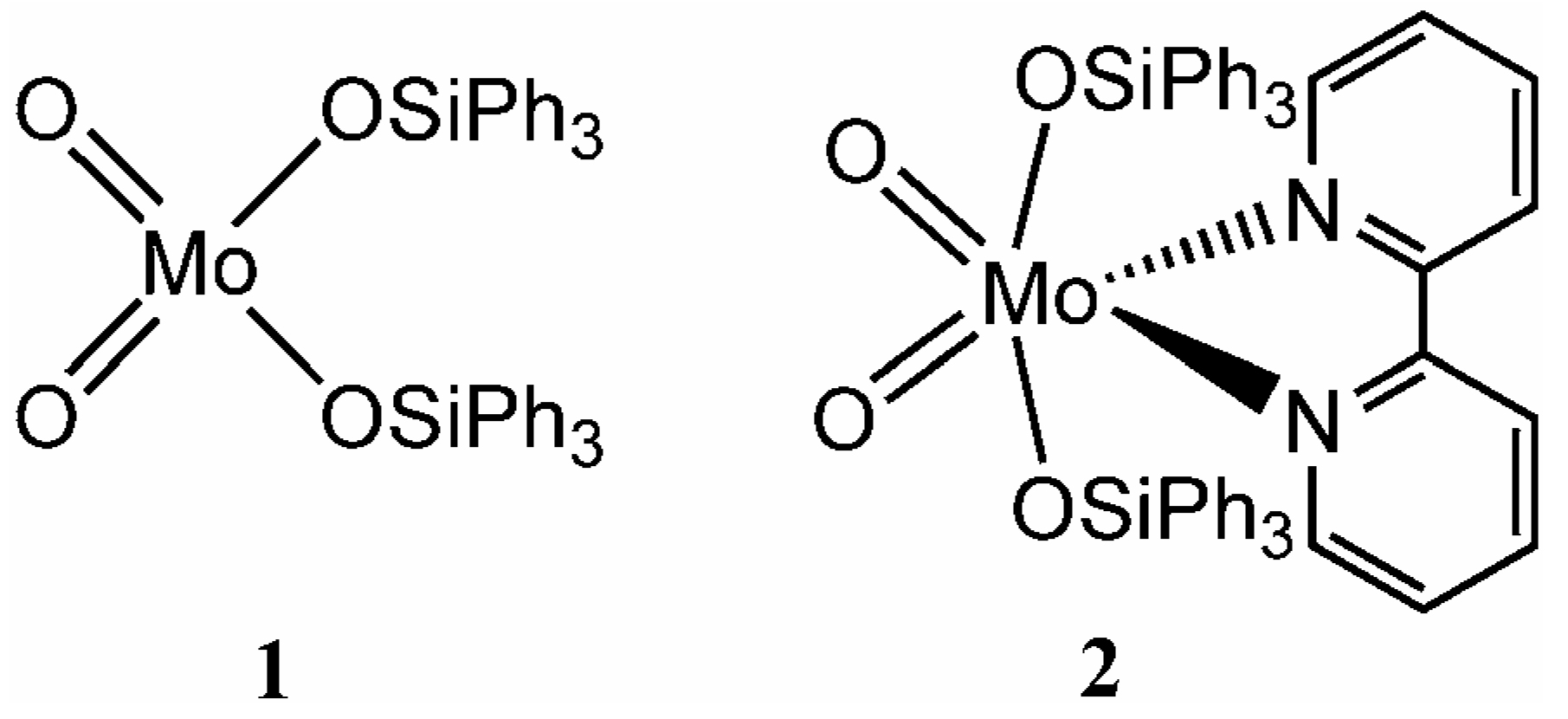
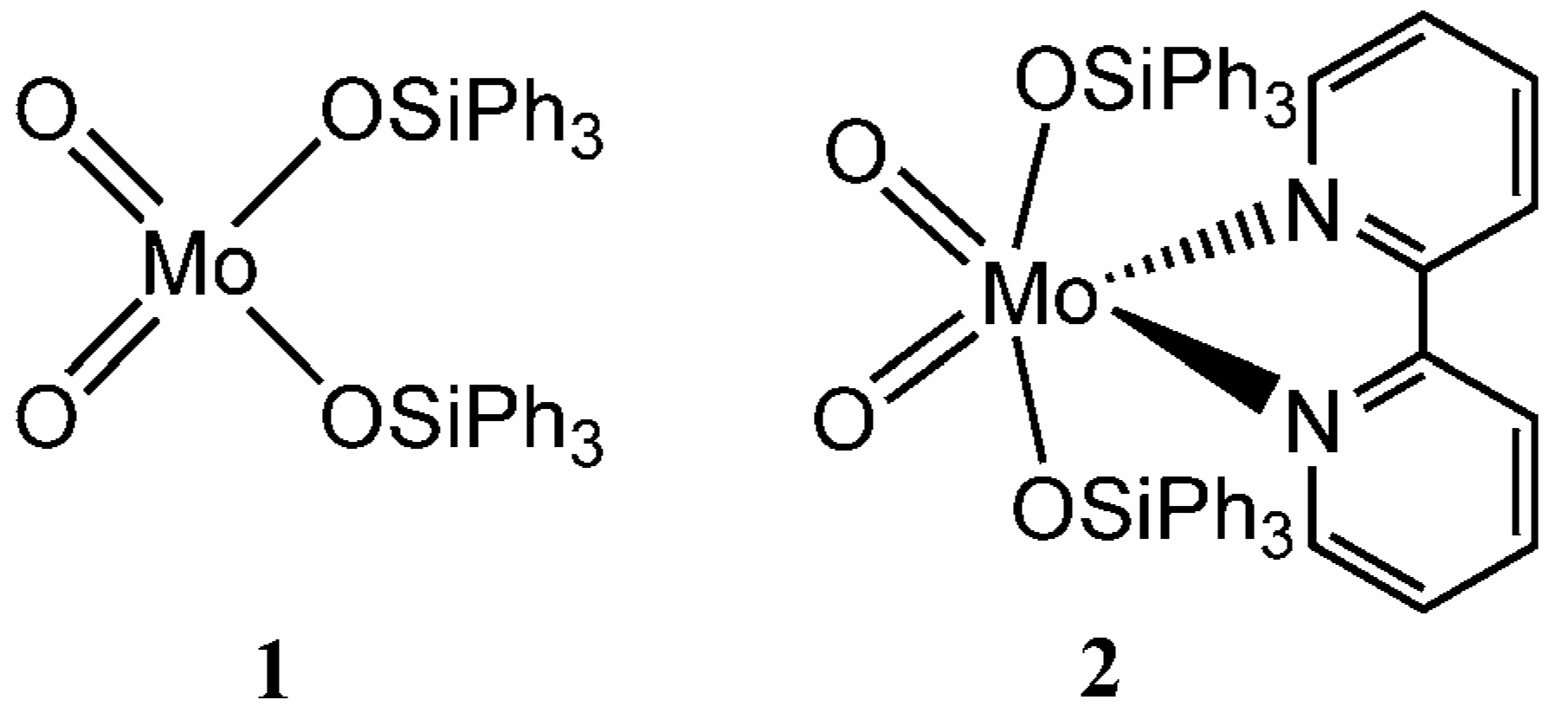
Synthesis and characterisation
The triphenylsiloxy complex MoO
2(OSiPh
3)
2 (
1) was prepared as described previously by the reaction of silver molybdate with two equivalents of Ph
3SiCl [
5]. The adduct MoO
2(OSiPh
3)
2(bpy) (
2) was obtained either by treatment of a solution of previously prepared and isolated
1 with one equivalent of 2,2'-bipyridine, or directly in one step by the addition of bpy at the end of the reaction used to prepare
1. Compounds
1 and
2 were characterised by elemental analysis,
1H-NMR, IR and Raman spectroscopy, and thermogravimetric analysis. The assignment of the vibrational spectra was supported by
ab initio calculations (
Table 1). Molybdenum(VI) complexes with the
