Application of the Solid-Phase Julia–Lythgoe Olefination in Vitamin D Side-Chain Construction

{kind=link}
{kind=link}
{kind=link}
Abstract
:Introduction
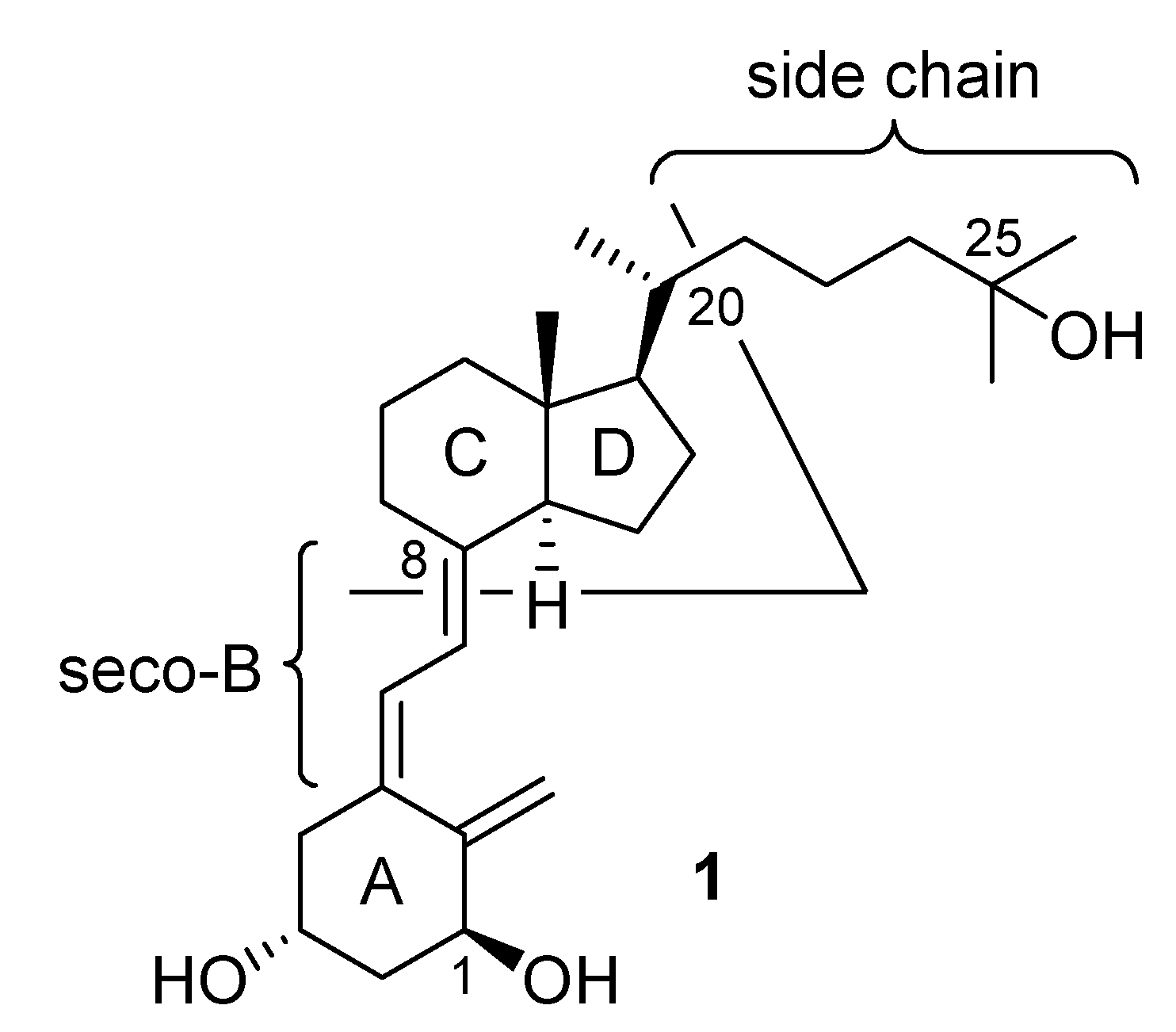
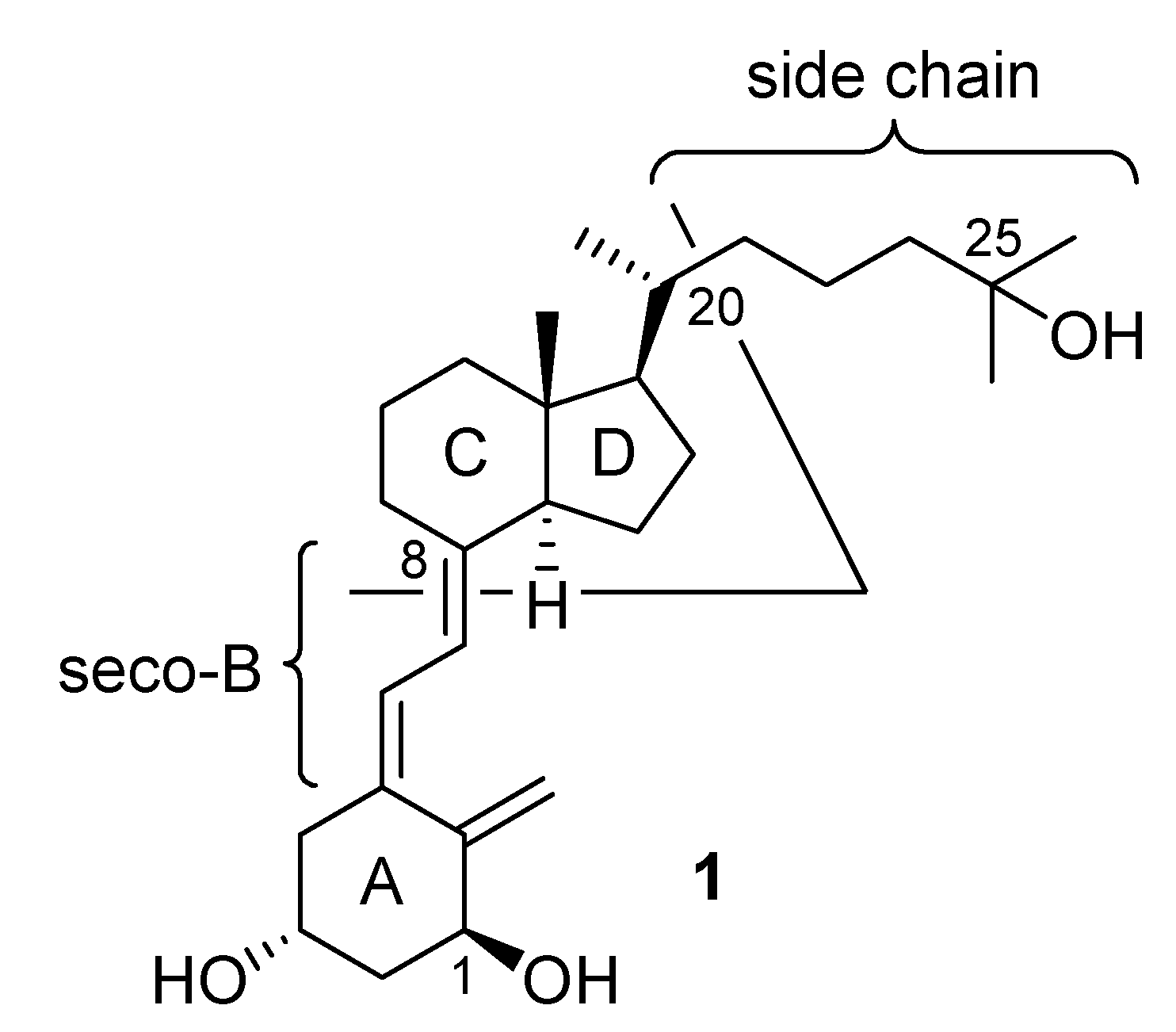
Results and Discussion
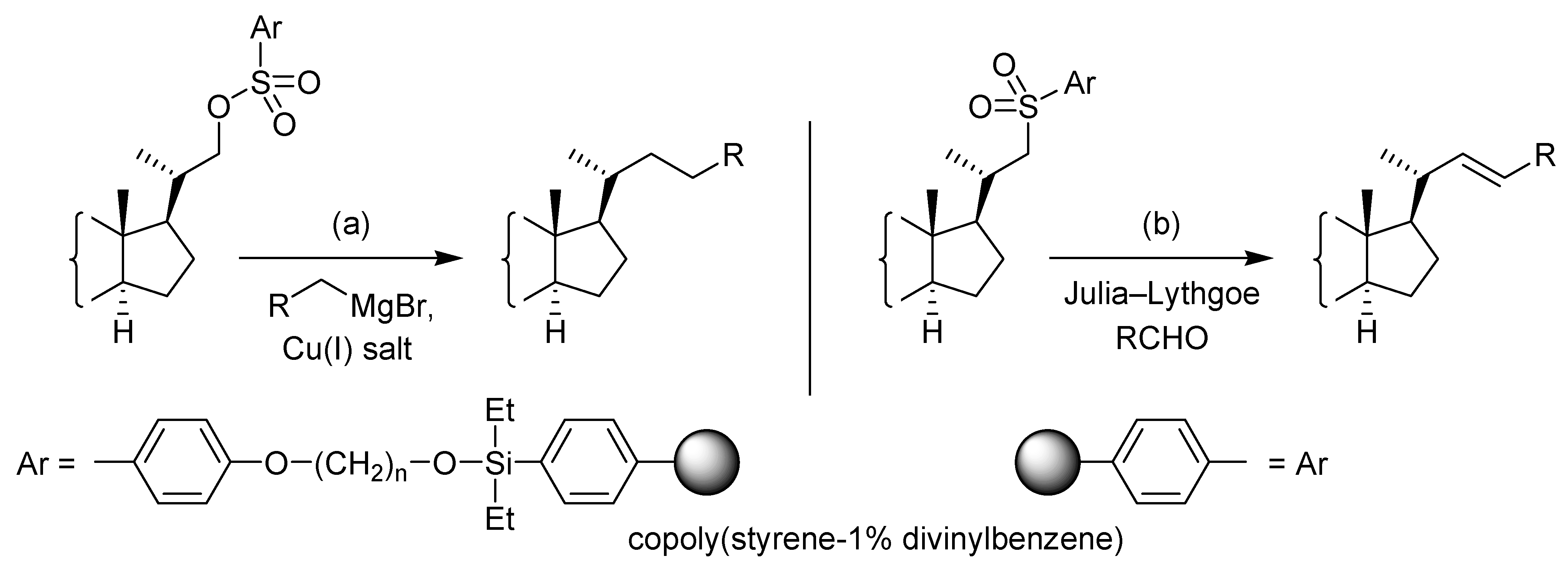
