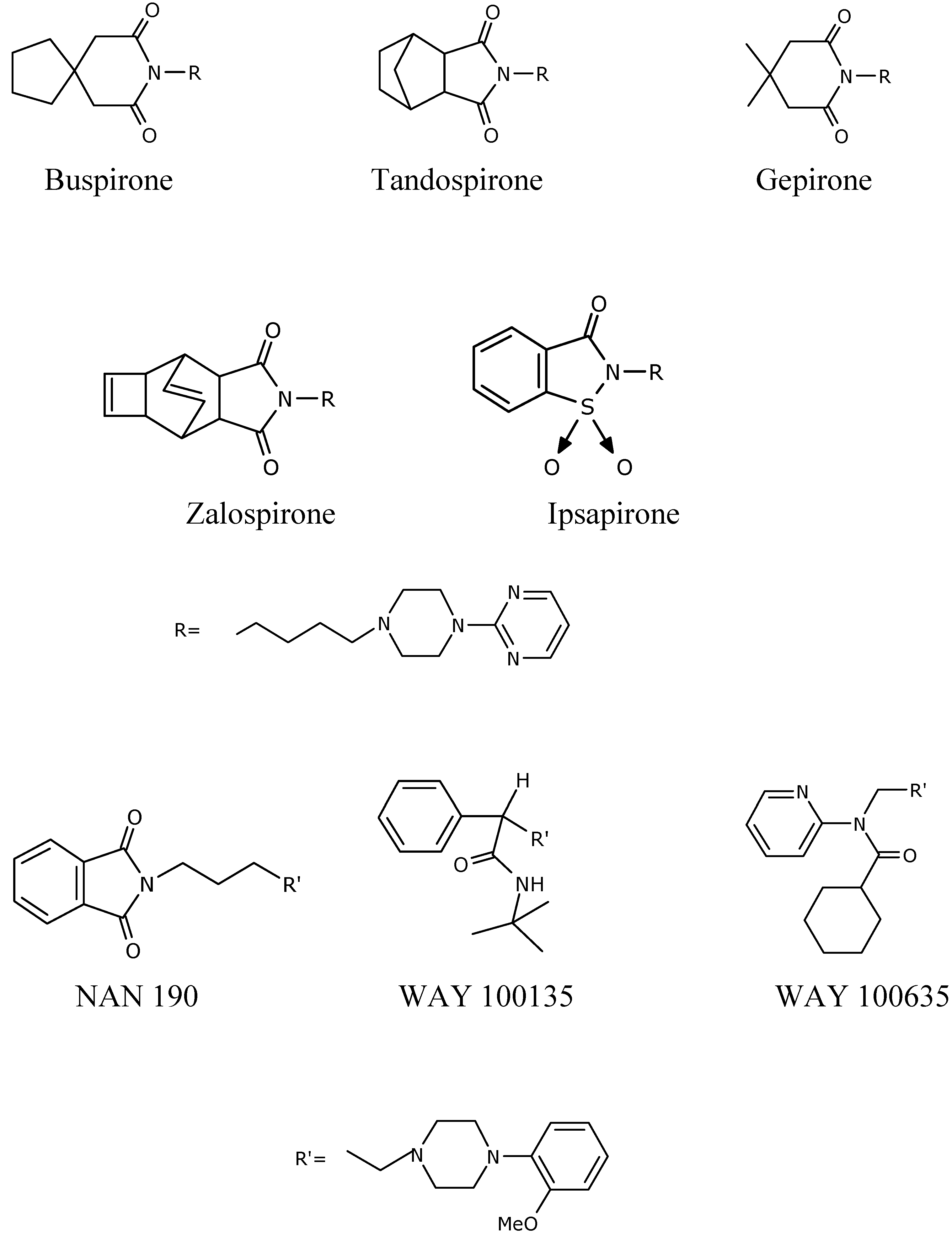
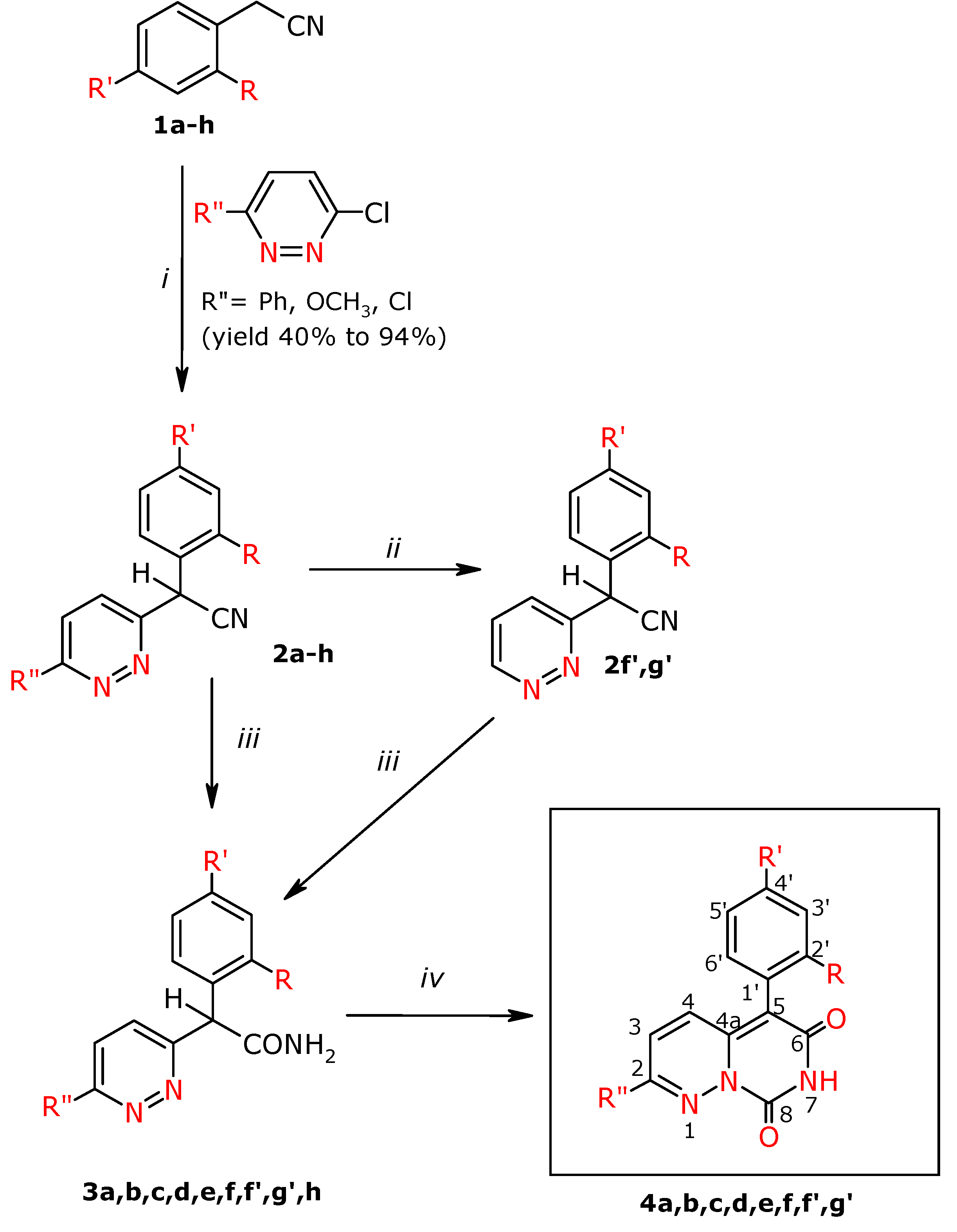
Experimental
General
Melting points of the substances were determined on a Mel-Temp® 3.0 (Barnsted/Thermolyne, USA) apparatus and are uncorrected. Elemental analysis was performed on a Perkin-Elmer CHN model 2400 analyzer. The IR spectra (KBr tablets) were performed on a Shimadzu FT IR-8300 apparatus.