cis-dioxo unit typically show two very strong IR peaks in the range 905–940 cm
–1 [
11], assigned to the Mo=O stretching modes. In the case of the starting material, Na
2MoO
4∙2H
2O, strong bands for the Mo=O stretching modes are observed in the IR spectrum at 897 and 834 cm
–1. These bands are shifted to higher frequencies (948 and 932 cm
–1) for compound
1, indicating a strengthening of the Mo=O bonds due to the formation of the O–SiPh
3 bonds. For the Mo–O bond, only a very weak band at 356 cm
–1 is observed, assigned to symmetric stretching. Compound
2 exhibits a pair of weak bands at 170 and 155 cm
–1 in the Raman spectrum, attributed to Mo–N asymmetric and symmetric stretching, respectively. As expected, the bidentate coordination of 2,2'-bipyridine, which implies a change from distorted tetrahedral to distorted octahedral geometry, weakens the MoO
4 bonds of the complex. As shown in
Table 1, the Mo=O bands are shifted to lower frequencies and in the Raman spectrum the Mo=O
sym is not observed. In contrast to compound
1, the asymmetric stretching band for the Mo–O bond appears with medium intensity at 377 cm
–1, while the symmetric stretching band is not observed.
Table 1.
Selected IR and Raman stretching frequencies of Na2MoO4∙2H2O, MoO2(OSiPh3)2 (1) and MoO2(OSiPh3)2(bpy) (2), and calculated (B3LYP) frequencies of the three compounds.
Table 1.
Selected IR and Raman stretching frequencies of Na2MoO4∙2H2O, MoO2(OSiPh3)2 (1) and MoO2(OSiPh3)2(bpy) (2), and calculated (B3LYP) frequencies of the three compounds.
