Two New Triterpenoids from Photinia serrulata

Abstract
:Introduction
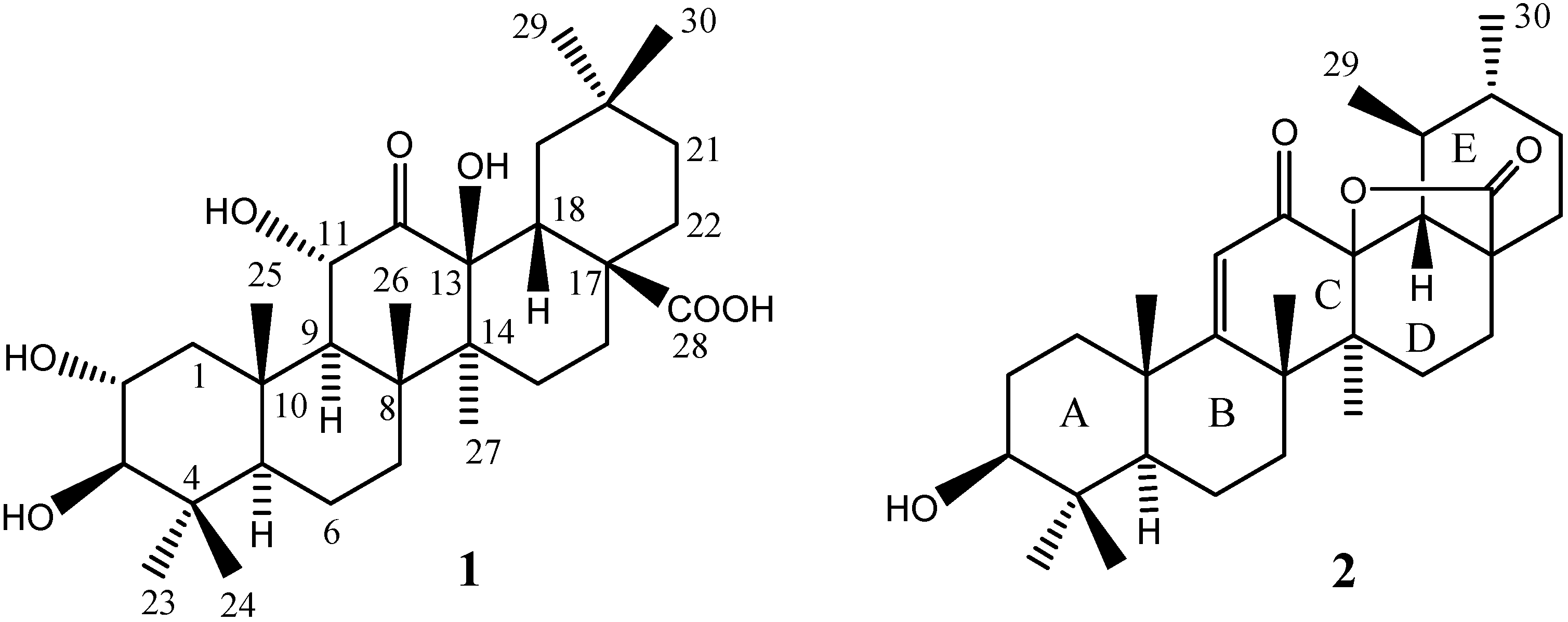
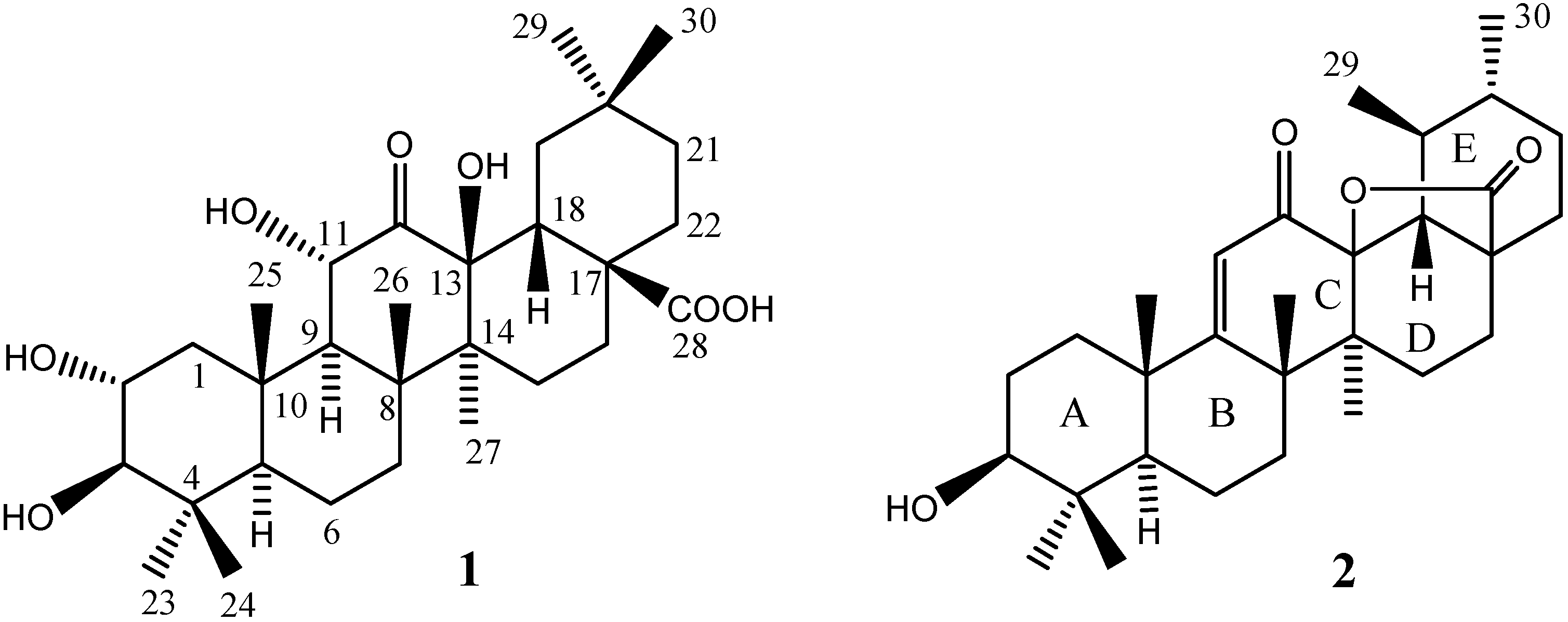
Results and Discussion

{kind=link}
| Position | 1 | 2 | ||
|---|---|---|---|---|
| δH (J=Hz) | δC | δH (J=Hz) | δC | |
| 1 | 2.18 m; 1.30 m | 47.0 t | 2.04 m; 1.42 m | 37.0 t |
| 2 | 3.71 m | 68.6 d | 1.72 m | 28.4 t |
| 3 | 3.05 d (9.5) | 83.2 d | 3.18 m | 77.6 d |
| 4 | 39.1 s | 39.9 s | ||
| 5 | 0.91 m | 55.0 d | 1.04 m | 50.8 d |
| 6 | 1.67 m; 1.55 m | 17.4 t | 1.74 m | 18.0 t |
| 7 | 1.31 m | 31.6 t | 1.76 m; 1.52 m | 34.5 t |
| 8 | 41.5 s | 46.6 s | ||
| 9 | 2.16 d (7.5) | 57.7 d | 185.1s | |
| 10 | 39.8 s | 41.3 s | ||
| 11 | 4.31 d (7.5) | 56.1 d | 6.00 s | 122.0 d |
| 12 | 200.2 s | 193.3 s | ||
| 13 | 90.0 s | 88.4 s | ||
| 14 | 41.7 s | 42.9 s | ||
| 15 | 1.66 m | 26.3 t | 1.80 m; 1.43 m | 26.5 t |
| 16 | 2.18 m; 1.38 m | 21.3 t | 2.18 m; 1.38 m | 22.7 t |
| 17 | 44.4 s | 45.6 s | ||
| 18 | 2.71 dd (13.5, 3.0) | 48.0 d | 2.42 d (12.0) | 55.5 d |
| 19 | 1.82 m; 1.42 m | 37.0 t | 1.80 m | 37.7 d |
| 20 | 31.6 s | 1.01 m | 40.5 d | |
| 21 | 1.33 m | 34.2 t | 1.58 m | 31.2 t |
| 22 | 1.75 m; 1.31 m | 26.8 t | 1.54 m | 32.1 t |
| 23 | 1.05 s | 28.3 q | 1.04 s | 28.5 q |
| 24 | 0.84 s | 16.3 q | 0.84 s | 16.1 q |
| 25 | 1.07 s | 18.0 q | 1.28 s | 24.9 q |
| 26 | 0.98 s | 19.7 q | 1.37 s | 30.8 q |
