Structural, Thermoanalytical and Molecular Modeling Studies on N-(3-hydroxypropyl) 3α,12α-Dihydroxy-5β-cholan-24-amide and Its Monohydrates

Abstract
:Introduction
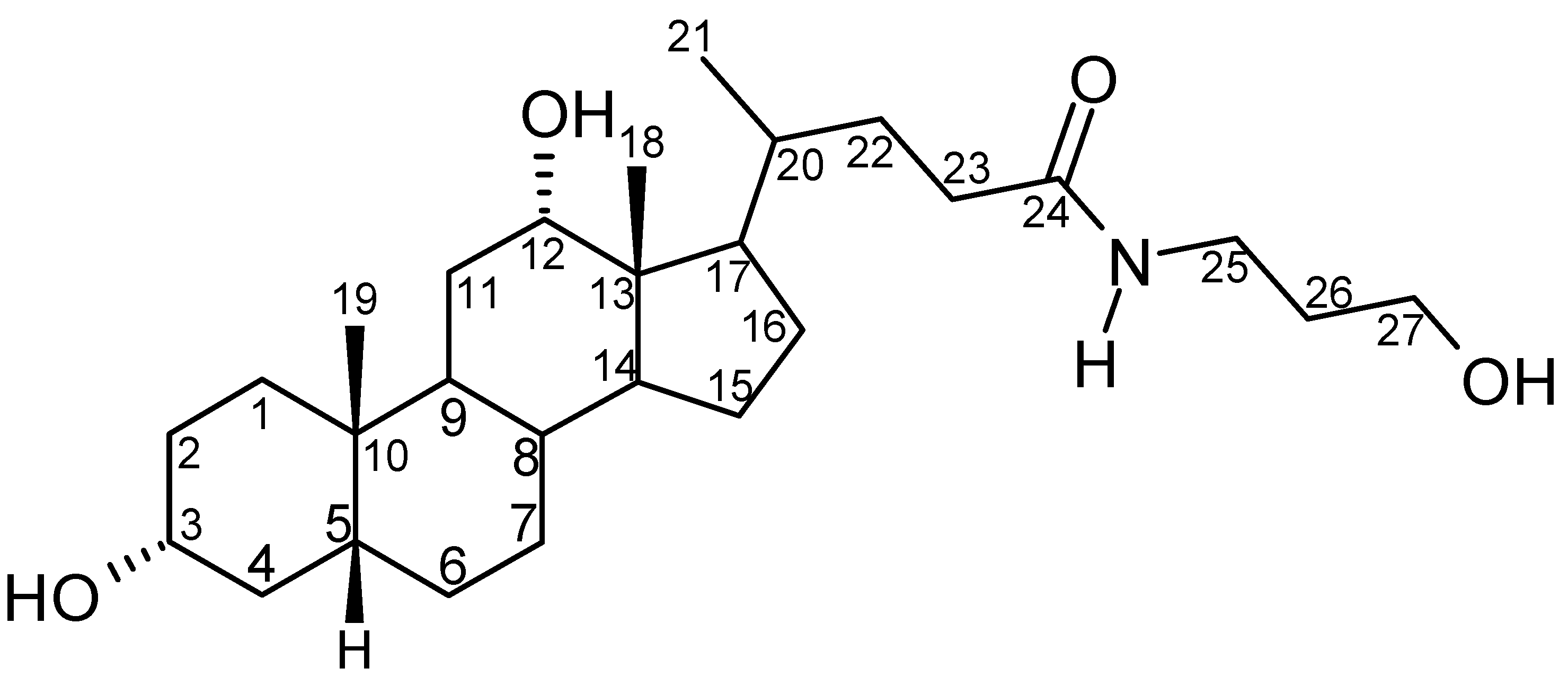
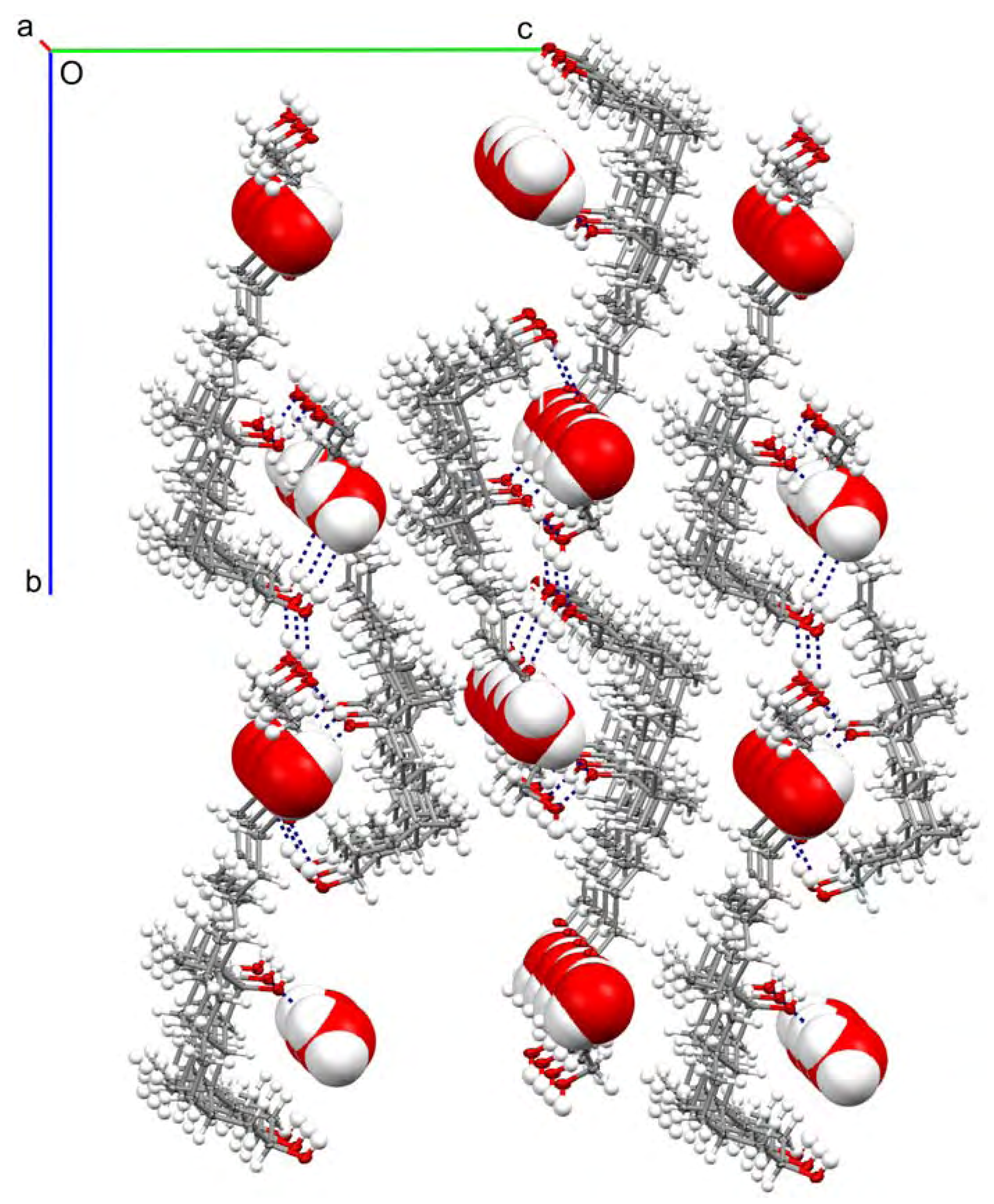
Results and Discussion
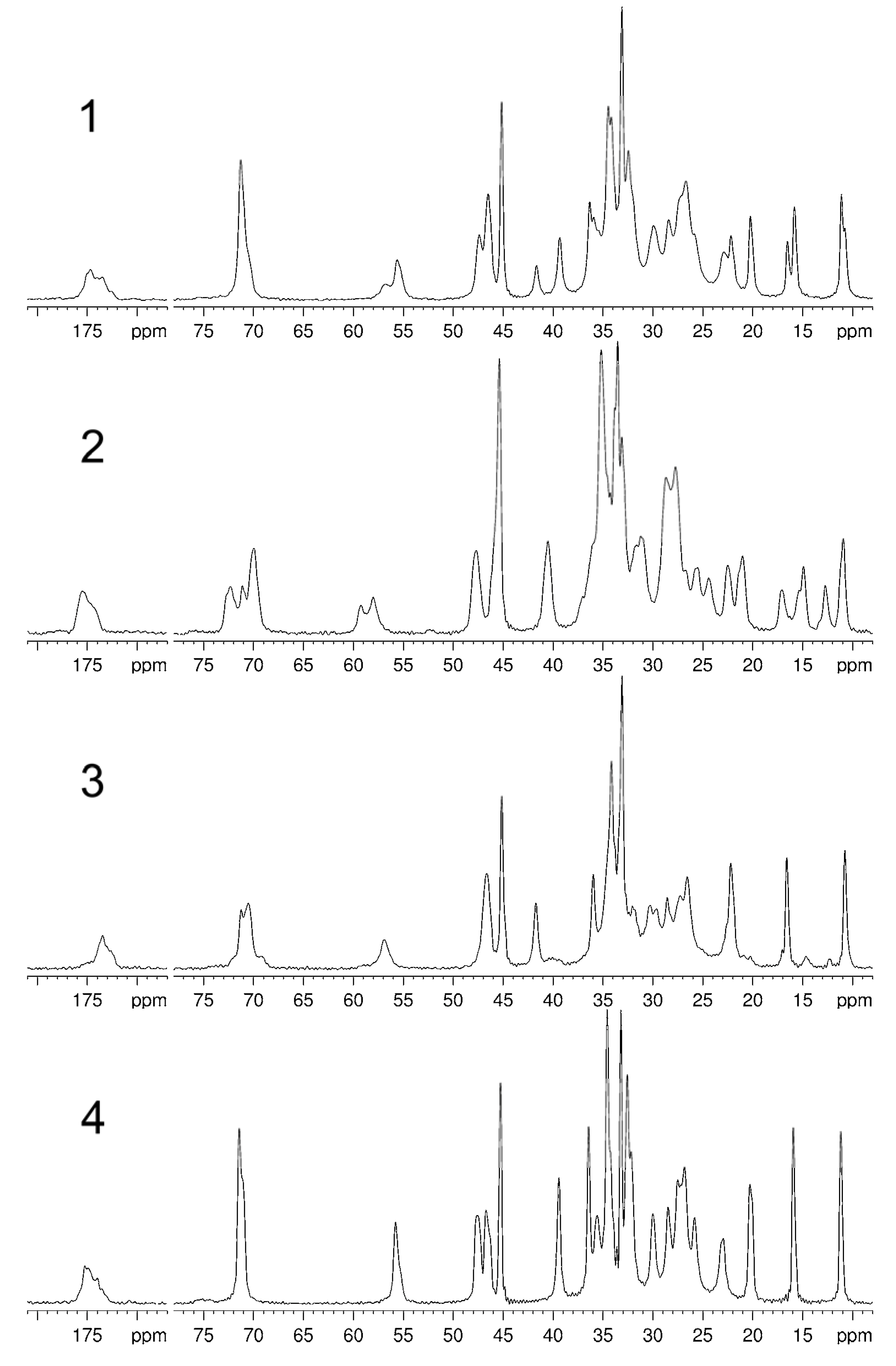

{kind=link}
{kind=link}
{kind=link}
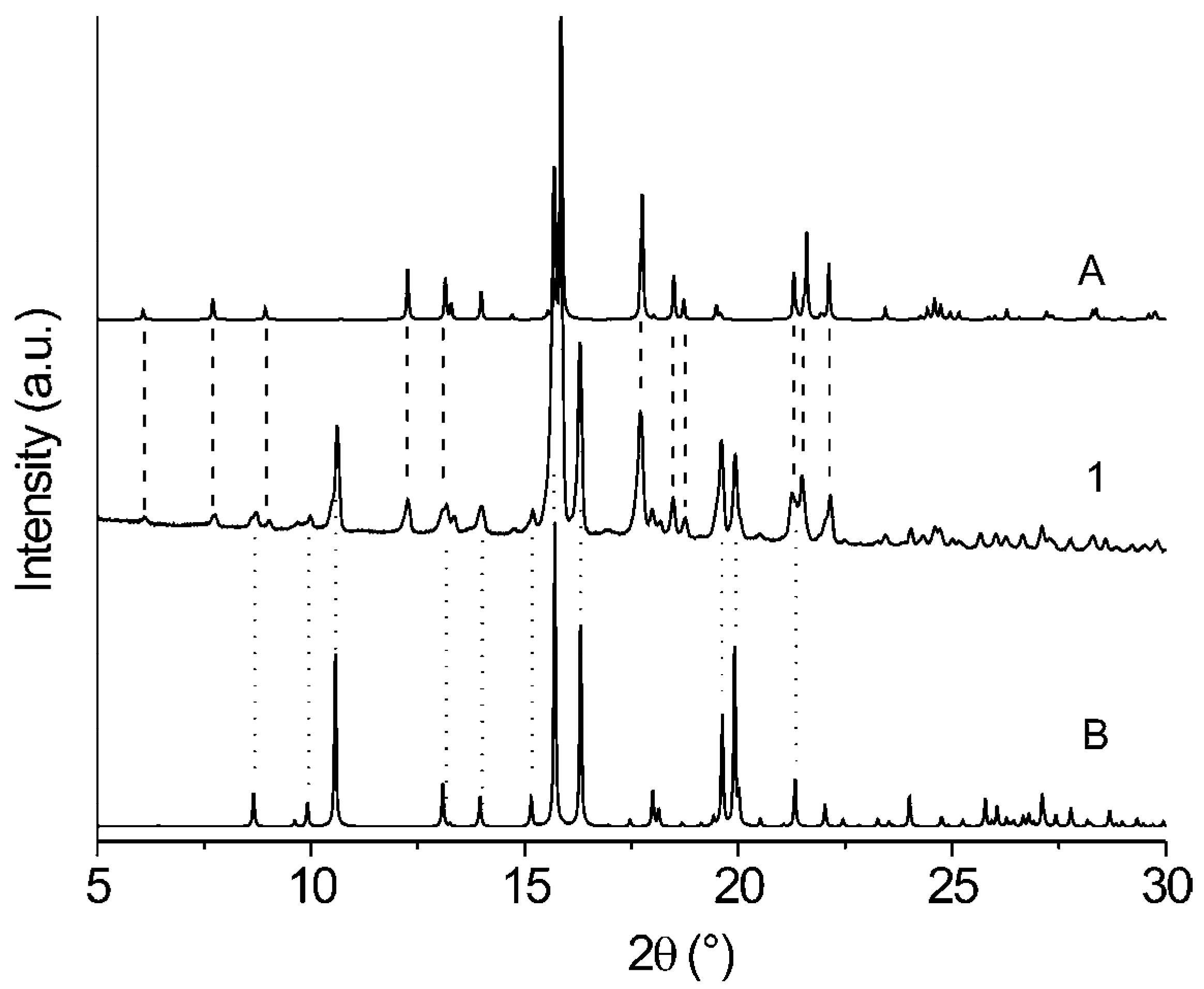{kind=link}
{kind=link}
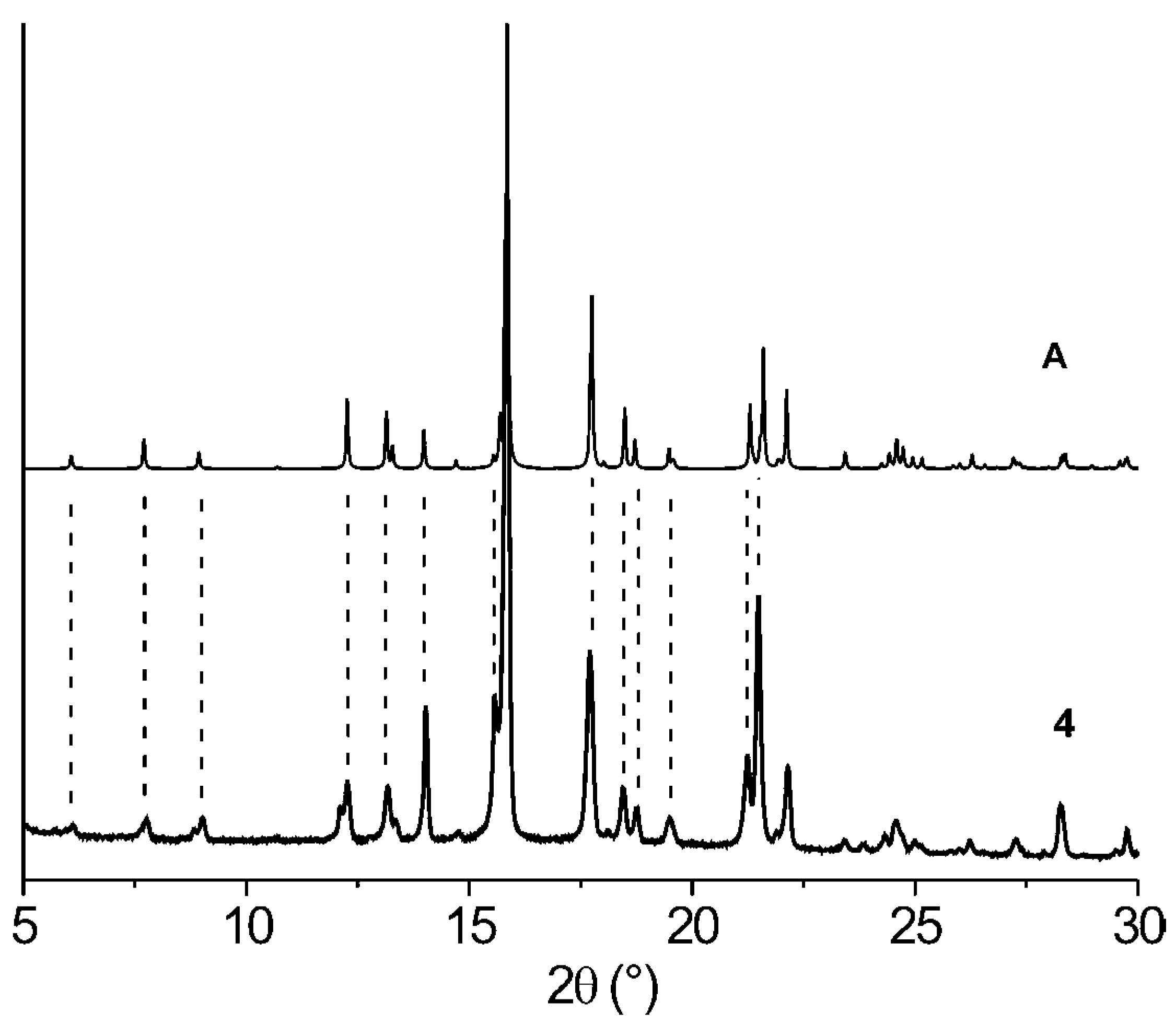{kind=link}
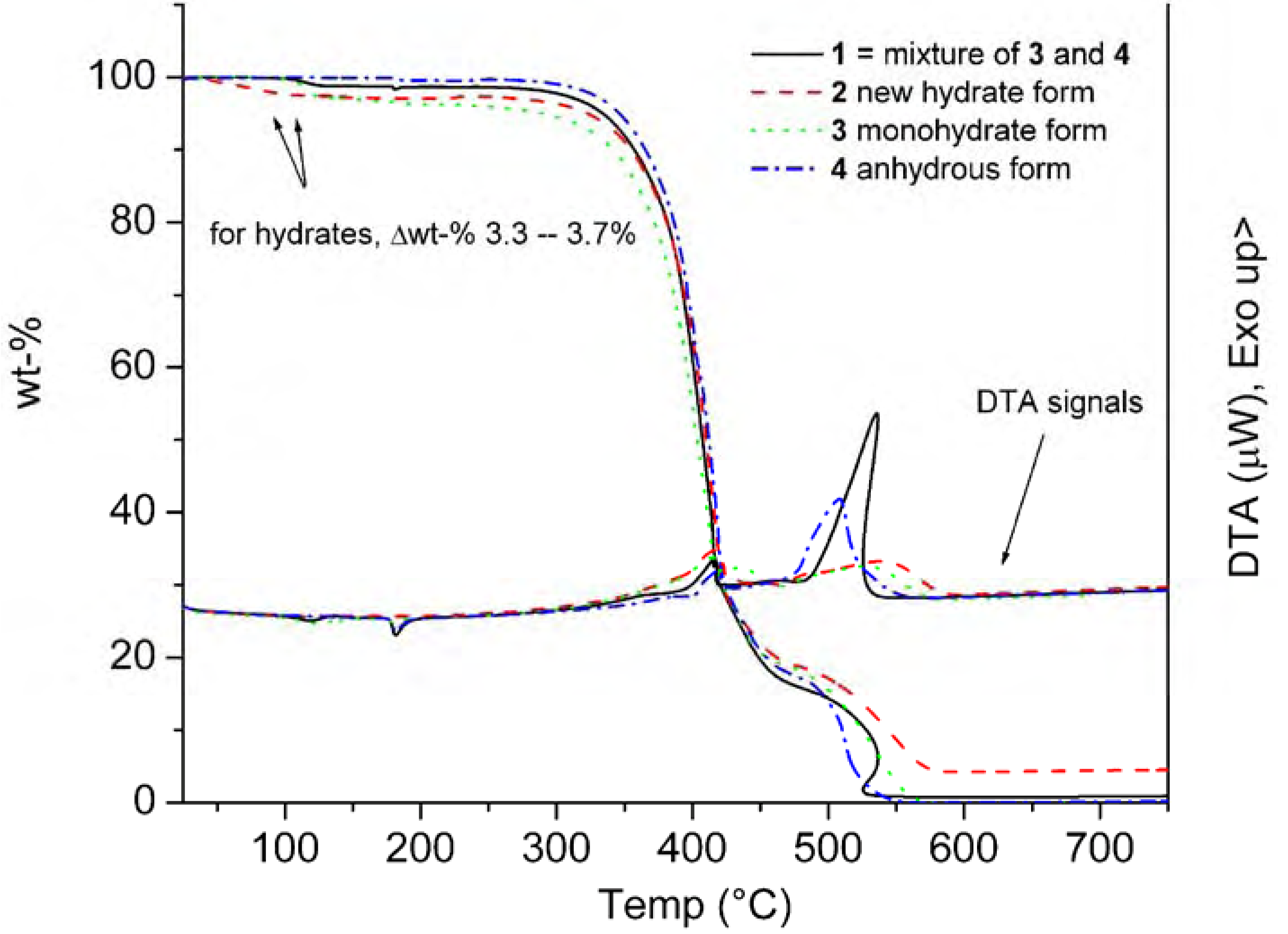{kind=link}
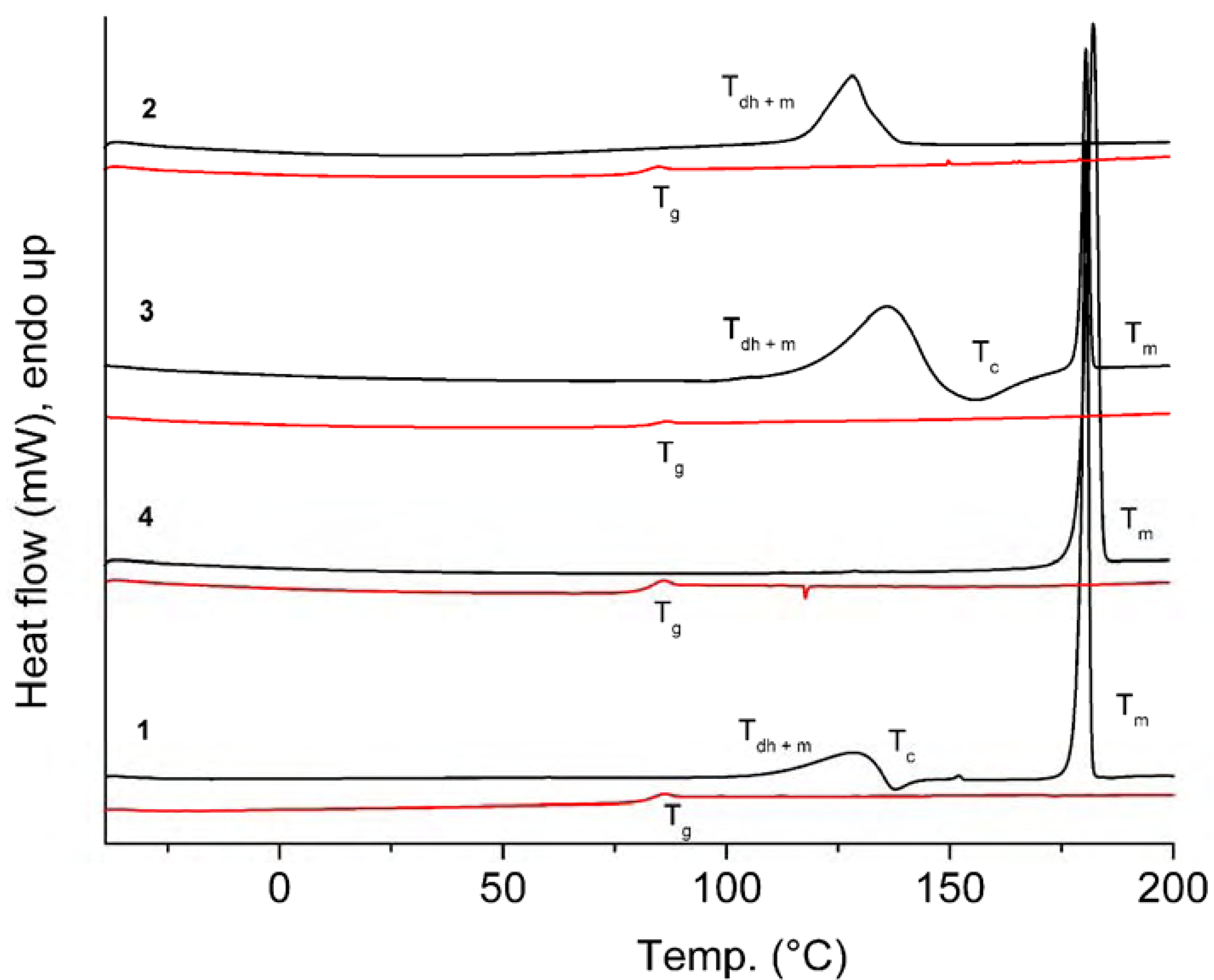{kind=link}
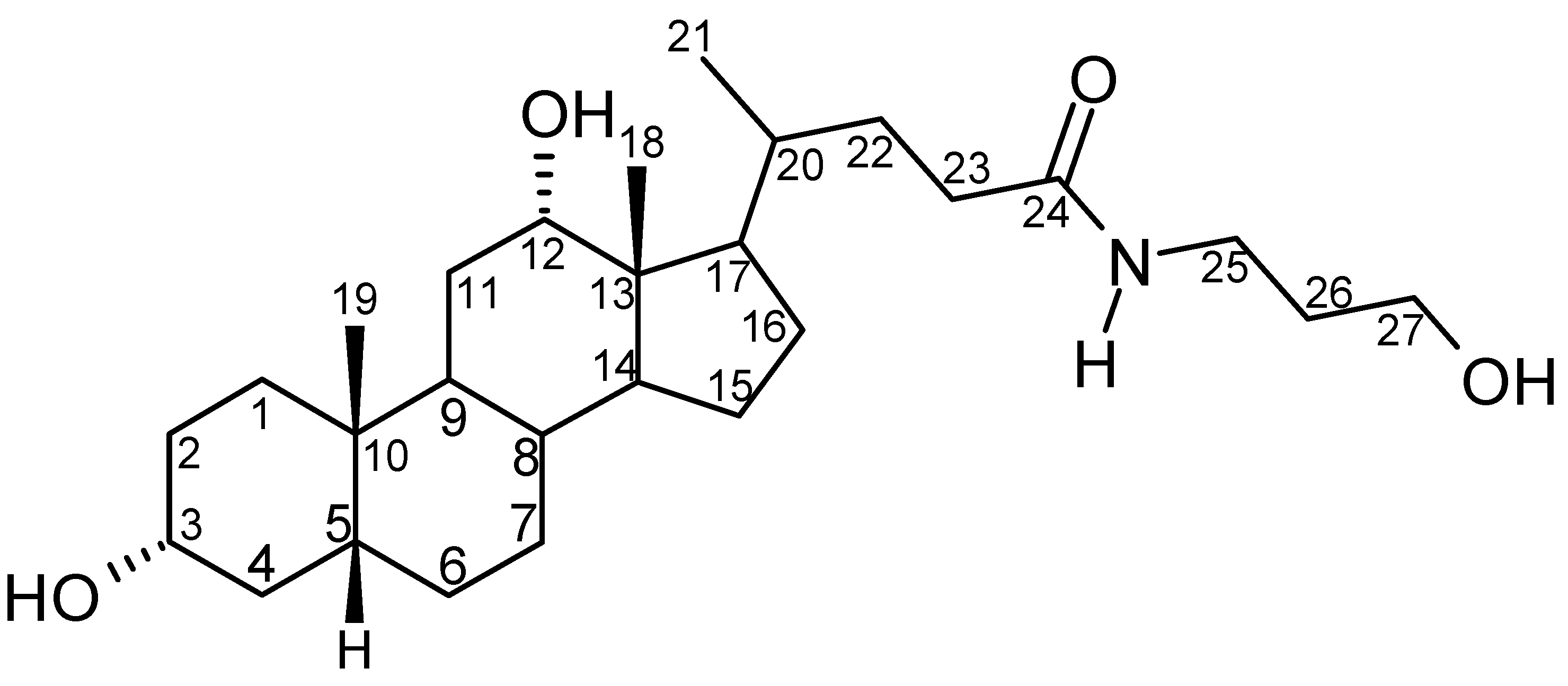{kind=link}
| C | 1a | 2 | 3 | 4 |
| 3 | 71.3c | 71.1d 70.0d | 70.5d | 71.4c |
| 12 | 71.3c | 72.3d | 71.2d | 71.4c |
| 18 | 11.1 10.8 | 12.7 10.9 | 10.8 | 11.2 |
| 19 | 22.2 20.2 | 22.5 21.0 | 22.2 | 20.3 |
| 21 | 16.5 15.8 | 17.0 14.9 | 16.6 | 15.9 |
| 24b | 174.7 173.5 | 175.5 | 173.5 | 175.2 |
| 27 | 56.8 55.6 | 59.2 58.0 | 56.9 | 55.8 |
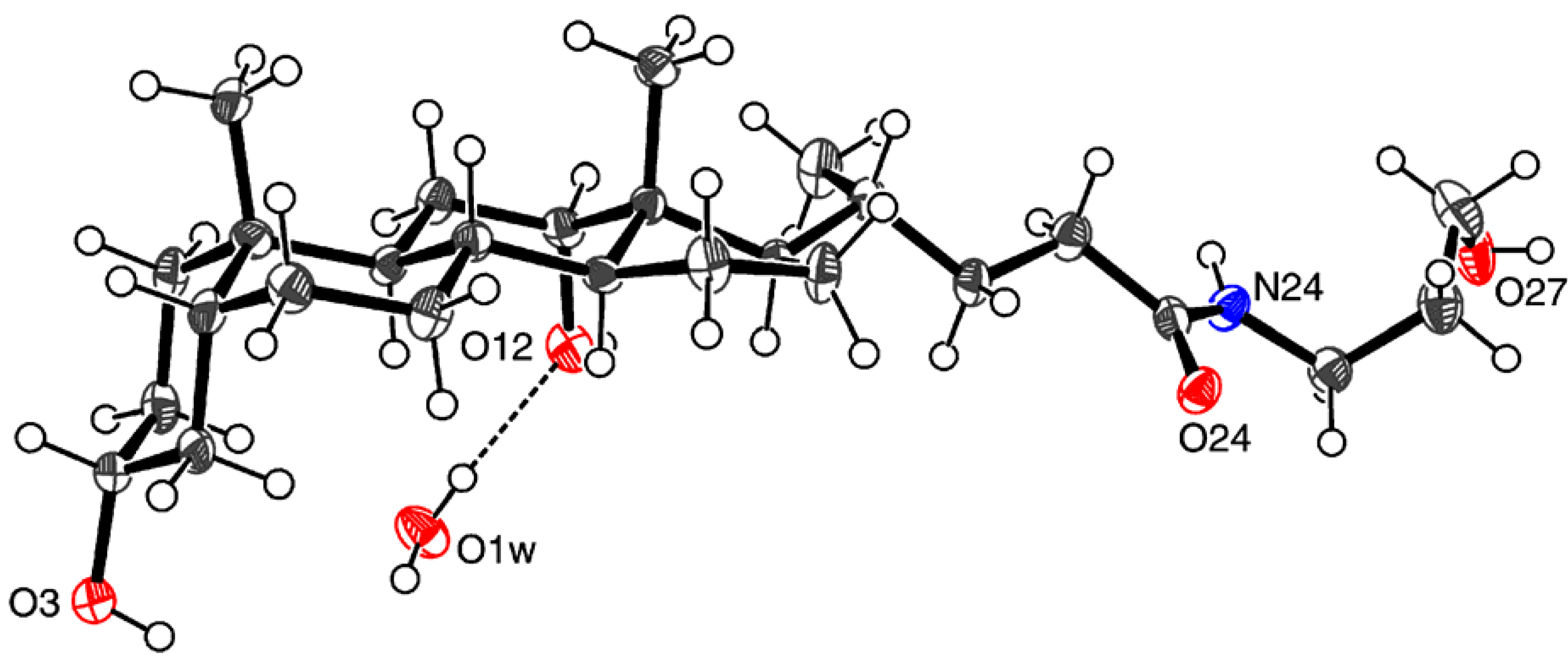
| D ─ H ··· A | D ─ H | H ··· A | D ··· A | D ─ H ··· A |
| O1w ─ H1wb ··· O12 | 0.85(2) | 2.00(2) | 2.837(6) | 172(7) |
| O3 ─ H3O ··· O24a | 0.84(2) | 2.11(3) | 2.932(5) | 165(6) |
| O1w ─ H1wa ··· O24a | 0.84(2) | 2.07(2) | 2.908(5) | 176(7) |
| O12 ─ H12O ··· O27b | 0.84(2) | 1.95(2) | 2.773(6) | 168(6) |
| N24 ─ H24 ··· O1wc | 0.90(2) | 2.06(2) | 2.960(6) | 174(5) |
| O27 ─ H27O ··· O3d | 0.83(2) | 1.97(2) | 2.791(6) | 169(7) |
| 3 | 4 | 3 | 4 | |||
|---|---|---|---|---|---|---|
| C3 – O3 | 1.434(5) | 1.444(3) | C20–C22–C23 | 115.4(4) | 113.1(2) | |
| C12 – O12 | 1.443(5) | 1.433(3) | C22–C23–C24 | 113.3(4) | 112.7(2) | |
| C24 – O24 | 1.237(5) | 1.244(3) | C23–C24–N24 | 114.4(4) | 116.4(2) | |
| C27 – O27 | 1.428(5) | 1.422(4) | C24–N24–C25 | 123.3(4) | 123.7(2) | |
| C24 – N24 | 1.329(5) | 1.332(3) | N24–C25–C26 | 113.4(4) | 112.8(2) | |