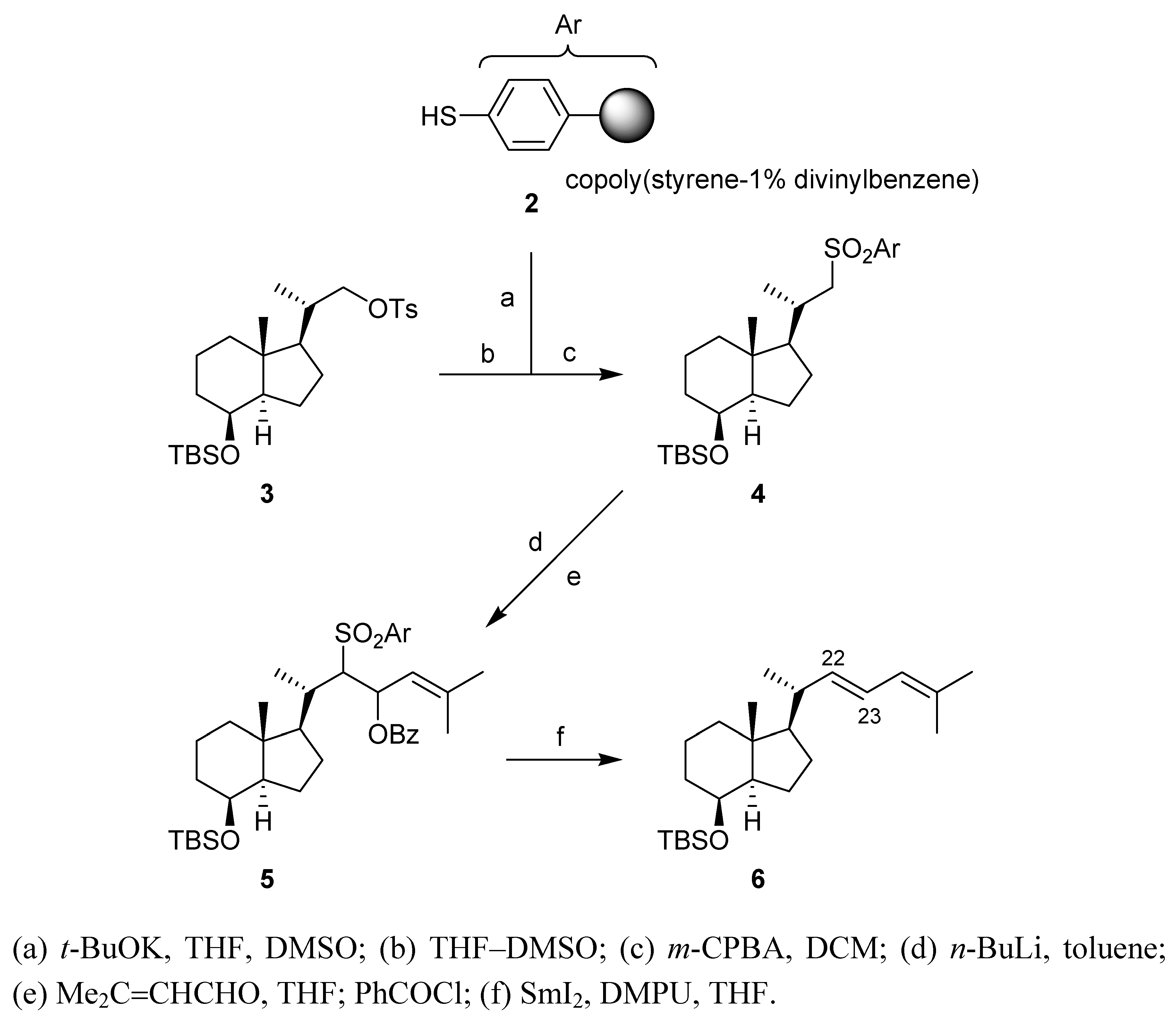
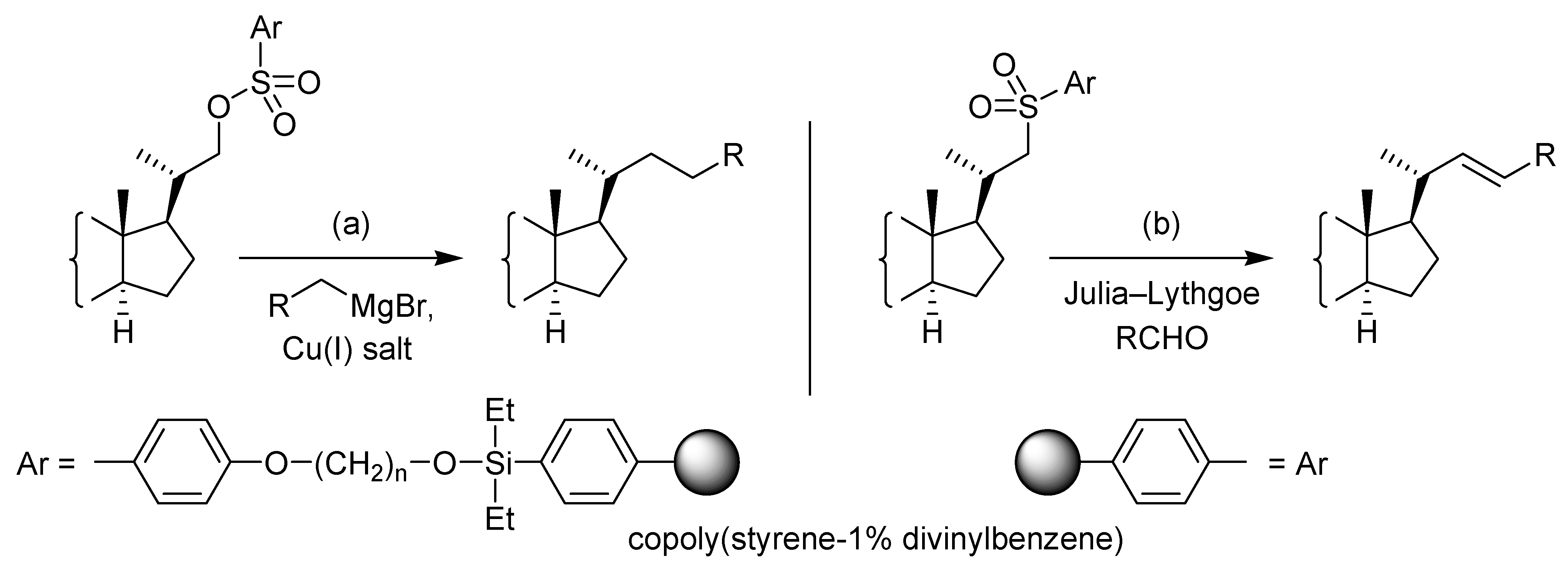
Conclusions
Experimental
General
Synthesis
(a) Potassium thiolate of thiophenol resin 2
(b) Substition of tosylate 3
(c) (1S,3aR,4S,7aS)-4-(tert-Butyldimethylsilyl)oxy-7a-methyl-1-