1H- and
13C-NMR spectra were recorded on the following spectrometers: a Bruker Avance DMX WB of basic frequency for the protons: 400.133 MHz, for nuclei
13C: 100.623 MHz and a Varian type Unity Plus of basic frequency for the protons: 500.605 MHz, for nuclei
13C: 125.877 MHz at room temperature. CDCl
3 was used as solvent and tetramethylsilane as internal standard. The chemical shifts of resonance signals are given in ppm, and the coupling constants are in Hz. Thin-layer chromatography (TLC) was performed on Merck DC-Platten Kiesegel 60 F
254 plates, developed in the dioxane, toluene, ethanol systems, 25% NH
4OH (6.0:3.2:0.5:0.2, v/v) or chloroform, methanol, diethyl ether, 25% NH
4OH (6.0:2.0:1.8:0.2, v/v) and visualized on a UV lamp. Analytical samples of the compounds were obtained by purification on the chromatographic column using the flash technique and Merck Kieselgel 60 (230-400 mesh) filling. They were eluted with methylene chloride/methanol mixtures (99:1; 97:3; 95:5, v/v). The starting phenylacetonitrile derivatives, 3,6-dichloropyridazine and 3-chloro-6-methoxypyridazine were commercial products purchased from Aldrich and used without further purification. The other starting reagent 3-chloro-6-phenylpyridazine was prepared by the reported procedure [
18,
19].
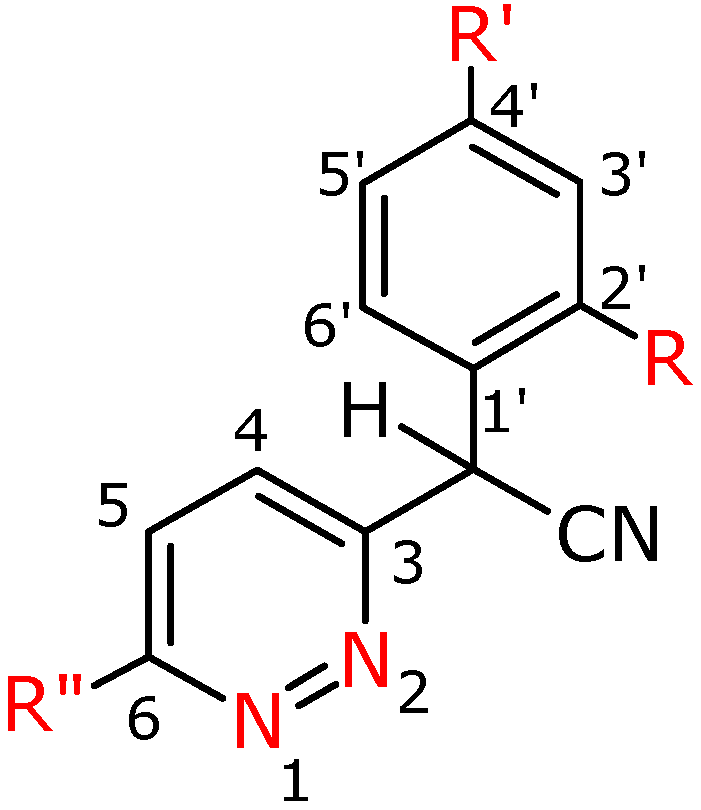
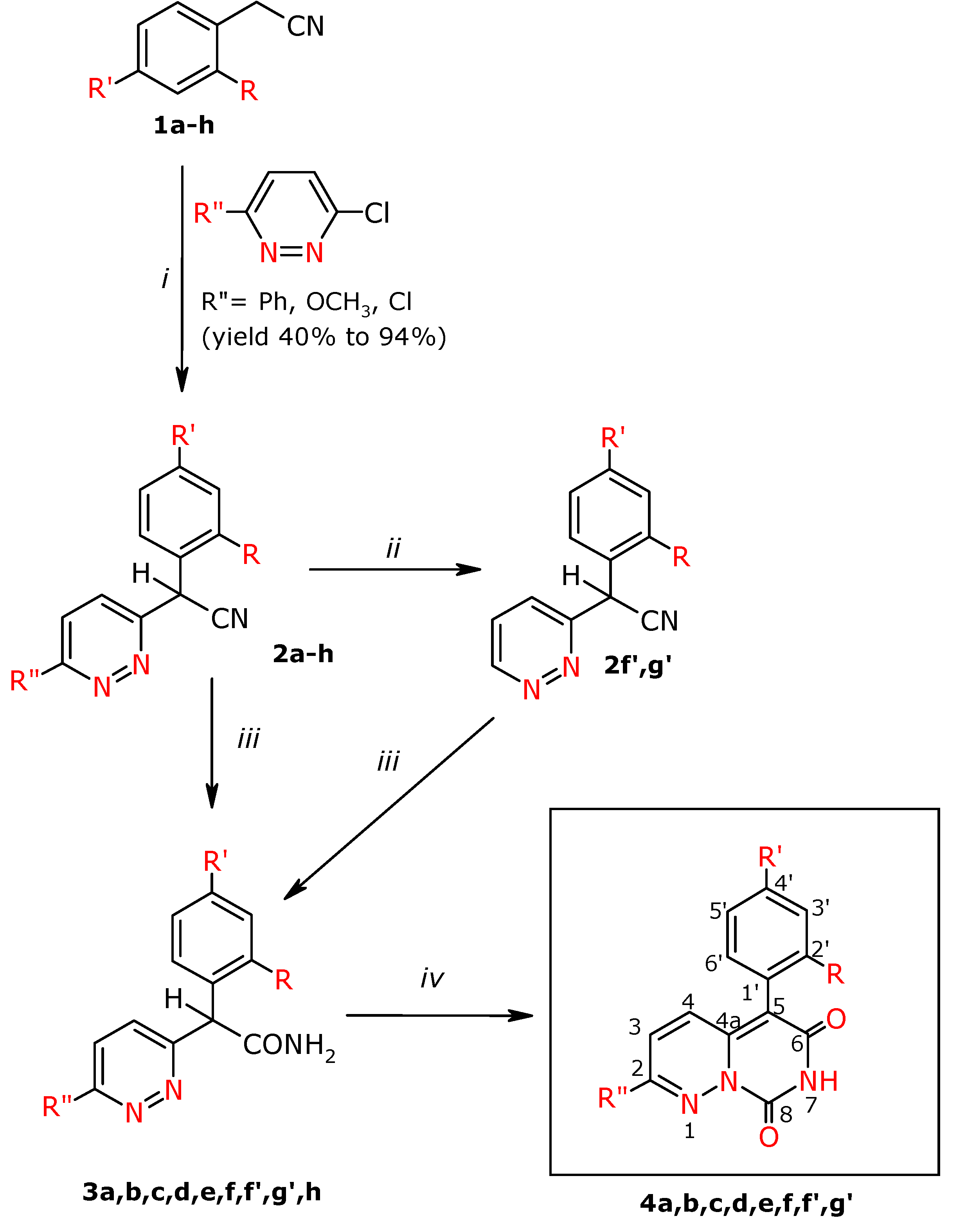
General procedure for the preparation of α-aryl-α-(3-pyridazin-3-yl)acetonitriles 2a-h
To DMSO (32.5 mL) was added KOH (16.5 g, 0.29 mol) and the mixture was stirred for 0.5 h at room temperature. Next an appropriate phenylacetonitrile derivative (0.11 mol) in DMSO (10 mL) was added dropwise and stirring was continued for another 0.5 h at room temperature. Next the appropriate 3-chloropyridazine derivative (0.07 mol) was added portionwise to the mixture, which was stirred at a temperature of 50 °C for 12 h. Next the post-reaction mixture was poured into ice water (1000 mL). The separated precipitates were filtered off. The obtained crude products 2c, 2d, 2e, 2f, 2f′ were purified by flash chromatography using CH2Cl2/MeOH (97:3 v/v), and then CH2Cl2/MeOH (99:1 v/v) as eluents, and then compounds 2c crystallized from absolute EtOH, 2d and 2f′ from EtOH. Compounds 2a, 2h, 2g′ were macerated with the ethyl acetate – hexane (1:1 v/v) mixture, and were then crystallized from EtOH. Compound 2b was crystallized from EtOH, 2g with ethyl acetate.