| C25 – N24 | 1.445(6) | 1.459(3) | C25–C26–C27 | 113.5(4) | 113.5(2) | |
| C17 – C20 | 1.535(6) | 1.534(3) | C26–C27–O27 | 111.8(4) | 112.7(2) | |
| C20 – C22 | 1.542(6) | 1.537(3) | ||||
| C22 – C23 | 1.517(5) | 1.524(3) | C17–C20–C22–C23 | –178.1(4) | –172.3(2) | |
| C23 – C24 | 1.516(6) | 1.505(4) | C20–C22–C23–C24 | 177.6(4) | 177.1(2) | |
| C25 – C26 | 1.523(6) | 1.521(4) | C22–C23–C24–N24 | 152.3(4) | 122.5(3) | |
| C26 – C27 | 1.504(6) | 1.524(4) | C23–C24–N24–C25 | 175.3(4) | –174.8(2) | |
| C24–N24–C25–C26 | –95.5(5) | –97.7(3) | ||||
| N24–N25–C27–C27 | –71.5(5) | 59.5(3) | ||||
| C25–C26–C27–O27 | –66.2(5) | 55.4(3) |
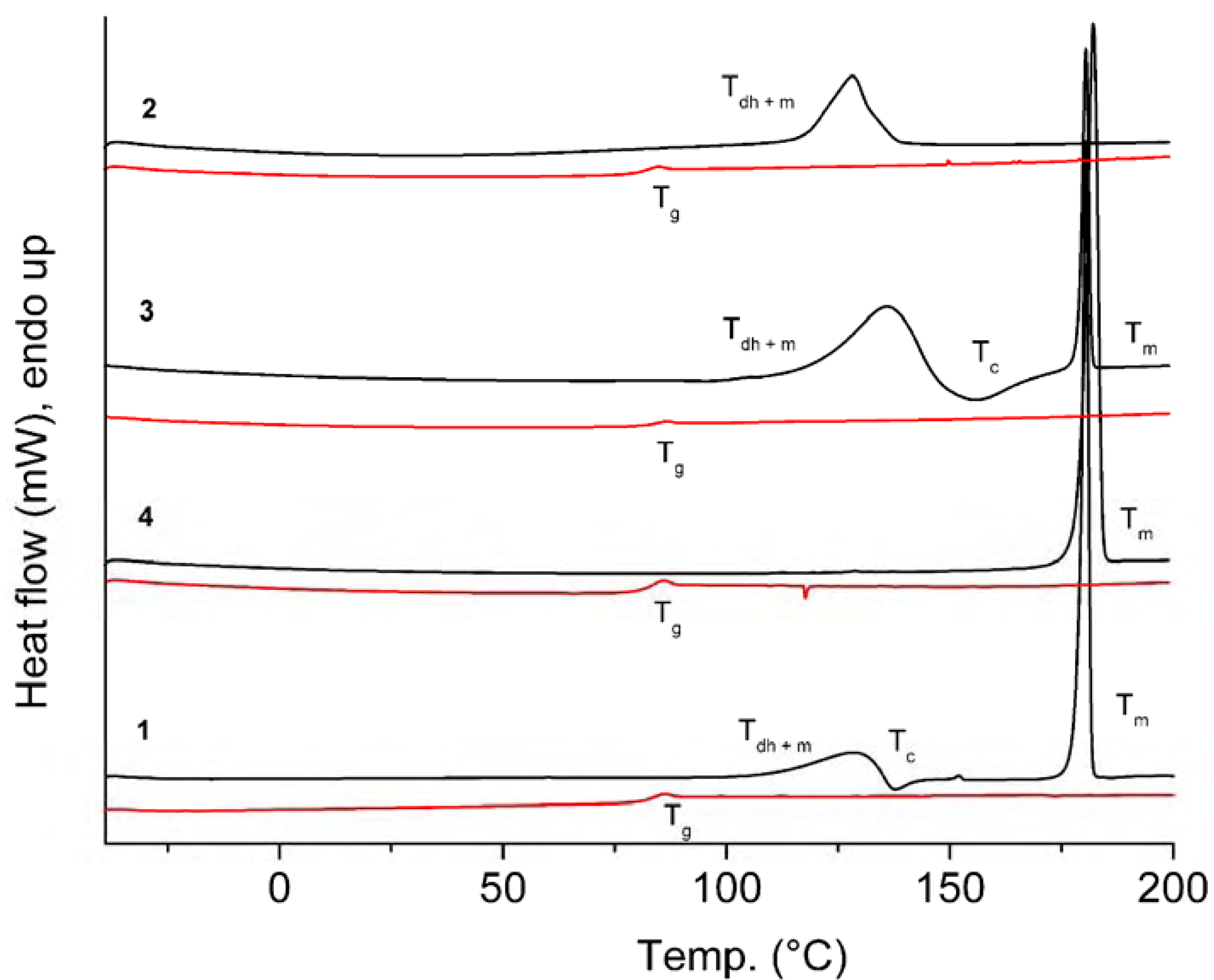
| Comp. | 1st Heating Tm, Tdh, Tc, (ΔH): °C, (J g–1) Tg, [ΔCp] : °C, [J g–1 °C–1] | 2nd heating Tg, [ΔCp] : °C, [J g–1 °C–1] | Dec. Td: °C |
| 1 | Tdh+ Tm 110.1 (30.92) Tc 135.5 (-5.17) Tm 179.0 (98.05) | Tg 81.6 [0.52] | 321 |
| 2 | Tdh + Tm 116.3 (88.16) {Tm of hydrate 142.7} | Tg 80.1 [0.56] | 323 |
| 3 | Tdh+ Tm 115.4 (147.78) Tc 144.0 (-35.97) Tm 178.7 (88.51) {Tm of hydrate 139.4} | Tg 82.5 [0.58] | 313 |
| 4 | Tm 179.2 (96.12) | Tg 81.5 [0.49] | 326 |
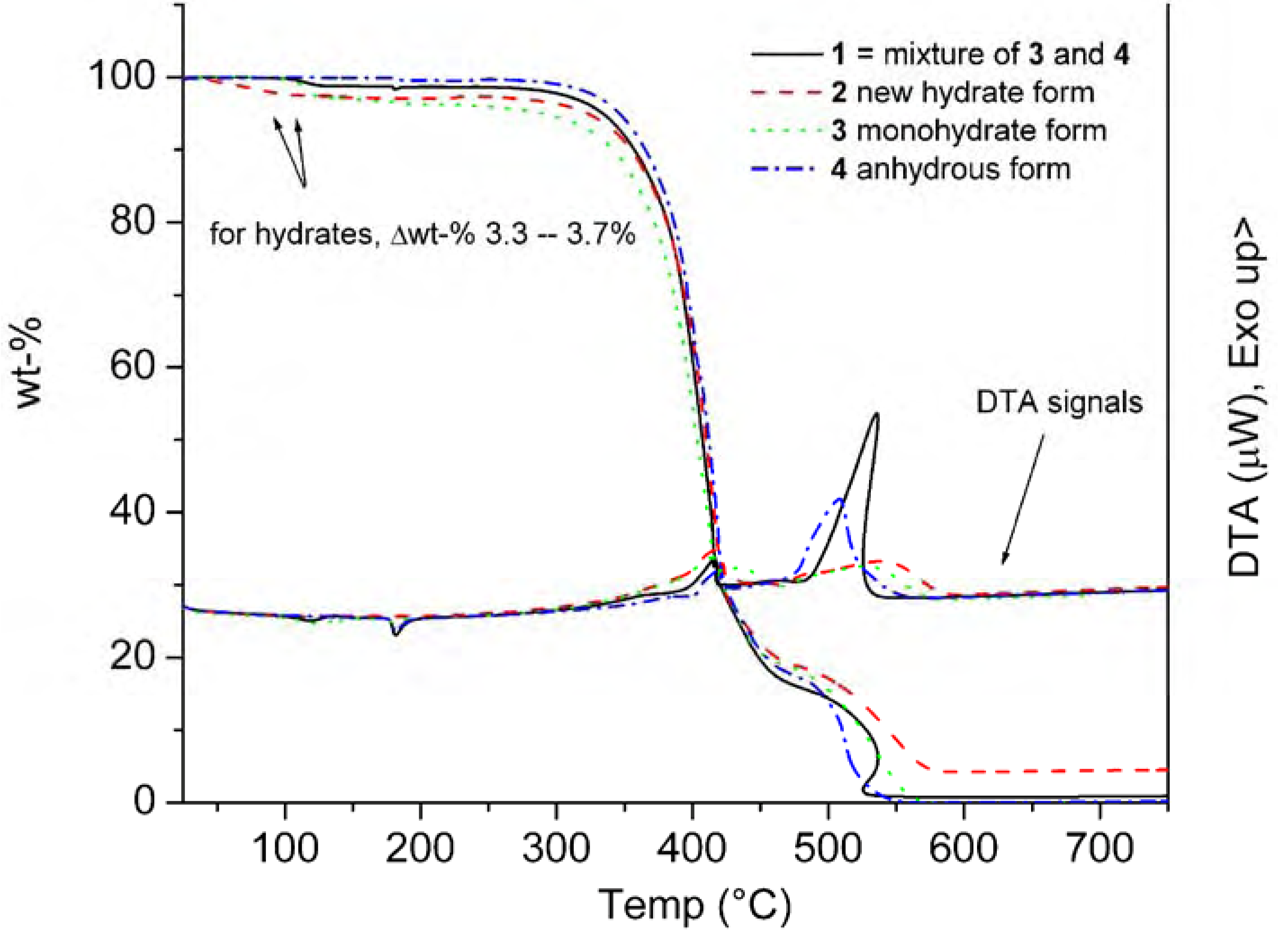
| C | δtheor 3 | δtheor 4 | δexp(solid) 3 | δexp(solid) 4 | δexp(CDCl3) 1 |
| 3 | 71.57 | 73.77 | 70.5 | 71.4 | 71.89 |
| 12 | 77.23 | 75.71 | 71.2 | 71.4 | 73.24 |
| 18 | 11.51 | 11.41 | 10.8 | 11.2 | 12.81 |
| 19 | 21.26 | 22.45 | 22.2 | 20.3 | 23.18 |
| 21 | 16.85 | 16.47 | 16.6 | 15.9 | 17.54 |
| 24 | 168.15 | 171.83 | 173.5 | 175.2 | 174.58 |
| 27 | 59.86 | 57.35 | 56.9 | 55.8 | 59.70 |
Conclusions
Experimental Section
Compounds
Spectroscopy
Thermal properties
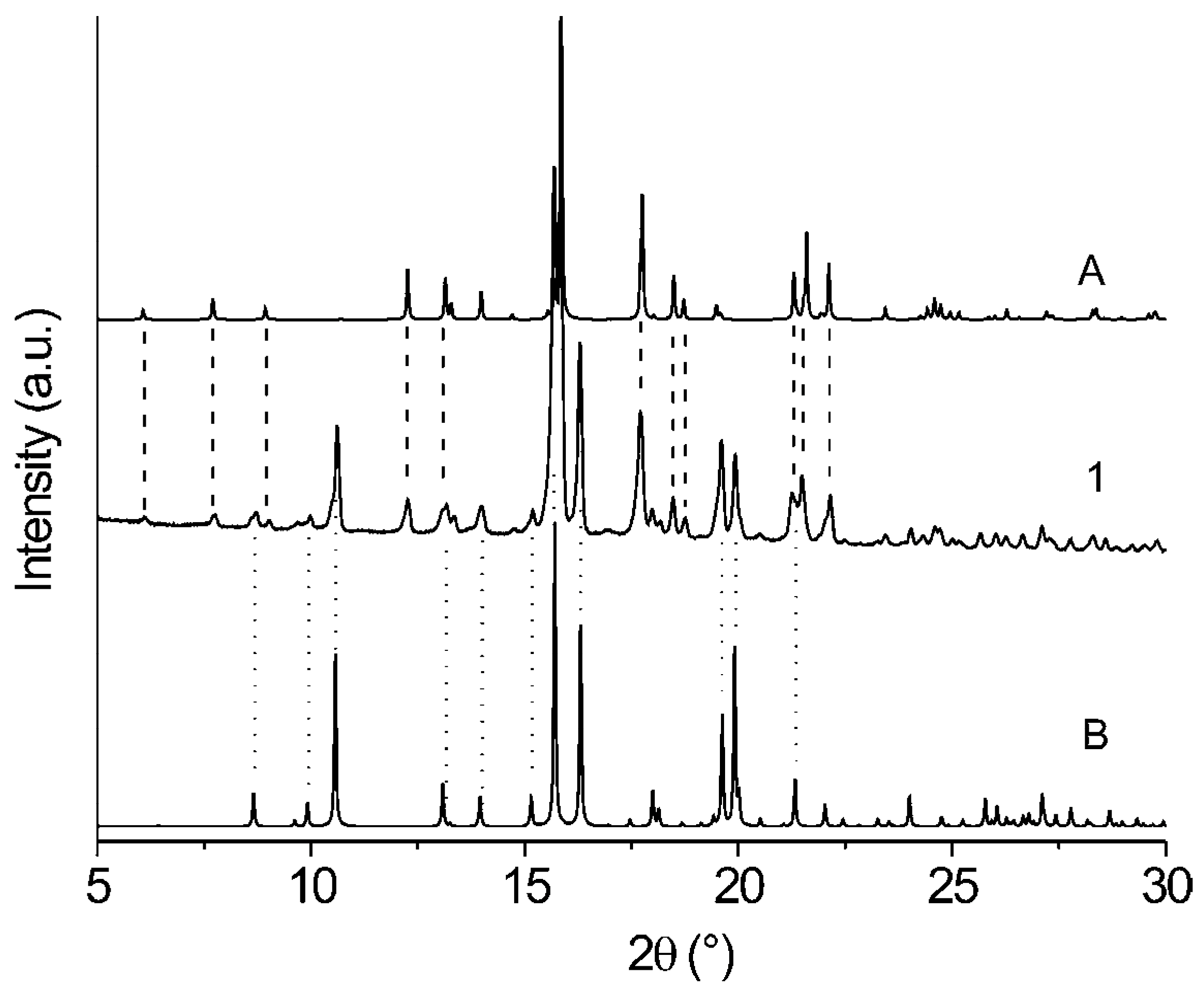
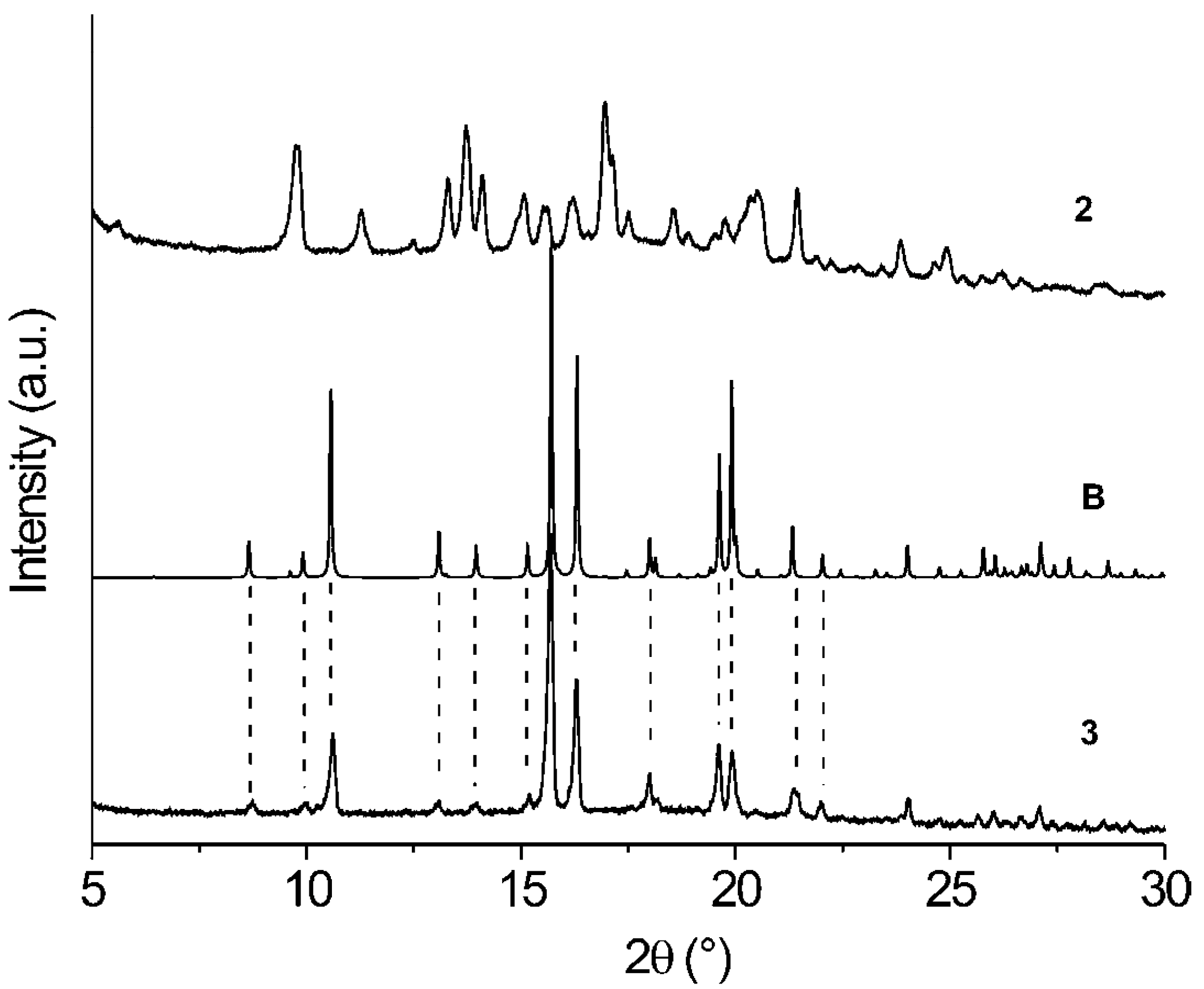
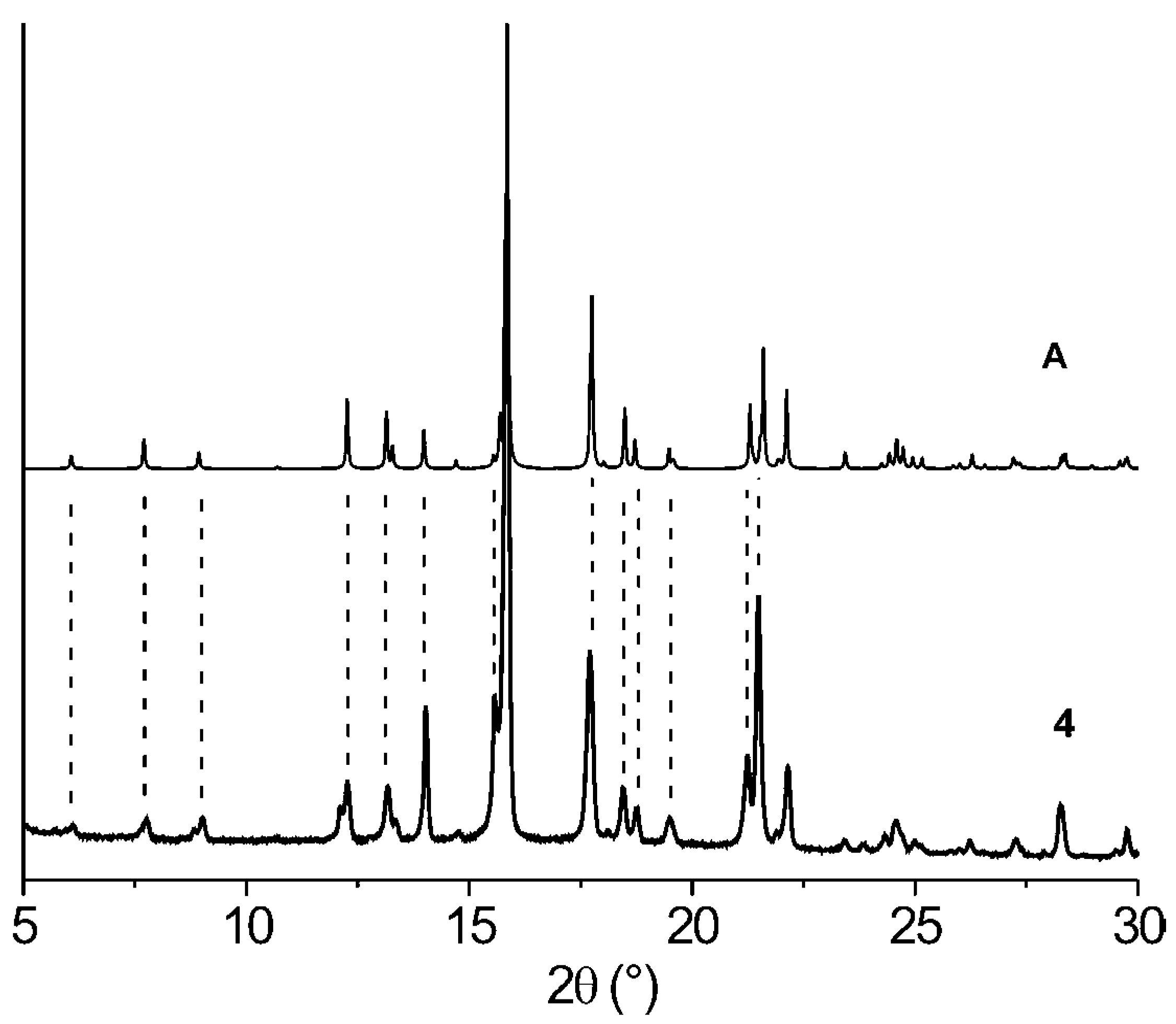
X-ray powder diffraction analysis
X-ray single crystal diffraction analysis [32]
| Empirical formula | C27H49NO5 |
