Leufolins A and B, Potent Butyrylcholinesterase-inhibiting Flavonoid Glucosides from Leucas urticifolia

Abstract
:Introduction
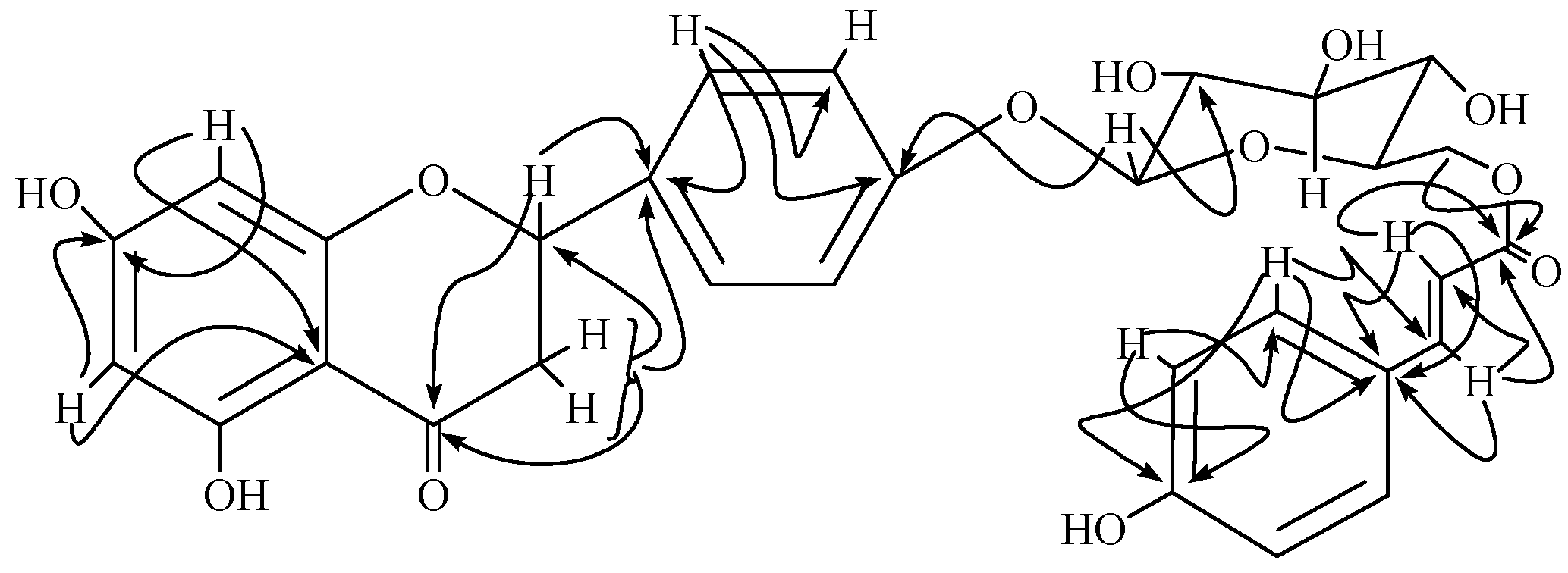
Results and Discussion
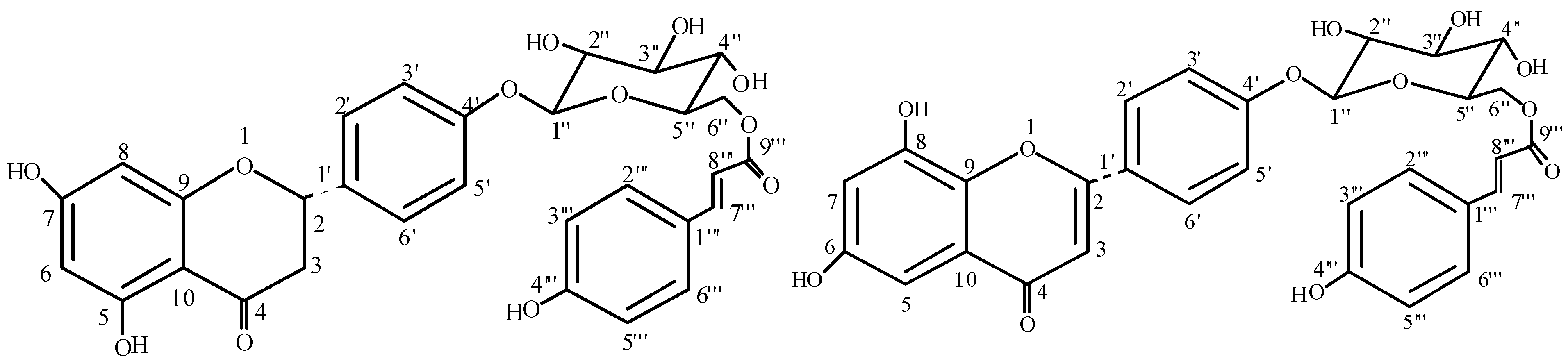

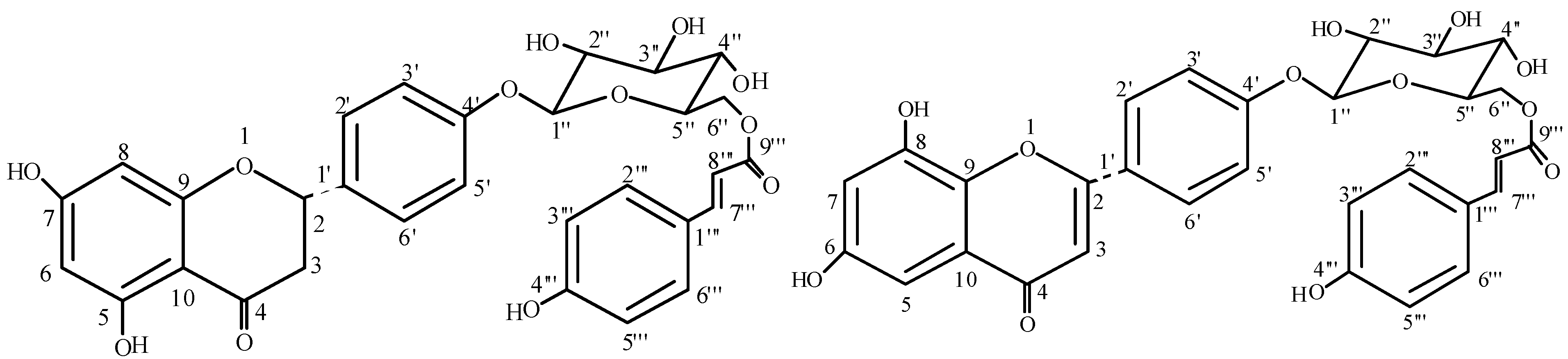{kind=link}
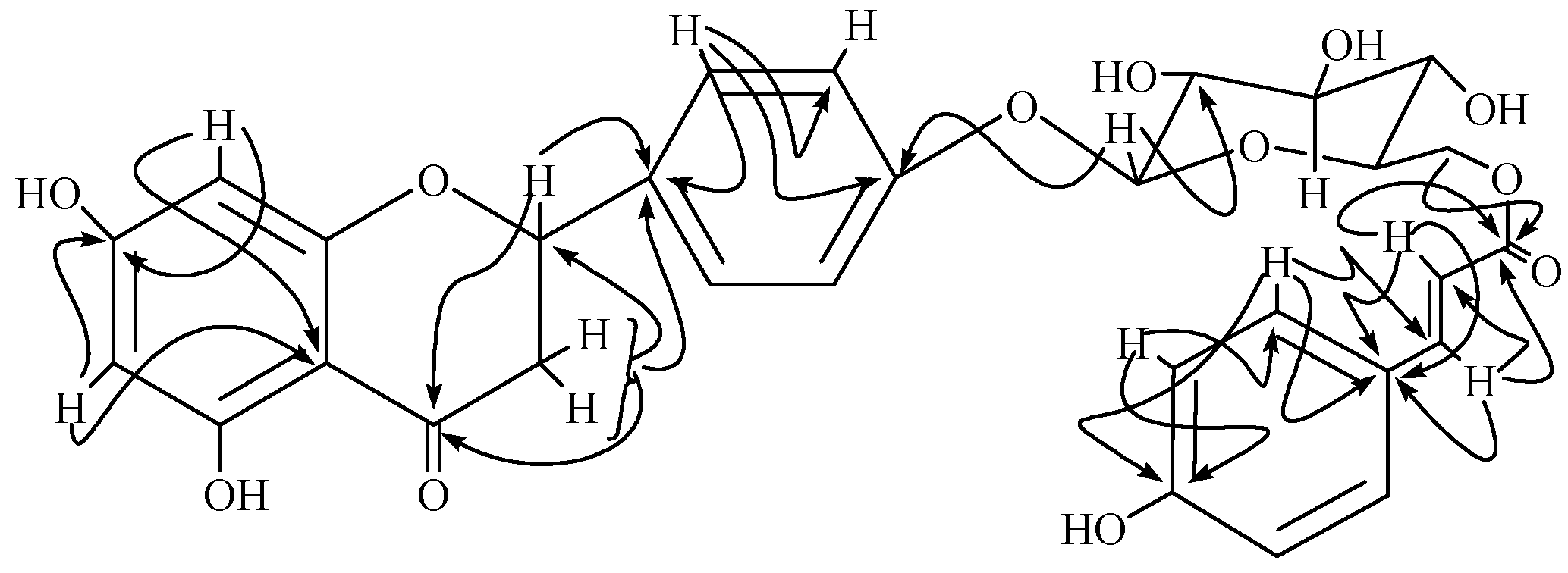{kind=link}
| 1 | 2 | |||
|---|---|---|---|---|
| C/H | δ H | δ C | δ H | δ C |
| 1 | - | - | ||
| 2 | 5.28 (1H, dd, 12.3, 2.7) | 80.3 | 164.9 | |
| 3 | 3.12 (1H, dd, 17.1, 12.3) 2.71 (1H, dd, 17.1, 2.7) | 44.0 | 6.89 (1H, s) | 104.7 |
| 4 | 198.4 | 182.8 | ||
| 5 | 164.9 | 6.90 (1H, d, 1.8) | 114.7 | |
| 6 | 6.21 (1H, d, 1.8) | 97.9 | - | 162.6 |
| 7 | 166.8 | 6.81 (1H, d, 1.8) | 101.6 | |
| 8 | 6.16 (1H, d, 1.9) | 97.1 | 150.0 | |
| 9 | 158.9 | 144.6 | ||
| 10 | 104.9 | 115.2 | ||
| 1' | 130.8 | 122.0 | ||
| 2' | 7.26 (1H, d, 8.5) | 128.9 | 7.07 (1H, d, 9.7) | 123.1 |
| 3' | 6.69 (1H, d, 8.5) | 115.7 | 7.25 (1H, d, 9.7) | 116.7 |
| 4' | 164.3 | 163.8 | ||
| 5' | 6.69 (1H, d, 8.5) | 115.7 | 7.25 (1H, d, 9.7) | 116.7 |
| 6' | 7.26 (1H, d, 8.5) | 128.9 | 7.07 (1H, d, 9.7) | 123.1 |
| 1'' | 4.97 (1H, d, 7.0) | 101.1 | 5.78 (1H, d, 7.3) | 101.7 |
| 2'' | 3.33 (1H, m) | 74.5 | 3.66 (1H, m) | 74.6 |
| 3'' | 3.45 (1H, m) | 77.8 | 3.94 (1H, m) | 78.3 |
| 4'' | 3.39 (1H, m) | 71.8 | 4.15 (1H, m) | 71.8 |
| 5'' | 3.75 (1H, t, 7.7) | 75.6 | 4.17 (1H, m) | 75.7 |
| 6'' | 4.55 (1H, d, 11.7) 4.28 (1H, dd, 11.7, 7.2) | 64.6 | 4.85-4.89 (2H, m) | 64.8 |
| 1''' | 127.2 | 134.0 | ||