| 27 | 1.38 s | 19.7 q | 1.21 s | 20.8 q |
| 28 | 177.6 s | 178.4 s | ||
| 29 | 0.90 s | 33.2 q | 0.76 d (6.4) | 18.7 q |
| 30 | 0.99 s | 23.3 q | 0.92 d (6.4) | 19.4 q |
Experimental
General
Plant Material
Extraction and Isolation
Cytotoxic Assay
Acknowledgments
References
- Beijing Institute of Botany, Chinese Academy of Sciences. Flora Reipublicae Popularis Sinicae; Science Press: Beijing, 1974; Vol. 36, p. 216. [Google Scholar]
- Hou, J.; Sun, T.; Hu, J.; Chen, S. Y.; Cai, X. Q.; Zou, G. L. Chemical composition, cytotoxic and antioxidant activity of the leaf essential oil of Photinia serrulata. Food Chem. 2007, 103, 355–358. [Google Scholar] [CrossRef]
- López-Pérez, J. L.; Therón, R.; Del Olmo, E.; Diaz, D. NAPROC-13: a database for the dereplication of natural product mixtures in bioassay-guided protocols. Bioinformatics 2007, 23, 3256–3257. [Google Scholar] [CrossRef]
- Numata, A.; Yang, P. M.; Takahashi, C.; Fujiki, R.; Nabae, M.; Fujita, E. Cytotoxic triterpenes from a chinese medicine, goreishi. Chem. Pharm. Bull. 1989, 37, 648–651. [Google Scholar] [CrossRef]
- Heart, H. M.; Athukoralage, P. S.; Jamie, J. F. Two new oleanane triterpenoids from Gordonia ceylanica and their conversions to taraxarane triterpenoids. Phytochemistry 2000, 54, 823–827. [Google Scholar]