| Compound | Calcda | IR (cm–1) | Raman (cm–1) | Assignment |
|---|
| Na2MoO4∙2H2O | 838 | 897 | 896 | νMoO4 unit |
| | 802 | 834 | 834 | |
| MoO2(OSiPh3)2 (1) | 948 | 948m | 949vw | νsym(Mo=O) |
| | 892 | 932m | 933vw | νasym(Mo=O) |
| | 550 | — | — | νasym(Mo–O)b |
| | 439 | 356vw | 358w | νsym(Mo–O)b |
| MoO2(OSiPh3)2(bpy) (2) | 944 | 933vs | — | νsym(Mo=O) |
| | 913 | 908vs | 909vs | νasym(Mo=O) |
| | 361 | 377m | 378w | νasym(Mo–O) |
| | 345 | — | — | νsym(Mo–O) |
| | 143 | — | 170w | νasym(Mo–N) |
| | 141 | — | 155w | νsym(Mo–N) |
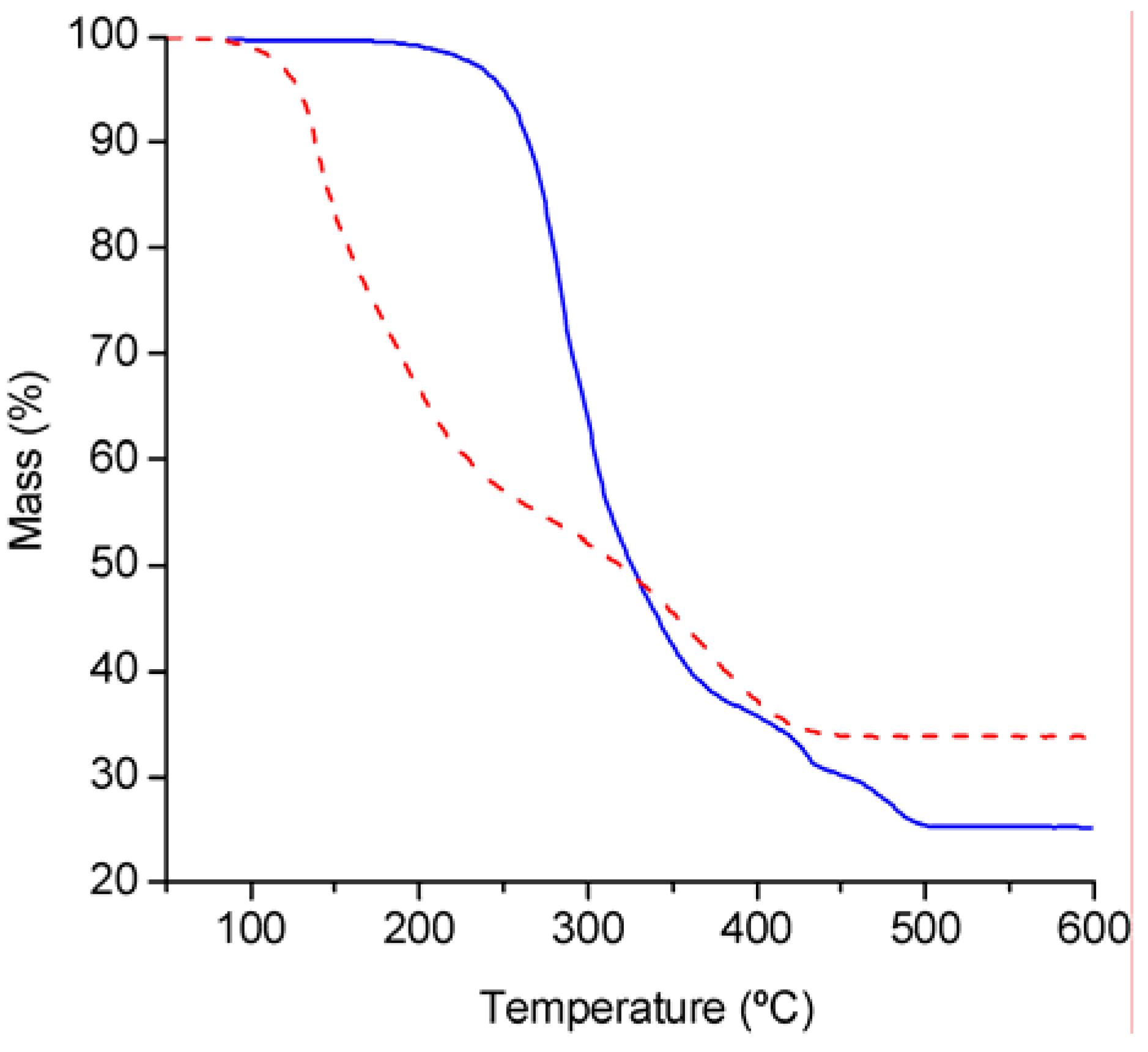
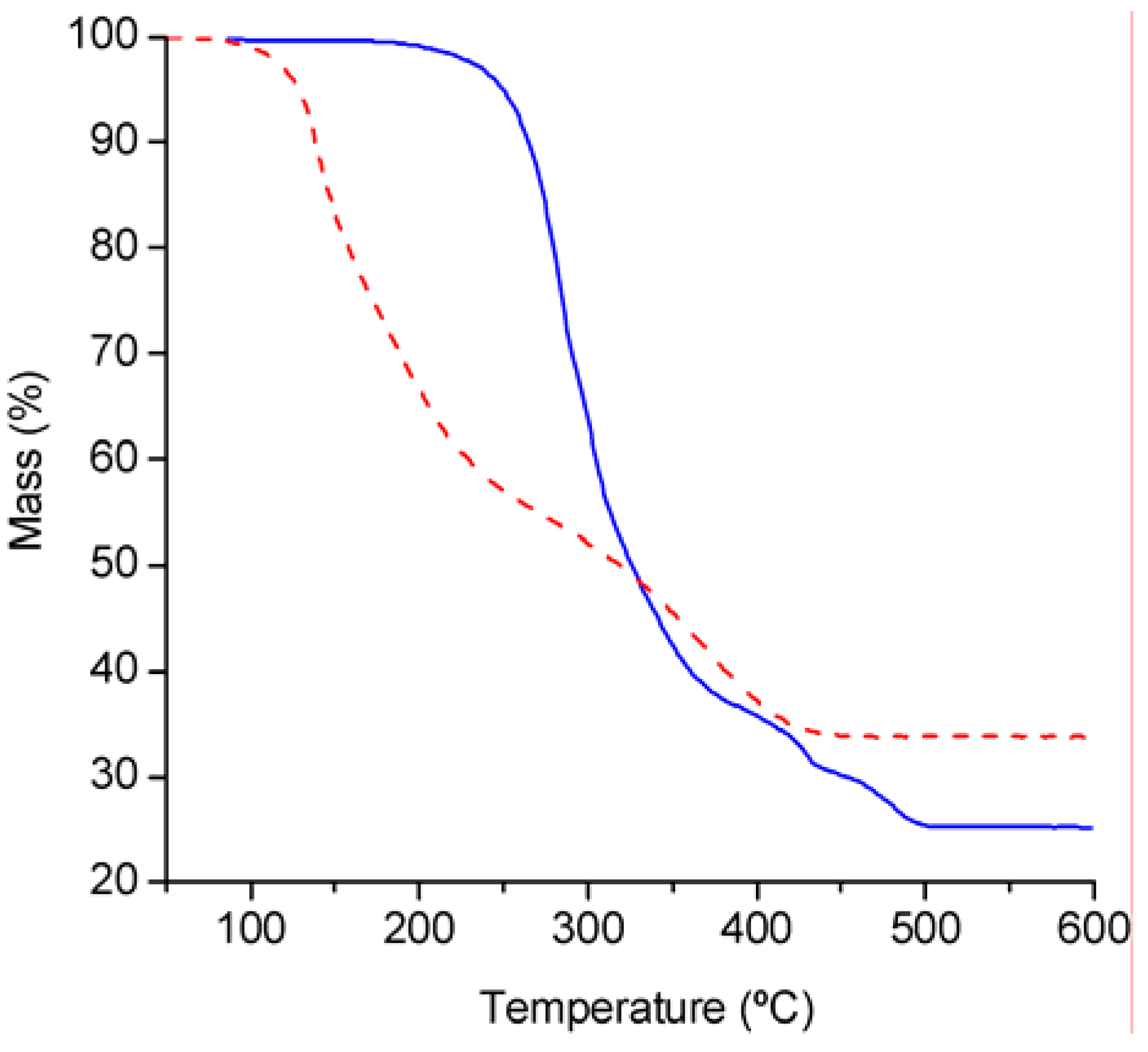
Thermogravimetric analysis under air (
Figure 2) showed that compound
2 is significantly more stable than
1. The tetrahedral complex
1 decomposes in two main steps from 80 to 440 ºC, with 45% mass loss in the temperature range 80–270 ºC, and 21% mass loss in the temperature range 270–440 ºC. The observed ceramic yield at 600 °C is 33.8%, which is 5.1% lower than the calculated yield for MoO
3·2SiO
2 (38.9%). The TGA results for
2 reflect a higher onset temperature of ca. 180 °C and a weight loss that is complete by 500 °C. The ceramic yield of 25% is 6.6% lower than that calculated for stoichiometric formation of MoO
3·2SiO
2 (31.6%).
Figure 2.
TGA curves of MoO
2(OSiPh
3)
2 (
1) (
![Molecules 11 00298 i001]()
) and MoO
2(OSiPh
3)
2(bpy) (
2) (
![Molecules 11 00298 i002]()
).
Figure 2.
TGA curves of MoO
2(OSiPh
3)
2 (
1) (
![Molecules 11 00298 i001]()
) and MoO
2(OSiPh
3)
2(bpy) (
2) (
![Molecules 11 00298 i002]()
).
