Synthesis of Ring-Contracted Erythromycin A Derivatives via Microwave-Assisted Intramolecular Transesterification

Abstract
:Introduction
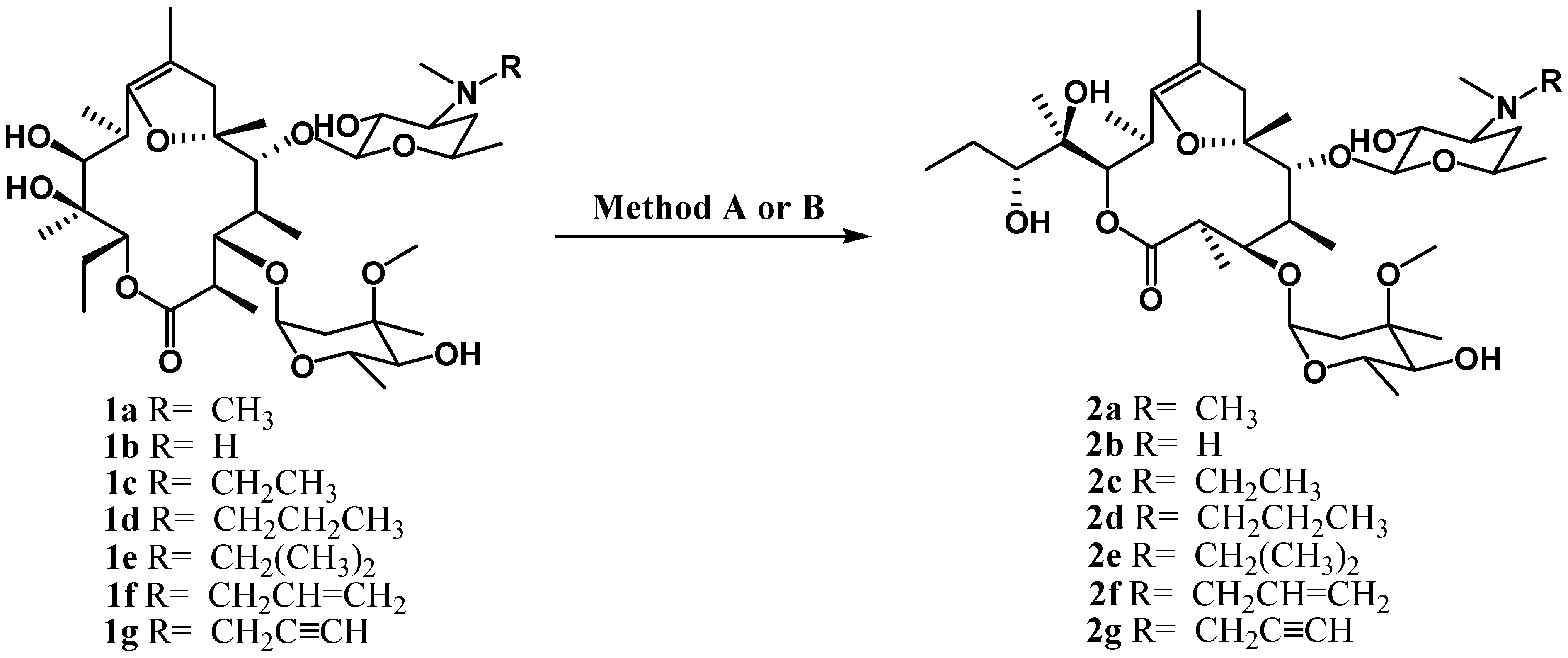
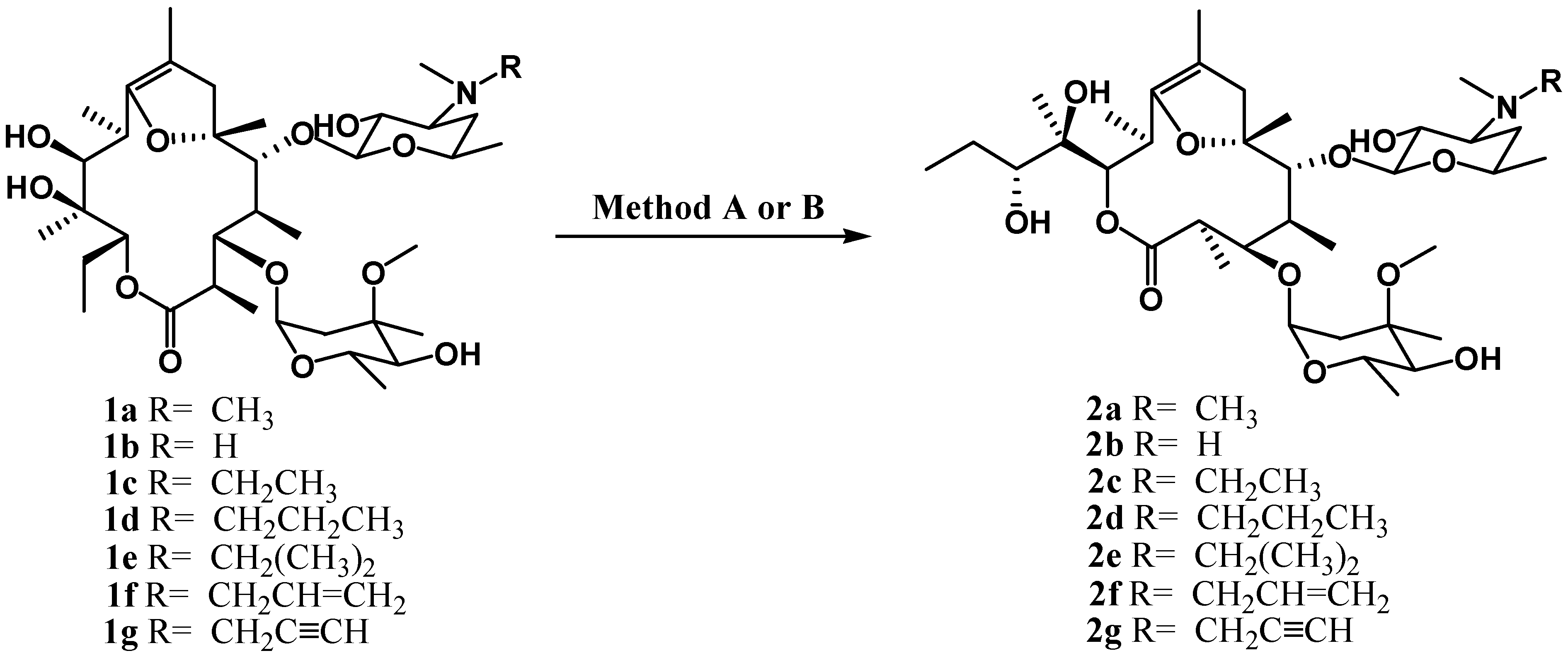
Results and Discussion

{kind=link}
| Method | Reagents | power (W) a | Temp.(°C) b | Time (min) | Yield (%) |
|---|---|---|---|---|---|
| A (solvent-containing) | K2CO3,TBAB, DMF | 200 | 130 | 10 | 58 |
| TBAB, DMF | 200 | 130 | 10 | 50 | |
| TBAF, DMF | 200 | 130 | 5 | 72 | |
| B (solvent-free) | Al2O3 (pH3.8-4.8) | 400 | 180 | 20 | trace |
| Al2O3 (pH6.5-7.5) | 400 | 180 | 20 | 63 | |
| Al2O3 (pH9.0-10) | 400 | 180 | 20 | trace | |
| Kieselguhr G | 400 | 180 | 20 | trace | |
| Silica gel | 400 | 180 | 10 | 33 | |
| 400 | 180 | 15 | 57 | ||
| 400 | 180 | 20 | 76 | ||
| 400 | 180 | 25 | 64 |
Conclusions
Experimental
General
Ring-contracted erythromycin (2a)
Analytical and spectral data for 2b -2g
Acknowledgments
References
- McGuire, J. M.; Bunch, R. L.; Anderson, R. C.; Boaz, H. E.; Flyn, E. H.; Powell, H. M.; Smith, J. W. Ilotycin, a new antibiotic. Antibiot. Chemother. 1952, 2, 281–283. [Google Scholar]
- Depoortere, I.; Peeters, T. L.; VanTrappen, G. The erythromycin derivative EM-523 is a potent motilin agonist in man and in rabbit. Peptides 1990, 11, 515–519. [Google Scholar] [CrossRef]
- Plewig, G.; Schopf, E. Anti-inflammatory effects of antimicrobial agents: an in vivo study. J. Invest. Dermatol. 1975, 65, 532–536. [Google Scholar]
- Kibwage, I. O.; Busson, R.; Janssen, G.; Hoogmartens, J.; Vanderhaeghe, H.; Bracke, J. Translactonization in erythromycins. J. Org. Chem. 1987, 52, 990–996. [Google Scholar] [CrossRef]