| 2''' | 7.37 (1H, d, 8.5) | 131.2 | 7.45 (1H, d, 8.4) | 130.6 |
| 3''' | 6.77 (1H, d, 8.5) | 116.4 | 7.23 (1H, d, 8.4) | 116.9 |
| 4''' | - | 161.1 | 162.8 | |
| 5''' | 6.77 (1H, d, 8.5) | 116.4 | 7.23 (1H, d, 8.4) | 116.9 |
| 6''' | 7.37 (1H, d, 8.5) | 131.2 | 7.45 (1H, d, 8.4) | 130.6 |
| 7''' | 7.59 (1H, d, 15.8) | 146.8 | 6.75 (1H, d, 12.9) | 144.6 |
| 8''' | 6.33 (1H, d, 15.8) | 114.9 | 6.09 (1H, d, 12.9) | 115.6 |
| 9''' | 169.0 | 166.6 | ||
Biological Activity
| Compounds | IC50 ± SEM[μM] |
|---|---|
| Leufolin A (1) | 1.6±0.98 |
| Leufolin B (2) | 3.6±1.7 |
| Eserine * | 0.93±0.3 |
Experimental
General
Plant Material
Extraction and Isolation
Acid Hydrolysis
In vitro Butyrylcholinesterase inhibition assay
Determination of IC50 values
References
- Jafri, S. M. H. Flora of Karachi; The Book Corporation Karachi: Karachi, Pakistan, 1966; p. 391. [Google Scholar]
- Watt, G. Dictionary of the Economic Products of India; Cosmo Publications: Delhi, India, 1890; Vol. 6, p. 632. [Google Scholar]
- Mhaskar, K. S.; Blatter, E.; Caius, J. F. Indian Medicinal Plants; SriSatguru Publications: Delhi, India, 1935; Vol. 9, p. 2778. [Google Scholar]
- Shinoda, J. A new biologically active flavone glycoside from the roots of Cassia fistula Linn. J. Pharm. Soc. Jpn. 1928, 48, 214–220. [Google Scholar]
- Chen, Y.; Lin, J.; Davidson, R. S.; Howarth, W. O. Isolation and structure of clemantine, A new flavanone glycoside from Clematis armandii Franch. Tetrahedron 1993, 49, 5169–5176. [Google Scholar] [CrossRef]