| Formula weight | 467.67 |
| Temperature (K) | 173 |
| Wavelength (Å) | 0.71073 |
| Crystal system | Orthorhombic |
| Space group | P212121 |
| Unit cell dimensions (Å) | a = 7.1151(2) b = 18.1803(4) c = 20.1803(5) |
| Volume (Å3) | 2610.42(11) |
| Z | 4 |
| Densitycalc (Mg/m3) | 1.190 |
| Absorption coefficient (mm-1) | 0.080 |
| F(000) | 1032 |
| Crystal size (mm) | 0.30 × 0.10 × 0.10 |
| Index ranges | -8≤h≤8; -21≤k≤21; -23≤l≤23 |
| Reflections collected/unique | 17693/2649 [Rint = 0.1650] |
| Data/restraints/parameters | 2649/6/319 |
| Goodness-of-fit on F2 | 1.070 |
| Final R indices [I > 2σ(I)] | R1 = 0.0595; wR2 = 0.0960 |
| R indices (all data) | R1 = 0.0870; wR2 = 0.1062 |
| Largest diff. peak and hole (eÅ-3) | 0.202 and -0.227 |
Molecular modeling
Acknowledgments
References and Notes
- Davis, A.P. Cholaphanes et al.; steroids as structural components in molecular engineering. Chem. Soc. Rev. 1993, 22, 243–253. [Google Scholar] [CrossRef]
- Enhsen, A.; Kramer, W.; Wess, G. Bile acids in drug discovery. Drug Discov. Today 1998, 3, 409–418. [Google Scholar] [CrossRef]
- Yoswathananont, N.; Miyata, M.; Nakano, K.; Sada, K. Perspectives in Supramolecular Chemistry; Toda, F., Bishop, R., Eds.; Wiley: New York, 2004; Vol 8, Separations and Reactions in Organic Supramolecular Chemistry; pp. 87–122, and references cited therein. [Google Scholar]
- Bortolini, O.; Fantin, G.; Fogagnolo, F. Bile acid derivatives as enantiodifferentiating host molecules in inclusion processes. Chirality 2005, 17, 121–130, and references cited therein. [Google Scholar] [CrossRef]
- Valkonen, A.; Lahtinen, M.; Virtanen, E.; Kaikkonen, S.; Kolehmainen, E. Bile acid amidoalcohols: simple organogelators. Biosens. Bioelectron. 2004, 20, 1233–1241. [Google Scholar] [CrossRef]