[(1S)-1-methyl-2-(phenylsulfonyl)-ethyl]octahydro-1H-indene (4) [13]
(d) α-Sulfonyl carbanion of sulfone 4
(e) (1R,3aR,4S,7aR)-1-[(1S)-3-Benzoyloxy-1,5-dimethyl-2-(phenylsulfonyl)hex-4-en-1-yl]-4-(tert-butyldimethylsilyl)oxy-7a-methyloctahydro-1H-indene (5)
(f) (1R,3aR,4S,7aR)-4-(tert-Butyldimethylsilyl)oxy-1-[(1R,2E)-1,5-dimethylhexa-2,4-dien-1-yl]-7a-methyloctahydro-1H-indene (6)
Acknowledgements
References and Notes
- Proceedings of the 12th Workshop on Vitamin, D.; Bouillon, R.; Norman, A. W.; Pasqualini, J. R. J. Steroid Biochem. Mol. Biol. 2004, 89–90, 1–633, and the previous 11 volumes in this series. [CrossRef](b)Vitamin D.; Feldman, D.; Glorieux, F. H.; Pike, J. W. (Eds.) Academic Press: San Diego, 1997.
- Zhu, G.-D.; Okamura, W. H. Chem. Rev. 1995, 95, 1877–1952. [CrossRef]Dai, H.; Posner, G. H. Synthesis 1994, 1383–1398.