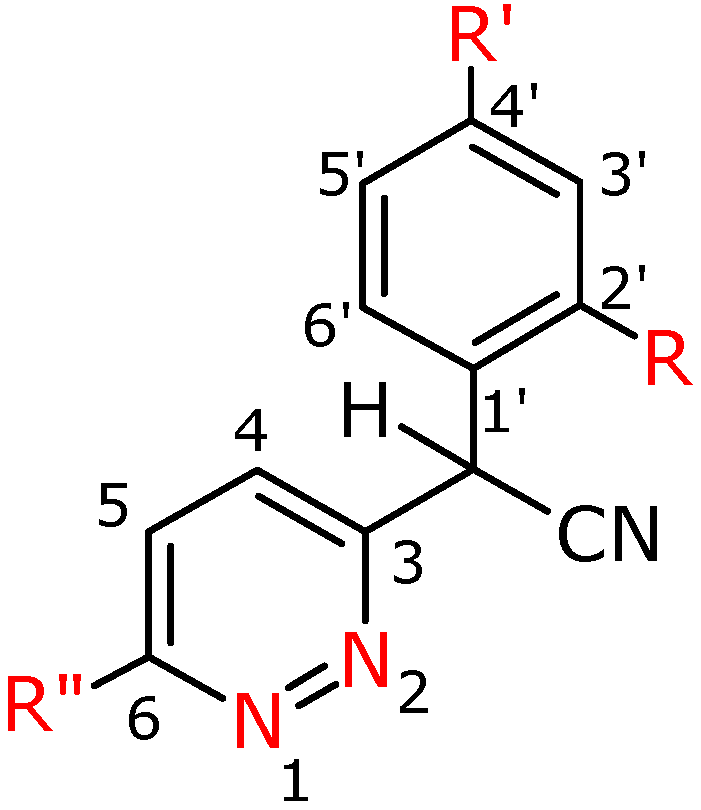
Figure 2.
Numbering system for compounds 2a-h.
Figure 2.
Numbering system for compounds 2a-h.
α-Phenyl-α-(6-phenylpyridazin-3-yl)-acetonitrile (
2a). Yield 65%; white crystals; m.p. 206.0-207.0ºC; {lit. [
15], 201-203ºC}; IR (cm
-1): 2246 (CN);
1H-NMR (500 MHz) δ: 5.73 (s, 1H, CH), 7.36 (tt,
3J=7.0,
4J=1.0, 1H, C4’H), 7.41 (td,
3J=7.5,
4J=1.5, 2H, C3’H, C5’H), 7.49- 7.56 (m, 5H, C2”H-C6”H), 7.63 (d,
3J=9.0, 1H, C4H), 7.88 (d, 1H, C5H), 8.07 (dd,
3J=7.5,
4J=2.0, 2H, C2’H, C6’H);
13C-NMR (125 MHz) δ: 43.6 (CH), 118.1 (CN), 125.1 (C5), 125.8 (C4), 127.2 (C3”, C5”), 127.6 (C2’, C6’), 128.9 (C4’), 129.2 (C3’, C5’), 129.5 (C2”, C6”), 130.6 (C4”), 133.5 (C1’), 135.4 (C1”), 157.0 (C3), 159.1 (C6); Anal. calcd. for C
18H
13N
3: C, 79.68; H, 4.83, N, 15.49; found: C, 79.62; H, 4.83;N, 15.44.
α-(4-Fluorophenyl)-α-(6-phenypyridazin-3-yl)-acetonitrile (2b). Yield 40%; white crystals; m.p. 200.8-201.2 ºC; IR (cm-1): 2248 (CN); 1H-NMR (500 MHz) δ: 5.70 (s, 1H, CH), 7.10 (m, 2H, C3’H, C5’H), 7.49-7.56 (m, 5H, C2”H-C6”H), 7.64 (d, 3J=9.0, 1H, C4H), 7.90 (d, 1H, C5H), 8.08 (m, 2H, C2’H, C6’H); 13C-NMR (125 MHz) δ: 42.9 (CH), 116.6 (d*,2J=22, C3’,C5’), 117.9 (CN), 125.2 (C4), 125.6 (C5), 127.2 (C2”, C6”), 129.2 (C3”, C5”), 129.4 (d*, 4J=3.6, C1’), 129.4 (d*, 3J=8.7, C2’,C6’), 130.6 (C4”), 135.3 (C1”), 156.7 (C3), 159.1 (C6), 162.9 (d*, 1J=249.1, C4’); Anal. calcd. for C18H12N3F: C, 74.73; H, 4.18, N, 14.52; found: C, 74.64; H, 4.08; N, 14.56.
α-(4-Tolyl)-α-(6-phenylpyridazin-3-yl)-acetonitrile (2c). Yield 71%; white crystals; m.p. 143.9-144.6 ºC; IR (cm-1): 2250 (CN); 1H-NMR (400 MHz) δ: 2.34 (s, 3H, CH3), 5.67 (s, 1H, CH), 7.20 (d, 2H, C3’H, C5’H), 7.40 (d, 3J=7.6, 2H, C2’H, C6’H), 7.51 (m, 3H, C3”H, C4”H, C5”H), 7.60 (d, 1H, C4H), 7.86 (d, 3J=8.8, 1H, C5H), 8.06 (m, 2H, C2”H, C6”H); 13C-NMR (100 MHz) δ: 21.3 (CH3), 43.4 (CH), 118.5 (CN), 125.3 (C5), 125.9 (C4), 127.3 (C2’, C6’), 127.6 (C3”, C5”), 129.3 (C2”, C6”), 130.3 (C3’, C5’), 130.7 (C4”), 131.8 (C1’), 135.6 (C4’), 139.0 (C1”), 157.3 (C6), 159.1 (C3); Anal. calcd. for C19H15N3: C, 79.98; H, 5.30, N, 14.72; found: C, 79.20; H, 5.26; N, 15.54.
α-(4-Methoxyphenyl)-α-(6-phenylpyridazin-3-yl)-acetonitrile (2d). Yield 94%; white crystals; m.p. 206.8-209.4 ºC; IR (cm-1): 2250 (CN); 1H-NMR (400 MHz) δ: 3.80 (s, 3H, OCH3), 5.66 (s, 1H, CH), 6.92 (d, 2H, C3’H, C5’H), 7.44 (d, 3J=8.4, 2H, C2’H, C6’H), 7.52 (m, 3H, C3”H, C4”H, C5”H), 7.61 (d, 1H, C4H), 7.88 (d, 3J=8.8, 1H, C5H), 8.08 (m, 2H, C2”H, C6”H); 13C-NMR (100 MHz) δ: 42.1 (CH), 55.6 (OCH3), 114.4 (C3’, C5’), 116.2 (C4), 117.9 (CN), 124.8 (C1’), 127.9 (C5), 127.9 (C2’, C6’), 128.4 (C6),129.1 (C2”, C6”), 129.6 (C3”, C5”), 131.7 (C4”), 133.7 (C1”), 157.3 (C3), 159.2 (C4’); Anal. calcd. for C19H15N3O: C, 75.73; H, 5.02, N, 13.94; found: C, 75.77; H, 5.42; N, 13.95.
α-(2-Methoxyphenyl)-α-(6-phenylpyridazin-3-yl)-acetonitrile (2e). Yield 85%; white crystals; m.p. 151.0-152.0 ºC; IR (cm-1): 2245 (CN); 1H-NMR (400 MHz) δ: 3.84 (s, 3H, OCH3), 5.93 (s, 1H, CH), 6.92 (d, 3J=8.4, 1H, C3’H), 7.04 (t, 3J=7.2, 1H, C5’H), 7.36 (t, 3J=7.6, 1H, C4’H), 7.51 (ps, 3H, C3”H, C4”H, C5”H), 7.57 (pt, 2H, C4H, C6’H), 7.85 (d, 3J=8.8, 1H, C5H), 8.08 (pd, 2H, C2”H, C6”H); 13C-NMR (100 MHz) δ: 38.4 (CH), 55.9 (OCH3), 111.5 (C3’), 118.4 (CN), 121.6 (C5’), 122.3 (C1’), 124.7 (C5), 126.2 (C4), 127.3 (C2”, C6”), 129.3 (C3”, C5”), 129.8 (C6’), 130.6 (C4”), 130.7 (C4’), 135.8 (C1”), 156.4 (C3), 156.8 (C6), 158.8 (C2’); Anal. calcd. for C19H15N3O: C, 75.73; H, 5.02, N, 13.94; found: C, 75.47; H, 5.10; N, 13.71.
α-Phenyl-α-(6-chloropyridazin-3-yl)-acetonitrile (
2f). Yield 69%; white crystals; m.p. 131.1-131.3 ºC; {lit. [
16], 125-126ºC}; IR (cm
-1): 2239 (CN);
1H-NMR (500 MHz) δ: 5.67 (s, 1H, CH), 7.38 (m, 1H, C4’H), 7.41 (m, 2H, C3’H, C5’H), 7.47 (m, 2H, C2’H, C6’H), 7.55 (m, 2H, C4H, C5H);
13C-NMR (125 MHz) δ: 43.1 (CH), 117.6 (CN), 127.5 (C2’, C6’), 127.7 (C5), 129.1 (C4’), 129.6 (C4), 129.7 (C3’, C5’), 132.8 (C1’), 157.0 (C3), 157.9 (C6); Anal. calcd. for C
12H
8N
3Cl: C, 62.75; H, 3.51, N, 18.30; found: C, 62.85; H, 3.43; N, 18.46.