- Mabry, T. J.; Markham, K. R.; Thomas, M. B. The systematic identification of flavonoids; Springer-Verlag: New York, 1970; p. 271. [Google Scholar]
- Ahmad, I.; Anis, I.; Malik, A.; Nawaz, S. A.; Choudhary, M. I. Cholinesterase inhibitory constituents from Onosma hispida. Chem. Pharm. Bull. 2003, 51, 412–414. [Google Scholar] [CrossRef] [PubMed]
- Iinuma, M.; Ohyama, M.; Tanaka, T.; Mizuno, M.; Hong, S. K. Three 2', 4', 6'- Trioxygenated flavanones in roots of Echinosophora koreensis. Phytochemistry 1992, 31, 665–669. [Google Scholar] [CrossRef]
- Sachdev, K.; Kulshrestha, D. K. Aliarin, a new flavonoid from Dodonaea viscose Linn. Indian J. Chem. B. 1982, 21, 798–799. [Google Scholar]
- Jiao, R. H.; Ge, H. M.; Shi, D. H.; Tan, R. X. An apigenin-derived xanthine oxidase inhibitor from Palhinhaea cernua. J. Nat. Prod. 2006, 69, 1089–1091. [Google Scholar] [CrossRef] [PubMed]
- Washington, J. S.; Saxena, V. K. A new acylated apigenin 4'-O-β-D-glucoside from the stems of Thymus serpyllum Linn. J. Inst. Chem. (India) 1985, 57, 153–154. [Google Scholar]
- Yu, S. Q.; Holloway, H. W.; Utsuki, T.; Brossi, A.; Greig, N. H. Synthesis of novel phenserine-based-selective inhibitors of butyrylcholinesterase for Alzheimer’s disease. J. Med. Chem. 1999, 42, 1855–1861. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, M.; Glick, D.; Loewensten, Y.; Soreq, H. Engineering of human cholinesterase explains and predict diverse consequences of administration of various drugs and poisons. Pharmacol. Ther. 1995, 67, 283–322. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, L. J.; Borradaile, N. M.; Huff, M. W. Antiatherogenic properties of naringenin, a citrus flavonoid. Cardiovasc. Drug Rev. 1999, 17, 160–178. [Google Scholar] [CrossRef]