- Fang, S. Y.; He, Z. S.; Fan, G. J. Triterpenoids from Adina rubella. J. Nat. Prod. 1996, 59, 304–307. [Google Scholar] [CrossRef]
- Dreiding, J.; Jeger, O.; Ruzicka, L. Zur Kenntnis der Triterpene. Ueberfuehrung der Ursolsaeure in 2 isomere acetoxy-lactone C32H46O5. Helv. Chim. Acta 1950, 33, 1325–1334. [Google Scholar] [CrossRef]
- Réthy, B.; Kovács, A.; Zupkó, I.; Forgo, P.; Vasas, A.; Falkay, G.; Hohmann, J. Cytotoxic phenanthrenes from the rhizomes of Tamus communis. Planta Med. 2006, 72, 767–770. [Google Scholar]
- Sample Availability: Samples of compounds 1 and 2 are available from the authors.
© 2007 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Song, Y.; Wang, Y.; Lu, Q.; Gao, J.; Bi, M.; Cheng, Y. Two New Triterpenoids from Photinia serrulata. Molecules 2007, 12, 2599-2604. https://doi.org/10.3390/12122599
Song Y, Wang Y, Lu Q, Gao J, Bi M, Cheng Y. Two New Triterpenoids from Photinia serrulata. Molecules. 2007; 12(12):2599-2604. https://doi.org/10.3390/12122599
Chicago/Turabian StyleSong, Yaling, Yuehu Wang, Qing Lu, Jinming Gao, Minggang Bi, and Yongxian Cheng. 2007. "Two New Triterpenoids from Photinia serrulata" Molecules 12, no. 12: 2599-2604. https://doi.org/10.3390/12122599
APA StyleSong, Y., Wang, Y., Lu, Q., Gao, J., Bi, M., & Cheng, Y. (2007). Two New Triterpenoids from Photinia serrulata. Molecules, 12(12), 2599-2604. https://doi.org/10.3390/12122599