α-(4-Fluorophenyl)-α-(6-chloropyridazin-3-yl)-acetonitrile (2g). Yield 75%; white crystals; m.p. 133.0-133.5 ºC; IR (cm-1): 2251 (CN); 1H-NMR (500 MHz) δ: 5.65 (s, 1H, CH), 7.08-7.11 (m, 2H, C2’H, C6’H), 7.44-7.47 (m, 2H, C3’H, C5’H), 7.55-7.66 (m, 2H, C4H, C5H); 13C-NMR (125 MHz) δ: 42.4 (CH), 116.8 (C3’, C5’), 117.5 (CN), 127.7 (C5), 129.4 (C2’, C6’), 129.5 (C1’), 129.7 (C4), 157.6 (C3), 157.7 (C6), 163.9 (C4’); Anal. calcd. for C12H7N3FCl: C, 58.20; H, 2.85; N, 16.97; found: C, 58,07; H, 2.86; N, 16,77.
α-Phenyl-α-(6-methoxypyridazin-3-yl)-acetonitrile (2h). Yield 53%; white crystals; m.p. 153.0-156.0 ºC; IR (cm-1): 2245 (CN); 1H-NMR (500 MHz) δ: 4.14 (s, 3H, OCH3 ), 5.59 (s, 1H, CH), 6.99 (d, 1H, C5H), 7.37 (dd, 3H, C3’H, C4’H, C5’H), 7.42 (d, 3J=9.0, 1H, C4H), 7.47 (d, 2H, 3J=8.0, C2’H, C6’H); 13C-NMR (125 MHz) δ: 43.1 (CH), 55.1 (OCH3), 118.1 (CN), 118.9 (C5), 127.5 (C2’, C6’), 128.2 (C4), 128.8 (C4’), 129.5 (C3’, C5’), 133.6 (C1’), 153.8 (C3), 164.8 (C6); Anal. calcd. for C13H11N3O: C, 69.32; H, 4.92, N, 18.65; found: C, 69.01; H, 5.31; N, 18.52.
Synthesis of α-phenyl-α-(pyridazin-3-yl)-acetonitrile (2f′) and α-(4-fluorophenyl)-α-(pyridazin-3-yl)-acetonitrile (2g′)
The appropriate nitrile 2f or 2g (0.06 mol) was placed in MeOH (300 mL), along with ammonium formate (18.9 g, 0.3 mol) and Pd/C (10%, 3.4g). The combined mixture was brought to boiling while stirring, the reaction was performed under nitrogen. For compound 2f′ the reaction time was 2 h, for compound 2g′ it was 1.5 h (TLC). After cooling the whole was filtered off from the catalyst and the filtrate was evaporated to dryness. The obtained precipitate was dissolved in CHCl3 – H2O mixture (1:1 v/v, 100 mL) and then extracted with CHCl3 (3 x 100 mL). The combined chloroform extracts were dried with anhydrous MgSO4. After filtering off the drying agent the filtrate was evaporated to dryness. The obtained precipitate 2 f′ was purified on the chromatographic column by flash technique, using CH2Cl2/MeOH eluents (97:3 v/v), and then it was crystallized from ethanol. Compound 2g′ was purified by maceration with the hexane : ethyl acetate mixture (1:1 v/v).
α-Phenyl-α-(pyridazin-3-yl)-acetonitrile (
2f′).Yield 57%; white crystals; m.p. 139.8-141.7 ºC; {lit. [
15,
16], 136-137 ºC; lit. [
17], 138-140 ºC}; IR (cm
-1): 2245 (CN);
1H-NMR (500 MHz) δ: 5.69 (s, 1H, CH), 7.37 (m, 1H, C4’H), 7.41 (m, 2H, C3’H, C5’H), 7.50 (m, 2H, C2’H, C6’H), 7.52 (m, 1H, C5H), 7.59 (m, 1H, C4H), 9.19 (m, 1H, C6H);
13C-NMR (125 MHz) δ: 44.2 (CH), 118.3 (CN), 125.7 (C4, C5), 127.9 (C2’, C6’), 129.3 (C4’), 129.9 (C3’, C5’), 133.7 (C1’), 151.4 (C6), 159.1 (C3); Anal. calcd. for C
12H
9N
3: C, 73.83; H, 4.65, N, 21.52; found: C, 73.62; H, 4.87; N, 21.46.
α-(4-Fluorophenyl)-α-(pyridazin-3-yl)-acetonitrile (2g′). Yield 58%; white crystals; m.p. 127.0-128.0 ºC; IR (cm-1): 2248 (CN); 1H-NMR (500 MHz) δ: 5.66 (s, 1H, CH), 7.07-7.11 (m, 2H, C2’H, C6’H), 7.46-7.61 (m, 4H, C4H, C5H, C3’H, C5’H), 9.18-9.20 (dd, 1H, C6H); 13C-NMR (125 MHz) δ: 43.2 (CH), 116.7 (C3’, C5’), 117.8 (CN), 127.7 (C5), 129.2 (C2’, C6’), 129.4 (C1’), 129.5 (C4), 151.2 (C6), 158.6 (C3), 163.9 (C4’); Anal. calcd. for C12H8N3F: C, 67.60; H, 3.78; N, 19.71; found: C, 66.41; H, 3.75; N, 19.45.
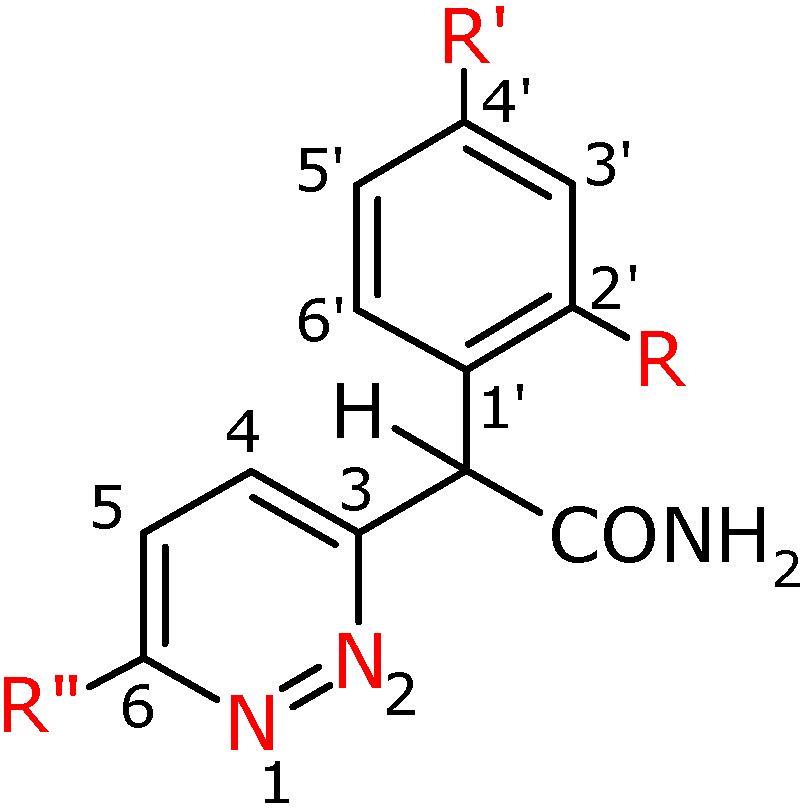
General procedure for the preparation of α-aryl- α-(pyridazin-3-yl)-acetamides 3a-f, 3h, 3f′, 3g′
To concentrated sulphuric acid (50 mL) was added the appropriate nitrile 2a, b, c, f, h, f′, g′ (0.03 mol). The mixture was stirred for 12 h at the temperature of 50 °C. Compounds 2e and 2d were hydrolyzed in the mixture of concentrated sulphuric acid (10 mL) and glacial acetic acid (30 mL) for 1 h at the temperature of 100 °C (2e) and for 10 h at the temperature of 50 oC (2d).