- Willemen, H.M.; Vermonden, T.; Marcelis, A.T.M.; Sudhölter, E.J.R. N-Cholyl amino acid alkyl esters - a novel class of organogelators. Eur. J. Org. Chem. 2001, 2329–2335. [Google Scholar]
- Willemen, H.M.; Vermonden, T.; Marcelis, A.T.M.; Sudhölter, E.J.R. Alkyl Derivatives of Cholic Acid as Organogelators: One-Component and Two-Component Gels. Langmuir 2002, 18, 7102–7106. [Google Scholar] [CrossRef]
- Sangeetha, N.M.; Balasubramanian, R.; Maitra, U.; Ghosh, S.; Raju, A.R. Novel Cationic and Neutral Analogues of Bile Acids: Synthesis and Preliminary Study of Their Aggregation Properties. Langmuir 2002, 18, 7154–7157. [Google Scholar] [CrossRef]
- Babu, P.; Sangeetha, N.M.; Maitra, U. Supramolecular chemistry of bile acid derivatives: formation of gels. Macromol. Symp. 2006, 241, 60–67, and references cited therein. [Google Scholar] [CrossRef]
- Terech, P.; Sangeetha, N.M.; Maitra, U. Molecular Hydrogels from Bile Acid Analogues with Neutral Side Chains: Network Architectures and Viscoelastic Properties. Junction Zones, Spherulites, and Crystallites: Phenomenological Aspects of the Gel Metastability. J. Phys. Chem. 2006, B110, 15224–15233. [Google Scholar]
- Mukhopadhyay, S.; Maitra, U.; Ira; Krishnamoorthy, G.; Schmidt, J.; Talmon, Y. Structure and Dynamics of a Molecular Hydrogel Derived from a Tripodal Cholamide. J. Am. Chem. Soc. 2004, 126, 15905–15914. [Google Scholar] [CrossRef]
- Maitra, U.; Babu, P. First synthesis of phosphonobile acids and preliminary studies on their aggregation properties. Steroids 2003, 68, 459–463. [Google Scholar] [CrossRef]
- Harris, R.K. NMR crystallography. The use of chemical shifts. Solid State Sci. 2004, 6, 1025–1037. [Google Scholar] [CrossRef]
- Harris, R.K.; Cadars, S.; Emsley, L.; Yates, J.R.; Pickard, C.J.; Jetti, R.K.R.; Griesser, U.J. NMR crystallography of oxybuprocaine hydrochloride, Modification II°. Phys. Chem. Chem. Phys. 2007, 9, 360–368. [Google Scholar] [CrossRef]
