 1,1’-diaminoferrocene. J. Chem. Soc. Dalton Trans. 2001, 1162–1164. [Google Scholar]
- Shafir, A.; Fiedler, D.; Arnold, J. Formation of 1:1 complexes of ferrocene-containing salen ligands with Mg, Ti and Zr. J. Chem. Soc. Dalton Trans. 2002, 555–560. [Google Scholar]
- Gregson, C.K.A.; Gibson, V.C.; Long, N.J.; Marshall, E.L.; Oxford, P.J.; White, A.J.P. Redox control within single-site polymerization catalysts. J. Am. Chem. Soc. 2006, 128, 7410–7411. [Google Scholar]
- Wölfle, H.; Kopacka, H.; Wurst, K.; Ongania, K.H.; Görtz, H.H.; Preishuber-Pflügl, P.; Bildstein, B. Planar chiral ferrocene salen-type ligands featuring additional central and axial chirality. J. Organomet. Chem. 2006, 691, 1197–1215. [Google Scholar] [CrossRef]
- Ballistreri, F.P.; Patti, A.; Pedotti, S.; Tomaselli, G.A.; Toscano, R.M. Synthesis of novel chiral ‘salen-type’ ferrocenyl ligands. Tetrahedron Asymmetry 2007, 18, 2377–2380. [Google Scholar] [CrossRef]
- Achard, T.; Belokon, Y.N.; Fuentes, J.A.; North, M.; Parsons, T. Influence of aromatic substituents on metal(II)salen catalysed, asymmetric synthesis of α-methyl α-amino acids. Tetrahedron 2004, 60, 5919–5930. [Google Scholar] [CrossRef]
- Malinowska, M.; Kwiatkowski, P.; Jurezac, J. The enantioselective high-pressure Diels-Alder reaction of 1-methoxybuta-1,3-diene with tert-butyldimethylsilyloxy-acetaldehyde catalyzed by (salen)Co(II) and (salen)Cr(III)Cl complexes. Tetrahedron Lett. 2004, 45, 7693–7696. [Google Scholar] [CrossRef]
- Pedotti, S.; Patti, A. Microwave-assisted synthesis of 1,5-dioxo-3-substituted [5]ferrocenophanes. J. Organomet. Chem. 2008, 693, 1375–1381. [Google Scholar] [CrossRef]
- Dalla Cort, A.; Pasquini, C.; Schiaffino, L. Nonsymmetrically substituted uranyl-salophen receptors: New opportunities for molecular recognition and catalysis. Supramol. Chem. 2007, 19, 79–87. [Google Scholar] [CrossRef]
- Cametti, M.; Dalla Cort, A.; Mandolini, L.; Nissinen, M.; Rissanen, K. Specific recognition of fluoride anion using a metallamacrocycle incorporatine a uranyl-salen unit. New J. Chem. 2008, 32, 1113–1116. [Google Scholar] [CrossRef]
- Pasini, A.; Gullotti, M.; Cesarotti, E. Uranyl complexes with tetradentate optically active schiff bases of salicylaldehyde. J. Inorg. Nucl. Chem. 1972, 32, 3821–3833. [Google Scholar]
- Kelly, S.M.; Jess, T.J.; Price, N.C. How to study proteins by circular dichroism. BBA-Proteins Proteomics 2005, 1751, 119–139. [Google Scholar] [CrossRef]
- Berova, N.; Di Bari, L.; Pescitelli, G. Application of electronic circular dichroism in configurational and conformational analysis of organic compounds. Chem. Soc. Rev. 2007, 36, 914–931. [Google Scholar] [CrossRef]
- Kypr, J.; Kejnovská, I.; Renčiuk, D.; Vorlíčková, M. Circular dichroism and conformational polymorphism of DNA. Nucl. Acids Res. 2009, 37, 1713–1725. [Google Scholar] [CrossRef]
- McCann, D.M.; Stephens, P.J. Determination of absolute configuration using density fuctional theory calculations of optical rotation and electronic circular dichroism: Chiral Alkenes. J. Org. Chem. 2006, 71, 6074–6098. [Google Scholar] [CrossRef]
- Bringmann, G.; Bruhn, T.; Maksimenka, K.; Hemberger, Y. The assignment of absolute stereostructures through quantum chemical circular dichroism calculations. Eur. J. Org. Chem. 2009, 2717–2727. [Google Scholar]
- Pasini, A.; Gullotti, M.; Ugo, R. Optically active complexes of Schiff bases. Part 4. An analysis of the circular dichroism spectra of some complexes of different coordination numbers with quadridentate Schiff bases of optically active diamines. J. Chem. Soc. Dalton Trans. 1977, 346–356. [Google Scholar]
- Hirotsu, M.; Nakajima, K.; Kojima, M.; Yoshikawa, Y. Manganese (III) complexes containing optically active tetradentate Schiff base ligands. Effect of phenyl substituents. Inorg. Chem. 1995, 34, 6173–6178. [Google Scholar] [CrossRef]
- Szlyk, E.; Surdykowski, A.; Barwiolek, M.; Larsen, E. Spectroscopy and stereochemistry of t6he optically active copper(II), cobalt(II) and nickel(II) complexes with Schiff bases N,N’-(1R,2R)-(-)-1,2-cyclohexylenebis(3-methylbenzylideneiminato) and N,N’-(1R,2R)-(-)1,2-cyclohexylene-bis-(5-methylbenzylideneiminato). Polyhedron 2002, 21, 2711–2717. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, H.; Li, L.; Hao, H.Q.; Wang, X.Y.; Chen, J.G. Synthesis and characterization of chiral nickel(II) Schiff base complexes and their CD spectra-absolute configuration correlations. Tetrahedron Asymmetry 2006, 17, 2059–2063. [Google Scholar] [CrossRef]
- Janowska, I.; Zakrzewski, J. Circular dichroism spectra of planar chiral 2-substituted ferrocenecarboxaldehydes and 2-ferrrocenyl-1,1-dicyanoethylenes. Tetrahedron Asymmetry 2003, 14, 3271–3273. [Google Scholar] [CrossRef]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).