Then the post-rection mixture was cooled and made alkaline at a temperature of 5 °C with concentrated NH4OH to pH~8. The separated reaction product was extracted with chloroform (3 x 150 mL). The combined chloroform extracts were dried with anhydrous MgSO4. After filtering the drying agent the solvent was distilled off and the crude product was macerated with acetonitrile 3c, 3d; hexane 3e and 3f, and the ethyl acetate- hexane mixture (1:1 v/v). After maceration for compounds 3d and 3e flash column chromatography was applied, using the following mixtures for elution: for 3d CHCl3-MeOH mixture (99:1 v/v), whereas for 3e CH2Cl2:CHOH (98:2 v/v). Next the compounds were purified by crystallization from EtOH 3a, 3b, 3c, 3d, 3f, 3h, 3f′, 3g′.
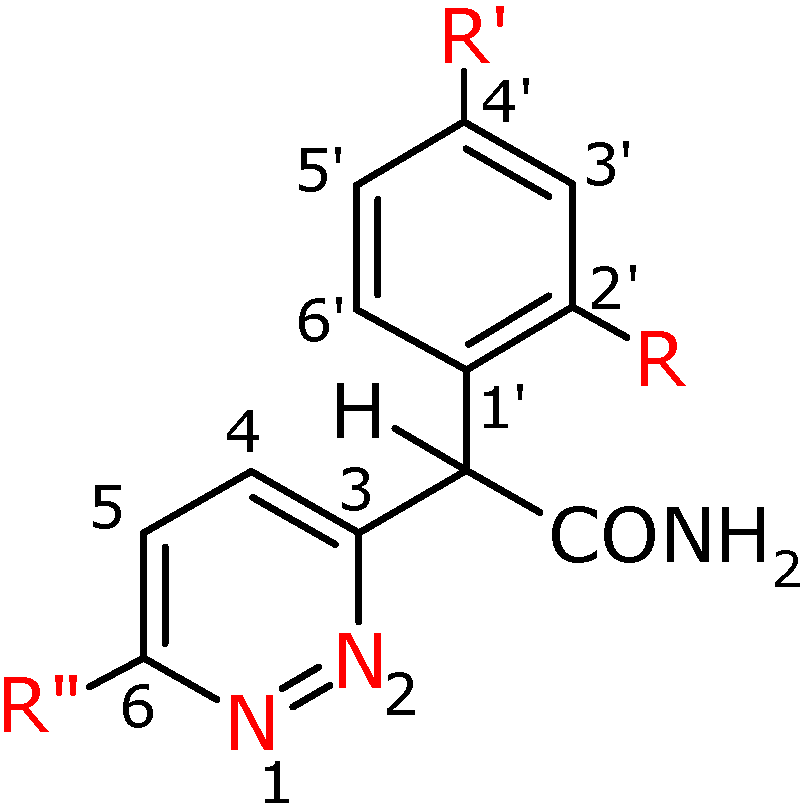
Figure 3.
Numbering system for compounds 3a-f, 3h, 3f′, 3g′.
Figure 3.
Numbering system for compounds 3a-f, 3h, 3f′, 3g′.
α-Phenyl-α-(6-phenylpyridazin-3-yl)-acetamide (3a). Yield 74%; white crystals; m.p. 177.7-179.8 ºC; IR (cm-1): 1655 (CO-NH2); 1H-NMR (500 MHz) δ: 5.36 (s, 1H, CH), 5.77 (bs, 1H, NH(2)), 7.17 (bs, 1H, NH (1)), 7.29 (tt, 3J=7.5, 4J=1.0, 1H, C4’H), 7.36 (td, 3J=7.5, 4J=1.5, 2H, C3’H, C5’H), 7.47 – 7.55 (m, 5H, C2”H-C6”H), 7.62 (d, 3J=8.5, 1H, C4H), 7.83 (d, 1H, C5H), 8.05 (dd, 3J=7.5, 4J=1.5, 2H, C2’H, C6’H); 13C-NMR (125 MHz) δ: 58.8 (CH), 124.7 (C5), 127.1 (C2’,C6’), 127.9 (C4), 128.2 (C4’), 128.5 (C3”, C5”), 129.0 (C3’, C5’), 129.1 (C2”, C6”), 130.2 (C4”), 135.9 ( C1’), 137.4 (C1”), 158.4 (C3), 159.8 (C6), 172.2 (CO); Anal. calcd. for C18H15N3O: C, 74.72; H, 5.23, N, 14.52; found: C, 74.74; H, 5.24; N, 14.48.
α-(4-Fluorophenyl)-α-(6-phenylpyridazin-3-yl)-acetamide (3b). Yield 63%; beige crystals; m.p. 199.5-200.7 ºC; IR (cm-1): 1686 (CO-NH2); 1H-NMR (500 MHz) δ: 5.31(s, 1H, CH), 6.82 (bs, 1H, NH (2)), 7.05 (m, 2H, C3’H, C5’H), 7.23 (bs, 1H, NH(1)), 7.46-7.56 (m, 5H, C2”H-C6”H), 7.60 (d, 3J=9.0 Hz, 1H, C4H), 7.85 (d, 1H, C5H), 8.05 (m, 2H, C2’H, C6’H); 13C-NMR (125 MHz) δ: 57.9 (CH), 116.0 (d*, 2J=22.0, C3’, C5’), 124.9 (C4), 127.1 (C2”, C6”), 128.1 (C5), 129.1 (C3”, C5”), 130.2 (d*, 3J=8.2, C2’, C6’), 130.3 (C4”), 133.2 (d*, 4J=3.3 ,C1’), 135.8 (C1”), 158.5 (C3), 159.6 (C6), 162.4 (d*, 1J=249.5, C4’), 171.9 (CO); Anal. calcd. for C18H14N3OF: C, 70.35; H, 4.59, N, 13.67; found: C, 70.30; H, 4.58; N, 13.77.
α-(4-Tolylo)-α-(6-phenylpyridazin-3-yl)-acetamide (3c). Yield 46 %; white crystals; m.p. 208.5-209.4 ºC; IR (cm-1): 1683 (CO-NH2); 1H-NMR (400 MHz) δ: 2.33 (s, 3H, CH3), 5.33 (s, 1H, CH), 5.64 (bs, 1H, NH(2)), 7.12 (bs, 1H, NH(1)), 7.18 (d, 2H, C3’H, C5’H), 7.40 (d, 3J=8.0, 2H, C2’H, C6’H), 7.53 (m, 3H, C3”H, C4”H, C5”H), 7.63 (d, 3J=9.2, 1H, C4H), 7.85 (d, 1H, C5H), 8.06 (dd, 3J=7.6, 4J=2.4, 2H, C2”H, C6”H); 13C-NMR (100 MHz) δ: 21.3 (CH3), 58.4 (CH), 125.3 (C5), 127.3 (C2’,C6’), 128.6 (C3”, C5”), 128.8 (C4), 129.4 (C3’, C5’), 130.1 (C2”, C6”), 130.6 (C4”), 134.5 (C1’), 136,0 (C1”), 138.2 (C4’), 158.6 (C3), 160.0 (C6), 172.4 (CO); Anal. calcd. for C19H17N3O: C, 75.22; H, 5.65, N, 13.85; found: C, 74.32; H, 5.51; N, 14.16.
α-(4-Methoxyphenyl)-α-(6-phenylpyridazin-3-yl)-acetamide (3d). Yield 70%; white crystals; m.p. 168.2-170.7 ºC; IR (cm-1): 1662 (CO-NH2); 1H-NMR (400 MHz) δ: 3.67 (s, 3H, OCH3), 5.32 (s, 1H, CH), 6.17 (s, 2H, NH2), 6.77 (m, 2H, C3’H, C5’H), 7.19 (m, 2H, C2’H, C6’H), 7.40 (m, 3H, C3”H, C4”H, C5”H), 7.57 (m, 1H, C4H), 7.71 (m, 1H, C5H), 7.93 (m, 2H, C2”H, C6”H); 13C-NMR (100 MHz) δ: 55.4 (OCH3), 57.7 (CH), 114.6 (C3’,C5’), 124.9 (C4), 127.2 (C2”, C6”), 128.2 (C5), 128.5 (C1”), 129.2 (C2’, C6’), 129.8 (C3”, C5”), 130.6 (C4”), 136.1 (C1’), 158.4 (C6), 159.3 (C3), 160.4 (C4’), 173.2 (CO); Anal. calcd. for C19H17N3O2: C, 71.46; H, 5.37, N, 13.16; found: C, 71.33; H, 5.57; N, 13.01.