- Bowles, R.K.; Morgan, K.R.; Furneaux, R.H.; Coles, G.D. 13C CP/MAS NMR study of the interaction of bile acids with barley β-D-glucan. Carbohydr. Polym. 1996, 29, 7–10. [Google Scholar] [CrossRef]
- Hoagland, P.D.; Pfeffer, P.E. Cobinding of bile acids to carrot fiber. J. Agric. Food Chem. 1987, 35, 316–319. [Google Scholar] [CrossRef]
- Heyes, S.J.; Dobson, C.M. Carbon-13 CP/MAS NMR study of the inclusion polymerization of 2,3-dimethylbutadiene in deoxycholic acid. Macromolecules 1992, 25, 3617–3623. [Google Scholar] [CrossRef]
- Nakaoki, T.; Sumida, T.; Takagi, M.; Takemoto, K. High resolution solid state 13C NMR investigation of the deoxycholic acid/ferrocene inclusion compound. Polym. Bull. 1999, 43, 365–369. [Google Scholar] [CrossRef]
- Imashiro, F.; Kuwahara, D.; Terao, T. Carbon-13 solid-state NMR study on populations, conformations, and molecular motions of γ-valerolactone enantiomers enclathrated in the chiral cholic acid host. J. Chem. Soc. Perkin Trans. 2 1993, 1759–1763. [Google Scholar] [CrossRef]
- Nakamura, S.; Imashiro, F.; Takegoshi, K.; Terao, T. Sequential Arrangement of γ-Valerolactone Enantiomers Enclathrated in Cholic Acid Channels as Studied by 13C Solid-State NMR: Elucidation of the Optical Resolution Mechanism. J. Am. Chem. Soc. 2004, 126, 8769–8776. [Google Scholar] [CrossRef]
- Hazra, B.G.; Pore, V.S.; Dey, S.K.; Datta, S.; Darokar, M.P.; Saikia, D.; Khanuja, S.P.S.; Thakur, A.P. Bile acid amides derived from chiral amino alcohols: novel antimicrobials and antifungals. Bioorg. Med. Chem. Lett. 2004, 14, 773–777. [Google Scholar] [CrossRef]
- Mosbach, E.H.; Ayengar, N.K.N.; McSherry, C.K. Synthesis of steroid compounds. U.S. Pat. 4460509, 1984. [Google Scholar]
- Mosbach, E.H.; McSherry, C.K.; Kuroki, S. Synthesis of 3α,12α-dihydroxy-7-methyl-5β-chol-6 and 7-en-24-oic acids and 7-methylene-5β-cholan-24-oic acid as intermediates for cholelithiasis therapeutics. U.S. Pat. 4648995, 1987. [Google Scholar]