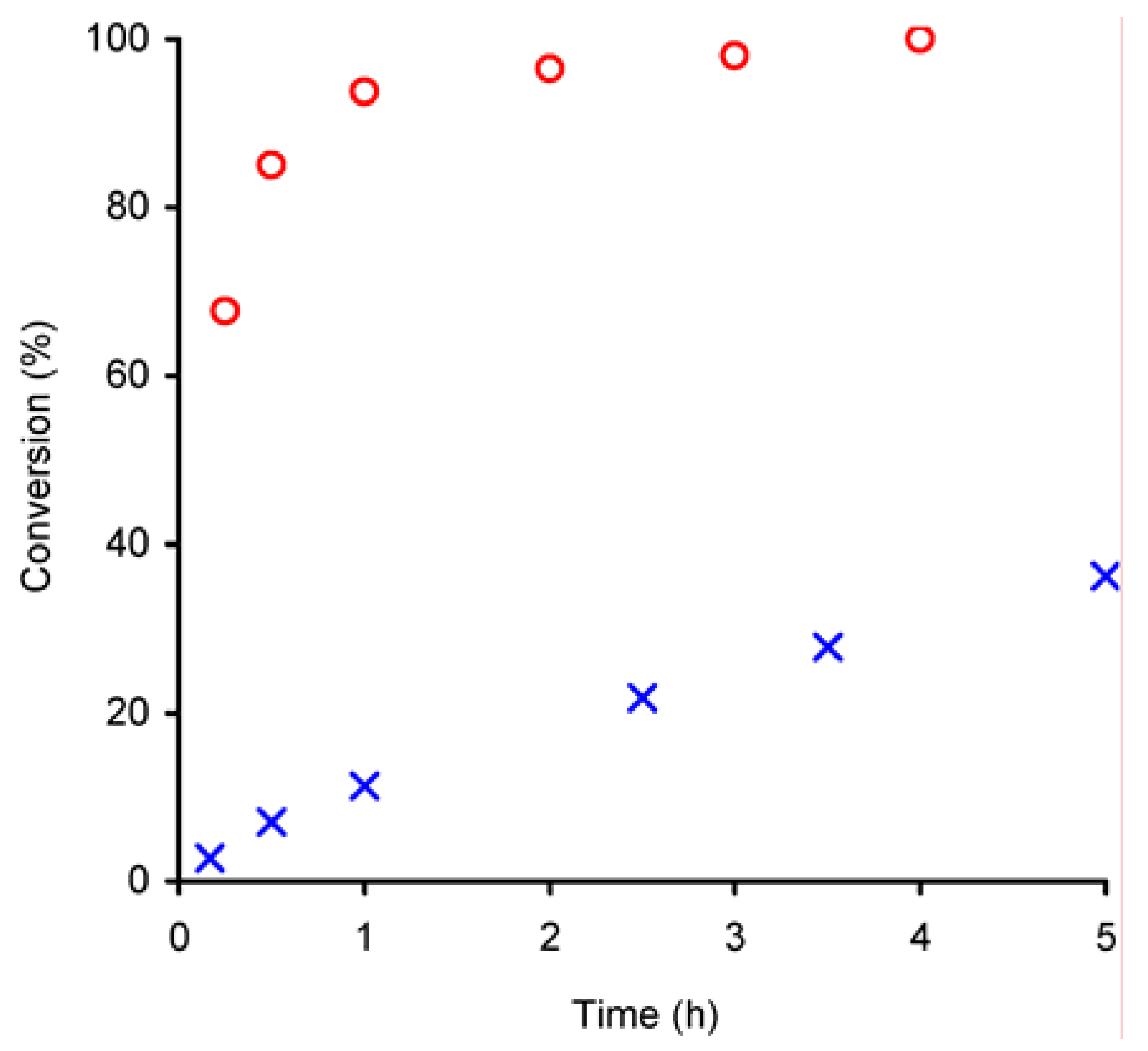
Catalysis
The performance of compounds
1 and
2 as epoxidation catalysts was studied using
cis-cyclooctene as a model substrate and
t-BuOOH as oxygen donor. In a control experiment, carried out without catalyst, no reaction occurred, whereas in the presence of compounds
1 or
2 the conversion of cyclooctene produced 1,2-epoxycyclooctane as the only product, confirming the catalytic role of these compounds (
Figure 3). Catalyst
1 (initial activity 272 mol·mol
Mo–1·h
–1) is much more active than
2 (initial activity 12 mol·mol
Mo–1·h
–1), and after 3 h the epoxide was obtained in quantitative yield in the presence of
1, whereas for
2 the conversion was still only 22%. The observed catalytic activity of
2 is comparable to that of other distorted octahedral Mo
VI complexes of the type MoO
2X
2L (X = Cl, Br) bearing bidentate N-donor ligands (L) such as ethylenediimine [
10], bipyridine [
11] and bipyrimidine [
12].