α-(2-Methoxyphenyl)-α-(6-phenylpyridazin-3-yl)-acetamide (3e). Yield 16%; white crystals; m.p. 183.0-184.0 ºC; IR (cm-1): 1686 (CO-NH2); 1H-NMR (400 MHz) δ: 3.82 (s, 3H, OCH3), 5.78 (bs, 1H, NH(1)), 5.88 (s, 1H, CH), 6.91 (d, 3J=8.4, 1H, C3’H), 6.97 (bs, 1H, NH(2)), 6.99 (t, 3J=7.6, 1H, C5’H), 7.31 (t, 3J=7.6, 1H, C4’H), 7.52 (m, 4H, C6’H, C3”H, C4”H, C5”H), 7.72 (d, 1H, C4H), 7.89 (d, 3J=8.8, 1H, C5H), 8.06 (dd, 3J=6.4, 4J=2.4, 2H, C2”H, C6”H); 13C-NMR (100 MHz) δ: 51.9 (CH), 55.8 (OCH3), 111.3 (C3’), 121.4 (C5’), 125.4 (C5), 125.7 (C1’), 127.3 (C2”, C6”), 129.3 (C4, C3”, C5”), 129.6 (C4’), 129.7 (C6’), 130.6 (C4”), 135.7 (C1”), 157.1 (C3), 158.3 (C6), 160.0 (C2’), 172.4 (CO); Anal. calcd. for C19H17N3O2: C, 71.46; H, 5.37, N, 13.16; found: C, 71.46; H, 5.45; N, 13.08.
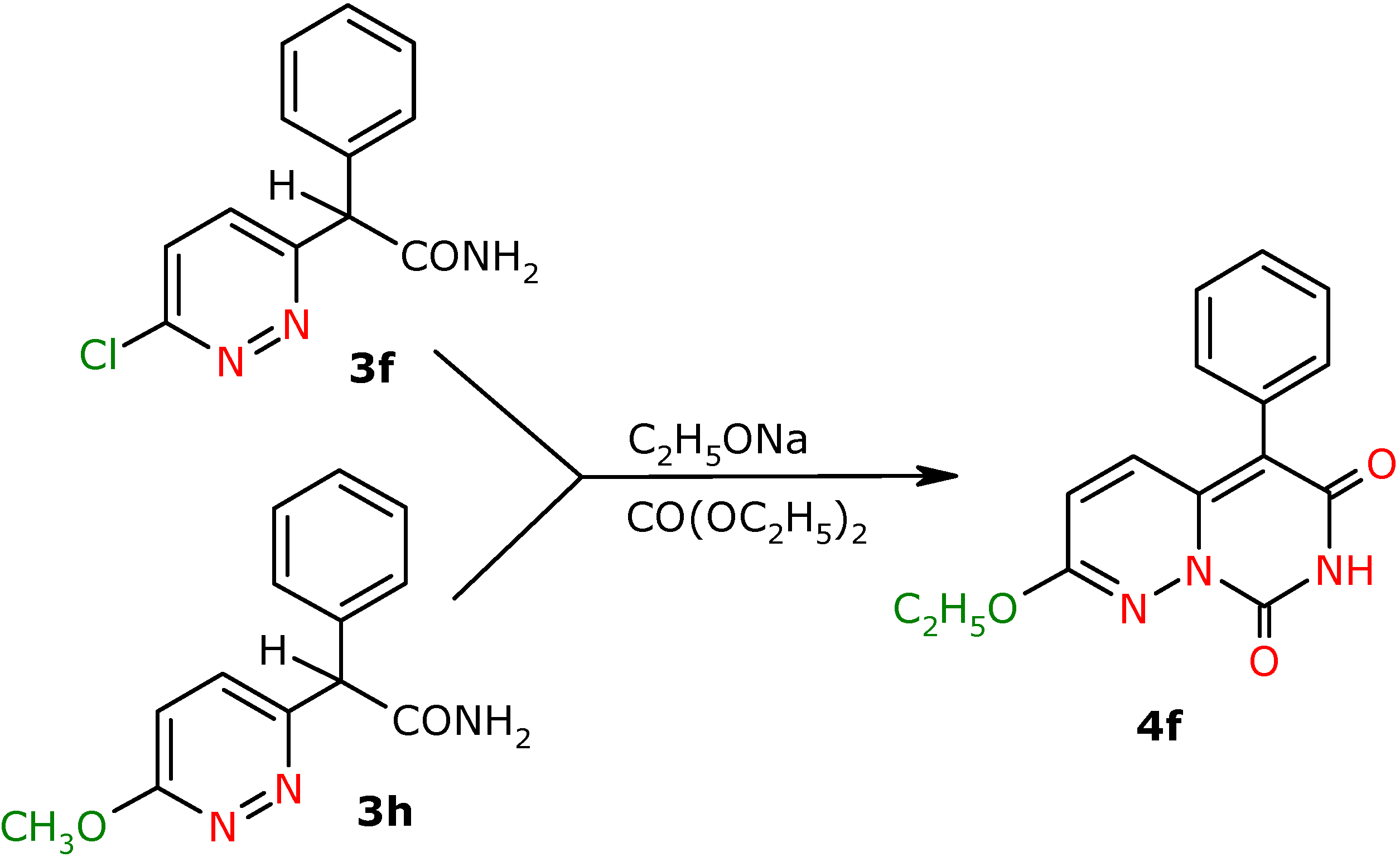
α-Phenyl-α-(6-chloropyridazin-3-yl)-acetamide (3f). Yield 29 %; white crystals; m.p. 140.0-141.0 ºC; IR (cm-1): 1685 (CO-NH2); 1H-NMR (500 MHz) δ: 5.45 (s, 1H, CH), 6.21 (bs, 1H, NH(2)), 6.83 (bs, 1H, NH(1)), 7.23-7.45 (m, 5H, 3J=8.8, 5H, C2’H, C3’H, C4’H, C5’H, C6’H), 7.44 (d, 1H, 3J=8.8, C5H), 7.64 (d, 1H, C4H); 13C-NMR (125 MHz) δ: 57.8 (CH), 128.1 (C5), 128.3 (C2’, C6’), 128.7 (C4’), 129.2 (C3’, C5’), 130.2 (C4), 136.8 (C1’), 156.1 (C3), 160.9 (C6), 175.0 (CO); Anal. calcd. for C12H10N3OCl: C, 58.19; H, 4.07, N, 16.97; found: C, 58.01; H, 4.02; N, 16.88.
α-Phenyl-α-(pyridazin-3-yl)-acetamide (3f′). Yield 47%; white crystals; m.p. 177.0-178.0 ºC; IR (cm-1):1679 (CO-NH2); 1H-NMR (500 MHz) δ: 5.61 (bs, 1H, NH(2)), 5.29 (s, 1H, CH), 6.96 (bs, 1H, NH(1)), 7.28-7.38 (m, 3J0=7.0, 4Jm=1.5, 3H, C3’H, C4’H, C5’H), 7.43-7.48 (m, 3H, C5H, C2’H, C6’H), 7.56 (dd, 3J0=8.5, 4Jm=1.5, 1H, C4H), 9.11 (dd, 3J0=5.0, 4Jm=2.0, 1H, C6H); 13C-NMR (125 MHz) δ: 59.1 (CH), 127.1 (C5), 127.8 (C4), 128.0 (C4’), 128.4 (C2’, C6’), 129.2 (C3’, C5’), 137.2 (C1’), 150.4 (C6), 161.4 (C3), 171.9 (CO); Anal. calcd. for C12H11N3O: C, 67.59; H, 5.20, N, 19.71; found: C, 67.52; H, 4.91; N, 19.78.
α-(4-Fluorophenyl)-α-(pyridazin-3-yl)-acetamide (3g′). Yield 66%; white crystals; m.p. 157.5-158.5 ºC; IR (cm-1): 1690 (CO-NH2); 1H-NMR (500 MHz) δ: 5,37 (s, 1H, CH), 5,94 (bs, 1H, NH(2)), 7,03 (t, 3J=8,4, 2H, C3’H, C5’H), 7,24 (bs, 1H, NH(1)), 7,40-7,52 (m, 3H, C5H, C2’H, C6’H), 7,62 (d, 3J=8,4, 1H, C4H), 9,12 (d, 1H, C6H); 13C-NMR (125 MHz) δ: 57.9 (CH), 116.0 (d*, 2J=21,6, C3’, C5’), 127,4 (C4), 127.8 (C5), 130,2 (d*, 3J=8,2, C2’, C6’), 132,9 (C1’), 150.5 (C6), 161.4 (C3), 162.4 (d*, 1J=246,4, C4’), 171.9 (CO); Anal. calcd. for C12H10N3FO: C, 62.33; H, 4.36; N, 18.17; found: C, 61,99; H, 4,31; N, 17,94.