- Della Valle, Fr.; Lorenzi, S.; Samson, J.C.J.J.; Della Valle, Fe. Pharmaceutical compositions containing N-acyl derivatives of aminoalcohols for the treatment of pathologies involving mast cells. Eur. Pat. Appl. EP 0550006, 1993. [Google Scholar]
- Della Valle, Fr.; Lorenzi, S.; Samson, J.C.J.J.; Della Valle, Fe. N-Acyl derivatives of amino alcohols active as local autacoids and useful in the therapy of autoimmune processes. U.S. Pat. 5506224, 1996. [Google Scholar]
- Della Valle, Fr.; Lorenzi, S.; Della Valle, Fe. Aminoalcohol N-acyl derivatives as therapeutic agents against the neurogenic endoneural edema of the peripheral nerve. U.S. Pat. Appl. US 5679667, 1997. [Google Scholar]
- Bundy, R.; Mauskopf, J.; Walker, J.T.; Lack, L. Interaction of uncharged bile salt derivatives with the ileal bile salt transport system. J. Lipid Res. 1977, 18, 389–395. [Google Scholar]
- Mukai, A.; Yazawa, K.; Mikami, Y.; Harada, K.; Gräfe, U. Biotransformation of bile acids by pathogenic actinomycetes Nocardia otitidiscaviarum and Amycolatopsis sp. strains. J. Antibiot. 2005, 58, 356–360. [Google Scholar] [CrossRef]
- Cambridge Structural Database (CSD, Version 5.28). CCDC: Cambridge, UK, 2006.
- Nakano, K.; Sada, K.; Kurozumi, Y.; Miyata, M. Importance of packing coefficients of host cavities in the isomerization of open host frameworks: guest-size-dependent isomerization in cholic acid inclusion crystals with monosubstituted benzenes. Chem. Eur. J. 2001, 7, 209–220. [Google Scholar] [CrossRef]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: visualization and analysis of crystal structures. J. Appl. Cryst. 2006, 39, 453–457. [Google Scholar] [CrossRef]
- CCDC-635027 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033; e-mail: deposit@ccdc.cam.ac.uk)