- Ellman, G. L.; Courtney, K. D.; Andres, V.; Featherstone, R. M. A New and rapid calorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Contact the authors.
© 2007 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Noor, A.-t.; Fatima, I.; Ahmad, I.; Malik, A.; Afza, N.; Iqbal, L.; Latif, M.; Khan, S.B. Leufolins A and B, Potent Butyrylcholinesterase-inhibiting Flavonoid Glucosides from Leucas urticifolia. Molecules 2007, 12, 1447-1454. https://doi.org/10.3390/12071447
Noor A-t, Fatima I, Ahmad I, Malik A, Afza N, Iqbal L, Latif M, Khan SB. Leufolins A and B, Potent Butyrylcholinesterase-inhibiting Flavonoid Glucosides from Leucas urticifolia. Molecules. 2007; 12(7):1447-1454. https://doi.org/10.3390/12071447
Chicago/Turabian StyleNoor, Atia-tun, Itrat Fatima, Ijaz Ahmad, Abdul Malik, Nighat Afza, Lubna Iqbal, Mehreen Latif, and Sher Bahadar Khan. 2007. "Leufolins A and B, Potent Butyrylcholinesterase-inhibiting Flavonoid Glucosides from Leucas urticifolia" Molecules 12, no. 7: 1447-1454. https://doi.org/10.3390/12071447
APA StyleNoor, A.-t., Fatima, I., Ahmad, I., Malik, A., Afza, N., Iqbal, L., Latif, M., & Khan, S. B. (2007). Leufolins A and B, Potent Butyrylcholinesterase-inhibiting Flavonoid Glucosides from Leucas urticifolia. Molecules, 12(7), 1447-1454. https://doi.org/10.3390/12071447