- Kirst, H. A.; Wind, J. A.; Paschal, J. W. Synthesis of ring-contracted derivatives of erythromycin. J. Org. Chem. 1987, 52, 4359–4362. [Google Scholar] [CrossRef]
- Kappe, C. O. Controlled Microwave Heating in Modern Organic Synthesis. Angew. Chem. Int. Ed. 2004, 43, 6250–6285. [Google Scholar] [CrossRef]
- Romanova, N. N.; Gravis, A. G.; Zyk, N. V. Microwave Irradiation in Organic Synthesis. Russ. Chem. Rev. 2005, 74, 969–1013. [Google Scholar] [CrossRef]
- Limousin, C.; Cleophax, J.; Loupy, A.; Petit, A. Synthesis of Benzoyl and Dodecanoyl Derivatives from Protected Carbohydrates under Focused Microwave Irradiation. Tetrahedron. 1998, 54, 13567–13578. [Google Scholar] [CrossRef]
- Kurath, P.; Jones, P. H.; Egan, R. S.; Perun, T. J. Acid degradation of erythromycin A and erythromycin B. Experentia. 1971, 27, 362–368. [Google Scholar] [CrossRef]
- Hoz, A.; Díaz-Ortiz, A.; Moreno, A. Microwaves in organic synthesis. Thermal and non-thermal microwave effects. Chem. Soc. Rev. 2005, 34, 164–178. [Google Scholar]
- Dandia, A.; Singh, R.; Khaturia, S.; Me′rienne, C.; Morgantc, G.; Loupy, A. Efficient microwave enhanced regioselective synthesis of a series of benzimidazolyl/triazolyl spiro[indole-thiazolidinones] as potent antifungal agents and crystal structure of spiro[3H-indole-3,20-thiazolidine]-30(1,2,4-triazol-3-yl)-2,40(1H)-dione. Bioorg. Med. Chem. 2006, 14, 2409–2417. [Google Scholar] [CrossRef]
- Miyashita, M.; Suzuki, T.; Yoshikoshi, A. Fluoride-promoted epoxidation of α,β-unsaturated compounds. Chem. Lett. 1987, 285–288. [Google Scholar] [CrossRef]
- Kabalka, G. W.; Pagni, R. M.; Wang, L.; Namboodiri, V.; Hair, C. M. Microwave-assisted, solventless Suzuki coupling reactions on palladium-doped alumina. Green Chem. 2000, 2, 120–122. [Google Scholar]