- COLLECT. Bruker AXS Inc.: Madison, WI, 2004.
- Otwinowski, Z.; Minor, W. Methods in Enzymology; Carter Jr, C.W., Sweet, R.M., Eds.; Academic Press: New York, 1997; vol. 276, Macromolecular Crystallography; Part A; pp. 307–326. [Google Scholar]
- Burla, M.C.; Camalli, M.; Carrozzini, B.; Cascarano, G.L.; Giacovazzo, C.; Polidori, G.; Spagna, J. SIR2002: the program. J. Appl. Cryst. 2003, 36, 1103. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXL-97; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Farrugia, L.J. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Cryst. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Farrugia, L.J. ORTEP-3 for Windows – a version of ORTEP-III with a Graphical User Interface (GUI). J. Appl. Cryst. 1997, 30, 565. [Google Scholar] [CrossRef]
- Wolinski, K.; Hinton, J.F.; Pulay, P.J. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, R.E.; Zakizewski, W.G.; Montgomery Jr., J.A.; Stratmann, R.E.; Burant, J.C.; Dapprich, S.; Millam, J.M.; Daniels, A.D.; Kudin, K.N.; Strain, M.C.; Farkas, O.; Tomasi, J.; Barone, V.; Cossi, M.; Cammi, R.; Mennuci, B.; Pomelli, C.; Adamo, C.; CliVord, S.; Ochterski, J.; Petersson, G.A.; Ayala, P.Y.; Cui, Q.; Morokuma, K.; Malick, D.K.; Rabuck, A.D.; Raghavachari, K.; Foresman, J.B.; Cioslowski, J.; Oritz, J.V.; Stefanov, B.B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Gomperts, R.; Martin, R.L.; Fox, D.J.; Keith, T.; AlLaham, M.A.; Peng, C.Y.; Nanayakkara, A.; Gonzalez, C.; Challacombe, M.; Gill, P.M.W.; Johnson, B.; Chen, W.; Wong, M.W.; Andres, J.L.; Head-Gordon, M.; Replogle, E.S.; Pople, J.A. Gaussian 98; Revision A.9; Gaussian: Pittsburg, PA, 1998. [Google Scholar]