α-Phenyl-α-(6-methoxypyridazin-3-yl)-acetamide (3h). Yield 69%; white crystals; m.p. 165-168 ºC; IR (cm-1): 1688 (CO-NH2); 1H-NMR (500 MHz) δ: 4.10 (s, 3H, OCH3), 5.28 (s, 1H, CH), 6.10 (s, 1H, NH(2)), 6.94 (d, 1H, C5H), 7.15 (bs, 1H, NH(1)), 7.27 (m, 1H, C4’H), 7.32 (m, 2H, C3’H, C5’H), 7.42 (m, 2H, C2’H, C6’H), 7.46 (d, 3J=9.0, 1H, C4H); 13C-NMR (125 MHz) δ: 54.8 (OCH3, J=1.0), 58.2 (CH), 118.2 (C5), 127.8 (C4), 128.4 (C2’,C6’), 129.1 (C3’, C5’), 130.6 (C4’), 137.6 (C1’), 156.7 (C3), 164.5 (C6), 172.6 (CO); Anal. calcd. for C13H13N3O2: C, 64.19; H, 5.39, N, 17.27; found: C, 64.59; H, 5.37; N, 17.36.
General procedure for the preparation of 7H,8H-pyrimido[1,6-b]pyridazin-6,8-dione derivatives 4a-f, 4f′, 4g′
To absolute ethanol (30 mL) was added sodium (0.18 mol) and the mixture was stirred until they had reacted. Next diethyl carbonate (0.17 mol) was added and the respective amide 3a-f, 3h, 3f′, 3g′ (0.1 mol) was added portionwise. The reaction was performed in boiling for 12 h. After cooling the post-reaction mixture was poured into distilled water (100 mL) and acidified with acetic acid to pH~3. The yellow precipitate separated and was filtered off. Compounds 4a, 4b, 4e were purified on a chromatographic column by the flash technique, using the CH2Cl2-MeOH (97:3 v/v) and CH2Cl2-MeOH (99:1 v/v) mixtures as eluents. Compound 4c was macerated with acetonitrile, 4d was crystallized from MeOH and 4f′ was crystallized from EtOH, and next was flash chromatographed using the CH2Cl2-MeOH (98:2 v/v) mixture as eluent. Compound 4g′ was crystallized from EtOH. Compound 4f was purified by flash chromatography with CHCl3-MeOH (97:3 v/v) and next was crystallized from MeOH.
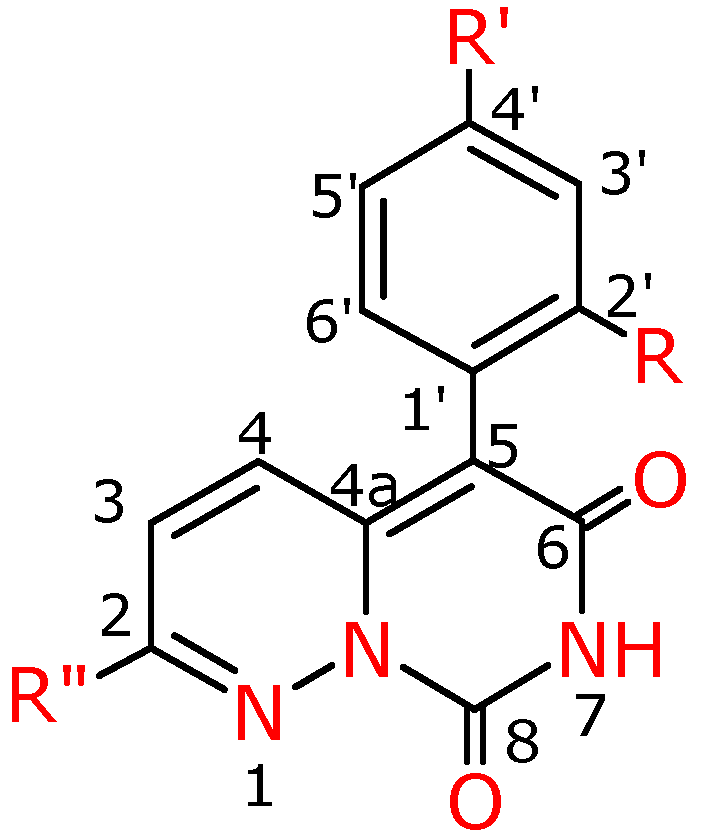
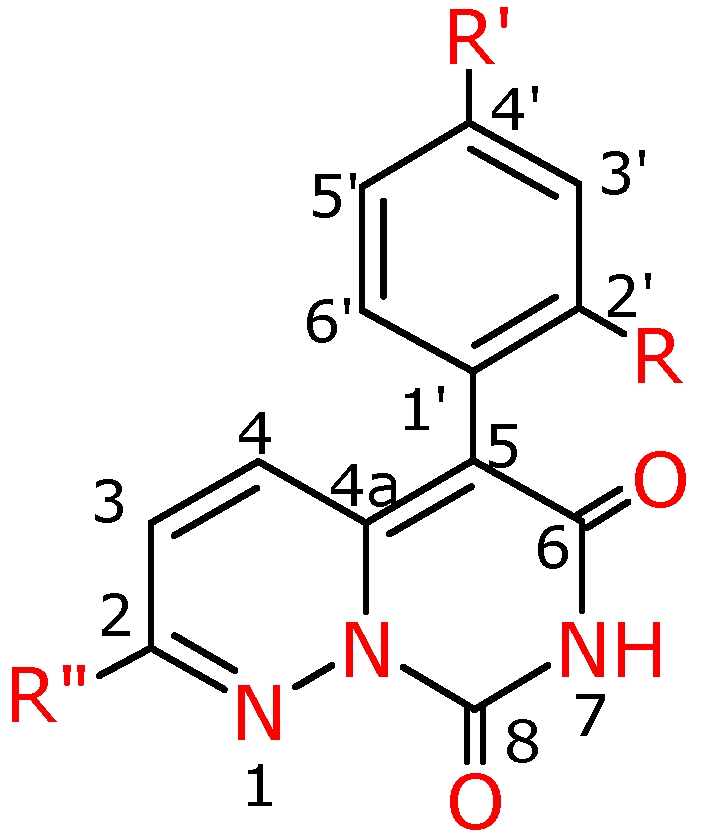
Figure 4.
Numbering system for compounds 4a-f, 4f′, 4g′.
Figure 4.
Numbering system for compounds 4a-f, 4f′, 4g′.
2,5-Diphenyl-7H,8H-pyrimido[1,6-b]pyridazin-6,8-dione (4a). Yield 94%; yellow crystals; m.p. 302-303 ºC; IR (cm-1): 1660 (CO), 1725 (CO); 1H-NMR (500 MHz) δ: 7.17(d, 3J=10.0 , 1H, C3H), 7.34 (dd, 3J=8.0, 4J=1.5 , 2H, C2’H, C6’H), 7.40 (d, 1H, C4H), 7.42 (tt, 3J=7.0, 4J=1.0 , 1H, C4”H), 7.46-7.54 (m, 5H, C3’H, C4’H, C5’H, C3”H, C5”H,), 7.92 (dd, 3J=6.4, 4J=1.6, 2H, C2”H, C6”H) 8.51 (bs, 1H, NH); 13C-NMR (125 MHz) δ: 107.5 (C5), 122.8 (C4), 126.7 (C3”, C5”), 128.7 (C4’), 129.0 (C2”, C6”), 129.2 (C3’, C5’), 130.6 (C1’), 131.01 (C2’, C6’), 131.03 (C4”), 131.1 (C3), 133.5 (C1”), 141.3 (C2), 147.5 (C8), 150.0 (C4a), 160.2 (C6); Anal. calcd. for C19H13N3O2: C, 72.37; H, 4.15, N, 13.33; found: C, 71.61; H, 4.15;N, 13.20.