- Tsuzuki, K.; Sunazuka, T.; Marui, S.; Toyoda, H.; Omura, S.; Inatomi, N.; Itoh, Z. Motilides, macrolides with gastrointestinal motor stimulating activity. Chem. Pharm. Bull. 1989, 37, 2687–2709. [Google Scholar]
- Kibwage, I. O.; Janssen, G.; Busson, R.; Hoogmartens, J.; Vanderhaeghe, H. Identification of novel erythromycin derivatives in mother liquor concentrates of Streptomyces erythraeus. J. Antibiot. 1987, 40, 1–6. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are available from the authors.
© 2007 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Bao, K.; Zhang, W.; Zhang, C.; Qu, Y.; Tian, L.; Wu, L.; Zhao, X.; Cheng, M. Synthesis of Ring-Contracted Erythromycin A Derivatives via Microwave-Assisted Intramolecular Transesterification. Molecules 2007, 12, 2123-2129. https://doi.org/10.3390/12092123
Bao K, Zhang W, Zhang C, Qu Y, Tian L, Wu L, Zhao X, Cheng M. Synthesis of Ring-Contracted Erythromycin A Derivatives via Microwave-Assisted Intramolecular Transesterification. Molecules. 2007; 12(9):2123-2129. https://doi.org/10.3390/12092123
Chicago/Turabian StyleBao, Kai, Weige Zhang, Chuanliang Zhang, Yingwei Qu, Liang Tian, Lan Wu, Xiang Zhao, and Maosheng Cheng. 2007. "Synthesis of Ring-Contracted Erythromycin A Derivatives via Microwave-Assisted Intramolecular Transesterification" Molecules 12, no. 9: 2123-2129. https://doi.org/10.3390/12092123
APA StyleBao, K., Zhang, W., Zhang, C., Qu, Y., Tian, L., Wu, L., Zhao, X., & Cheng, M. (2007). Synthesis of Ring-Contracted Erythromycin A Derivatives via Microwave-Assisted Intramolecular Transesterification. Molecules, 12(9), 2123-2129. https://doi.org/10.3390/12092123