Figure 3.
Kinetics of the epoxidation of cyclooctene with
t-BuOOH in decane at 55 ºC, catalysed by compounds
1 (
![Molecules 11 00298 i003]()
) and
2 (
![Molecules 11 00298 i004]()
).
Figure 3.
Kinetics of the epoxidation of cyclooctene with
t-BuOOH in decane at 55 ºC, catalysed by compounds
1 (
![Molecules 11 00298 i003]()
) and
2 (
![Molecules 11 00298 i004]()
).
For compound
1, the reaction rate decreases drastically with time. In general, the mechanisms proposed for
t-BuOOH-based epoxidations of olefins with Mo
VI complexes are heterolytic in nature, involving coordination of the oxidant to the metal centre (by the terminal oxygen of
–OO
tBu in the case of the complexes MoO
2X
2L with X = Cl, Br or Me, and L = Lewis base N- or O-ligand), which acts as a Lewis acid thereby increasing the oxidising power of the peroxo group, and subsequently the olefin is epoxidised by nucleophilic attack on an electrophilic oxygen atom of the oxidising species [
8,
11]. The rapid decrease in the olefin conversion rate has been attributed to the formation of
tert-butanol (
t-BuOH), a by-product of the epoxidation, that acts as a competitor to
t-BuOOH for coordination to the metal centre, leading to the formation of inactive species [
11,
12].
The solvent effect was studied for compounds
1 and
2 using 1,2-dichloroethane,
n-hexane or acetonitrile, at 55 ºC. No dependence of product selectivity on the solvent was observed. For both catalysts, conversion after 24 h reaction followed the order: no co-solvent ≥ dichloroethane > hexane > CH
3CN, and TOF (calculated at 30 min) of cyclooctene epoxidation was highest without a co-solvent or with dichloroethane (
Table 2). Acetonitrile has a negative effect on initial catalytic activity, which may be related to its ability to coordinate to the molybdenum centre. According to the above mechanistic assumptions, the competition between solvent and oxidant molecules for coordination to the metal centre may retard the reaction. However, the coordinating power of the solvent molecules does not solely explain the observed catalytic activity since when hexane, a non-coordinating solvent, is added to the reaction medium the epoxidation rate decreases significantly for both catalysts. Indeed, the TOF for
1 (calculated at 30 min) in the presence of CH
3CN is higher than that observed for hexane. Hence, the dependence of catalytic activity on the type of solvent may be related to solvent-oxidant competitive ligand effects as well as changes in the solubility of the catalyst. The somewhat higher activity of
2 in dichloroethane may be associated with the higher catalyst solubility in the reaction medium.
Table 2.
Catalytic activity of compounds 1 and 2 for cyclooctene epoxidation at 55 ºC, using different co-solvents.
Table 2.