- Nagpal, S.; Na, S.; Rathnachalam, R. Endocr. Rev. 2005, 26, 662–687. [CrossRef]Bouillon, R.; Okamura, W. H.; Norman, A. W. Endocr. Rev. 1995, 16, 200–257.
- Kabat, M. M.; Radinov, R. Curr. Opin. Drug Disc. 2001, 4, 808–833.Posner, G. H.; Kahraman, M. Eur. J. Org. Chem. 2000, 3889–3895.
- Cano, M.; Balasubramanian, S. Drug Future 2003, 28, 659–678. [CrossRef]Ganesan, A. Drug Discov. Today 2002, 7, 47–55. [CrossRef]Fruchtel, J. S.; Jung, G. Angew. Chem., Int. Ed. 1996, 35, 17–42. [CrossRef]
- Hijikuro, I.; Doi, T.; Takahashi, T. J. Am. Chem. Soc. 2001, 123, 3716–3722. [CrossRef]Doi, T.; Hijikuro, I.; Takahashi, T. J. Am. Chem. Soc. 1999, 121, 6749–6750.
- Baggiolini, E. G.; Iacobelli, J. A.; Hennessy, B. M.; Batcho, A. D.; Sereno, J. F.; Uskokovic, M. R. J. Org. Chem. 1986, 51, 3098–3108. [CrossRef]Lythgoe, B.; Moran, T. A.; Nambudiry, M. E. N.; Ruston, S.; Tideswell, J.; Wright, P. W. J. Chem. Soc., Perkin Trans. 1 1978, 590–595.
- Franzen, R. G. Tetrahedron 2000, 56, 685–691. [CrossRef]Schlosser, M. Angew. Chem., Int. Ed. 1974, 13, 701–706. [CrossRef]
- D'herde, J. N. P.; De Clercq, P. J. Tetrahedron Lett. 2003, 44, 6657–6659. [Google Scholar]
- Fréchet, J. M. J.; de Smet, M. D.; Farrell, M. J. Polymer 1979, 20, 675–680. [CrossRef]
- Fernández, B.; Martinez Pérez, J. A.; Granja, J. R.; Castedo, L.; Mouriño, A. J. Org. Chem. 1992, 57, 3173–3178. [CrossRef]Mascareñas, J. L.; Mouriño, A.; Castedo, L. J. Org. Chem. 1986, 51, 1269–1272. [CrossRef]
- Shabangi, M.; Sealy, J. M.; Fuchs, J. R.; Flowers II, R. A. Tetrahedron Lett. 1998, 39, 4429–4432. [CrossRef]
- In the compound chemical name of 4 and 5 the phenylsulfonyl group, although further connected via the 4-position to the solid phase (see Scheme 1), is named as such. The ACD/I-Lab Web service (ACD/IUPAC Name Free 8.05) was used to generate the core of the chemical names.
- Sample Availability: No samples available.
© 2006 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
D'herde, J.N.; De Clercq, P.J. Application of the Solid-Phase Julia–Lythgoe Olefination in Vitamin D Side-Chain Construction. Molecules 2006, 11, 655-660. https://doi.org/10.3390/11080655
D'herde JN, De Clercq PJ. Application of the Solid-Phase Julia–Lythgoe Olefination in Vitamin D Side-Chain Construction. Molecules. 2006; 11(8):655-660. https://doi.org/10.3390/11080655
Chicago/Turabian StyleD'herde, Jo N., and Pierre J. De Clercq. 2006. "Application of the Solid-Phase Julia–Lythgoe Olefination in Vitamin D Side-Chain Construction" Molecules 11, no. 8: 655-660. https://doi.org/10.3390/11080655
APA StyleD'herde, J. N., & De Clercq, P. J. (2006). Application of the Solid-Phase Julia–Lythgoe Olefination in Vitamin D Side-Chain Construction. Molecules, 11(8), 655-660. https://doi.org/10.3390/11080655