- Allinger, N.L. Conformational analysis. 130. MM2. A hydrocarbon force field utilizing V1 and V2 torsional terms. J. Am. Chem. Soc. 1977, 99, 8127–8134. [Google Scholar] [CrossRef] Allinger, N.L.; Yuh, Y.H. Quantum Chemistry Program Exchange. Bloomington IN; Program No. 395. Burkert, U.; Allinger, N.L. (Eds.) Molecular Mechanics; ACS Monograph 177; American Chemical Society: Washington, D.C., 1982.
- Stewart, J.J.P. Optimization of parameters for semiempirical methods. I. Method. J. Comput. Chem. 1989, 10, 209–220. [Google Scholar] [CrossRef]
- HyperChemTM, Release 7.0 for Windows; Hypercube, Inc.: Gainesville, FL, 2002.
- Sample Availability: Samples of the compounds 1 - 4 are available from authors.
© 2008 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Valkonen, A.; Kolehmainen, E.; Lahtinen, M.; Sievänen, E.; Noponen, V.; Tolonen, M.; Kauppinen, R. Structural, Thermoanalytical and Molecular Modeling Studies on N-(3-hydroxypropyl) 3α,12α-Dihydroxy-5β-cholan-24-amide and Its Monohydrates. Molecules 2007, 12, 2161-2178. https://doi.org/10.3390/12092161
Valkonen A, Kolehmainen E, Lahtinen M, Sievänen E, Noponen V, Tolonen M, Kauppinen R. Structural, Thermoanalytical and Molecular Modeling Studies on N-(3-hydroxypropyl) 3α,12α-Dihydroxy-5β-cholan-24-amide and Its Monohydrates. Molecules. 2007; 12(9):2161-2178. https://doi.org/10.3390/12092161
Chicago/Turabian StyleValkonen, Arto, Erkki Kolehmainen, Manu Lahtinen, Elina Sievänen, Virpi Noponen, Minna Tolonen, and Reijo Kauppinen. 2007. "Structural, Thermoanalytical and Molecular Modeling Studies on N-(3-hydroxypropyl) 3α,12α-Dihydroxy-5β-cholan-24-amide and Its Monohydrates" Molecules 12, no. 9: 2161-2178. https://doi.org/10.3390/12092161
APA StyleValkonen, A., Kolehmainen, E., Lahtinen, M., Sievänen, E., Noponen, V., Tolonen, M., & Kauppinen, R. (2007). Structural, Thermoanalytical and Molecular Modeling Studies on N-(3-hydroxypropyl) 3α,12α-Dihydroxy-5β-cholan-24-amide and Its Monohydrates. Molecules, 12(9), 2161-2178. https://doi.org/10.3390/12092161