2-Phenyl-5-(4-fluorophenyl)-7H,8H-pyrimido[1,6-b]pyridazin-6,8-dione (4b). Yield 91%; yellow crystals; m.p. 292.8-293.2 ºC; IR (cm-1):1654 (CO), 1709 (CO); 1H-NMR (400 MHz) δ: 7.23-7.30 (m, 3H, C3H, C3’H, C5’H), 7.30-7.40 (m, 2H, C2’H, C6’H), 7.50-7.64 (m, 4H, C4H, C3”H, C4”H, C5”H), 7.92-8.20 (m, 2H, C2”H, C6”H), 12.03 (s, 1H, NH); 13C-NMR (100 MHz) δ: 107.1 (C5), 117.4 (d*,2J=21.4, C3’, C5’), 125.0 (C4), 128.4 (C3”, C5”), 130.1 (d*, 4J=3.0, C1’), 131.1 (C2”, C6”), 132.7 (C4”), 132.8 (C3), 135.3 (d*, 3J=8.0, C2’, C6’), 135.6 (C1”), 143.0 (C2), 149.7 (C8), 150.6 (C4a), 162.8 (C6), 163.7 (d*, 1J=244.8, C4’); Anal. calcd. for C19H12N3O2F: C, 68.46; H, 3.63, N, 12.61; found: C, 68.84; H, 4.01; N, 13.00.
2-Phenyl-5-(4-tolyl)-7H,8H-pyrimido[1,6-b]pyridazin-6,8-dione (4c). Yield 92%; yellow crystals; m.p. 321.0-321.4 ºC; IR (cm-1): 1659 (CO), 1728 (CO); 1H-NMR (400 MHz) δ: 2.35 (s, 3H, CH3), 7.19 (d, 3J=7.6, 2H, C3’H, C5’H), 7.26(m, 3H, C3H, C2’H, C6’H), 7.55 (m, 4H, C4H, C3”H, C4”H, C5”H), 7.97 (m, 2H, C2”H, C6”H), 11.97 (s, 1H, NH); 13C-NMR (100 MHz) δ: 22.9 (CH 3), 108.1 (C5), 124.8 (C4), 128.4 (C3”,C5”), 130.8 (C1’), 131.1 (C2’, C6’, C2”, C6”), 132.8 (C4”), 132.9 (C3), 133.0 (C3’, C5’), 135.6 (C1”), 139.1 (C4’), 142.8 (C2), 149.7 (C8), 150.5 (C4a), 162.8 (C6); Anal. calcd. for C20H15N3O2: C, 72.94; H, 4.59; N, 12.76; found: C, 72.56; H, 4.45; N, 13.06.
2-Phenyl-5-(4-methoxyphenyl)-7H,8H-pyrimido[1,6-b]pyridazin-6,8-dione (4d). Yield 98%; yellow crystals; m.p. 306.6-308.4 ºC; IR (cm-1): 1654 (CO), 1717 (CO); 1H-NMR (400 MHz) δ: 3.86 (s, 3H, OCH3), 7.02 (d, 3J=10.0 , 2H, C3’H, C5’H), 7.17 (d, 3J=10.0 , 1H, C3H), 7.27 (d, 2H, C2’H, C6’H), 7.42 (d, 1H, C4H), 7.52 (m, 3H, C3”H, C4”H, C5”H), 7.92 (m, 2H, C2”H, C6”H), 8.60 (bs, 1H, NH); 13C-NMR (100 MHz) δ: 55.6 (OCH 3), 107.4 (C5), 114.7 (C3’, C5’), 122.8 (C4), 125.0 (C1’), 126.9 (C3”, C5”), 129.4 (C2”, C6”), 131.3 (C4”), 131.4 (C3), 132.4 (C2’, C6’), 133.7 (C1”), 141.3 (C2), 148.4 (C8), 150.2 (C4a), 155.5 (C4’), 159.9 (C6); Anal. calcd. for C20H15N3O3: C, 69.56; H, 4.38, N, 12.17; found: C, 69.46; H, 4.68; N, 11.78.
2-Phenyl-5-(2-methoxyphenyl)-7H,8H- pyrimido[1,6-b]pyridazin-6,8-dione (4e).Yield 60%; yellow crystals; m.p. 275.0-277.0 ºC; IR (cm-1): 1643 (CO), 1747 (CO); 1H-NMR (400 MHz) δ: 3.79 (s, 3H, OCH3), 7.02 (d, 3J=8.4 , 1H, C3’H), 7.07 (t, 3J=7.6, 1H, C5’H), 7.11 (d, 3J=10.0 , 1H, C3H), 7.16 (d, 1H, C4H), 7.25 (d, 1H, C6’H), 7.42 (td, 3J=8.0 , 1H, C4’H), 7.50 (m, 3H, C3”H, C4”H, C5”H), 7.92 (m, 2H, C2”H, C6”H), 8.52 (bs, 1H, NH); 13C-NMR (100 MHz) δ: 55.2 (OCH3), 103.3 (C5), 111.1 (C3’), 118.7 (C1’), 120.6 (C5’), 121.9 (C4), 126.3 (C2”, C6”), 128.7 (C3”, C5”), 130.0 (C4’), 130.5 (C4”), 131.2 (C3), 132.4 ( C6’), 133.2 (C1”), 141.1 (C2), 147.3 (C8), 149.3 (C4a), 157.3 (C2’), 159.7 (C6); Anal. calcd. for C20H15N3O3: C, 69.56; H, 4.38, N, 12.17; found: C, 69.36; H, 4.57; N, 11.26.
2-Ethoxy-5-phenyl-7H,8H-pyrimido[1,6-b]pyridazin-6,8-dione (4f). Yield 31% (from 3f), 72%(from 3h); yellow crystals; m.p. 272.0-274.0 ºC; IR (cm-1): 1636 (CO), 1738 (CO); 1H-NMR (500 MHz) δ: 1.42 (t, 3H, 3J=7.0 , C2Het), 4.40 (k, 2H, C1Het), 6.48 (d, 3J=9.5 , 1H, C3H), 7.25 (d, 1H, C4H), 7.28-7.33 (m, 2H, C3’H ,C5’H), 7.37-7.42 (m, 1H, C4’H), 7.42-7.48 (m, 2H, C2’H, C6’H), 8.82 (bs, NH); 13C-NMR (125 MHz) δ: 14.0 (C2et), 64.1 (C1et), 108.5 (C5), 121.0 (C3), 128.6 (C4’), 128.9 (C2’,C6’), 130.9 (C1’), 131.1 (C3’, C5’), 132.6 (C4), 141.0 (C4a), 147.3 ( C8), 156.5 (C6), 160.4 (C2); Anal. calcd. for C15H13N3O3: C, 63.60; H, 4.62, N, 14.83; found: C, 63.26; H, 4.60; N, 14.74.
5-Phenyl-7H,8H-pyrimido[1,6-b]pyridazin-6,8-dione (4f′). Yield 27 %; yellow crystals; m.p. 265.5-266.3 ºC; IR (cm-1): 1651 (CO), 1751 (CO); 1H-NMR (500 MHz) δ: 6.85 (dd, 3J1=12.0 , 3J2=4.5 , 1H, C3H), 7.27 (dd, 3J=12.0 , 4J=2.0 , 1H, C4H), 7.32 (dd, 3J=10.5 , 4J=2.0 , 2H, C2’H, C6’H), 7.40 (tt, 3J=8.0 , 4J=2.5 , 1H, C4’H), 7.47 (t, 3J=8.0 , 2H, C3’H, C5’H), 8.06 (m, 1H, C2H), 9.5 (bs, 1H, NH); 13C-NMR (125 MHz) δ: 105.0 (C5), 125.2 (C4), 129.6 (C4’), 130.0 (C3’, C5’), 131.7 (C3), 132.3 (C1’), 132.5 (C2’,C6’), 144.5 (C2), 147.0 (C8), 150.3 (C4a), 163.5 (C6); Anal. calcd. for C13H9N3O2: C, 65.27; H, 3.79, N, 17.56; found: C, 65.58; H, 3.82 N, 17.14.
5-(4-Fluorophenyl)-7H,8H-pyrimido[1,6-b]pyridazin-6,8-dione (4g’). Yield 69%; yellow crystals; m.p. 278.0-278.5 ºC; IR (cm-1): 1625 (CO), 1756 (CO); 1H-NMR (500 MHz) δ: 6.67-6.69 (dd, 1H, C4H), 7.13-7.18 (m, 2H, C2’H, C6’H), 7.25-7.30 (m, 3H, C3H, C3’H, C5’H), 7.98 (d, 1H, C2H), 8.65 (s, 1H, NH); 13C-NMR (125 MHz) δ: 106.8 (C5), 116.4 (C3’, C5’), 123.3 (C1’), 126.2 (C4), 130.5 (C2’, C6’), 132.8 (C3), 142.3 (C8), 142.4 (C2), 147.4 (C4a), 161.9 (C6), 163.8 (C4’); Anal. calcd. for C13H8N3O2F: C, 60.70; H, 3.13; N, 16.34; found: C, 60.83; H, 3.37; N, 16.56.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}