Catalytic activity of compounds 1 and 2 for cyclooctene epoxidation at 55 ºC, using different co-solvents.
| Solvent | Compound 1 | Compound 2 |
| TOFa (mol·molMo–1·h–1) | Conv.b (%) | TOFa (mol·molMo–1·h–1) | Conv.b (%) |
| none | 170 | 100c | 14 | 74 |
| 1,2-dichloroethane | 135 | 100c | 17 | 69 |
| n-hexane | 22 | 96 | 4 | 31 |
| Acetonitrile | 70 | 82 | 2 | 20 |
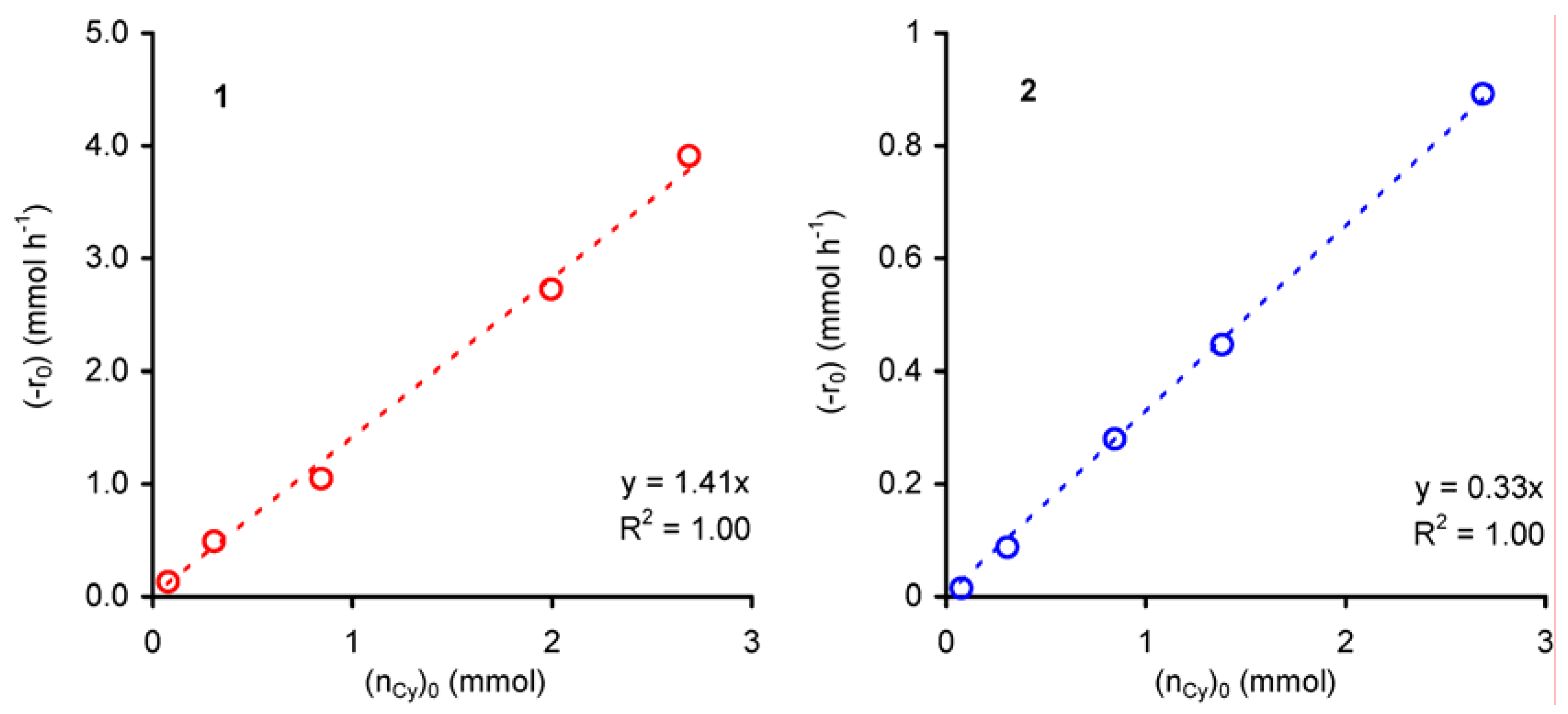
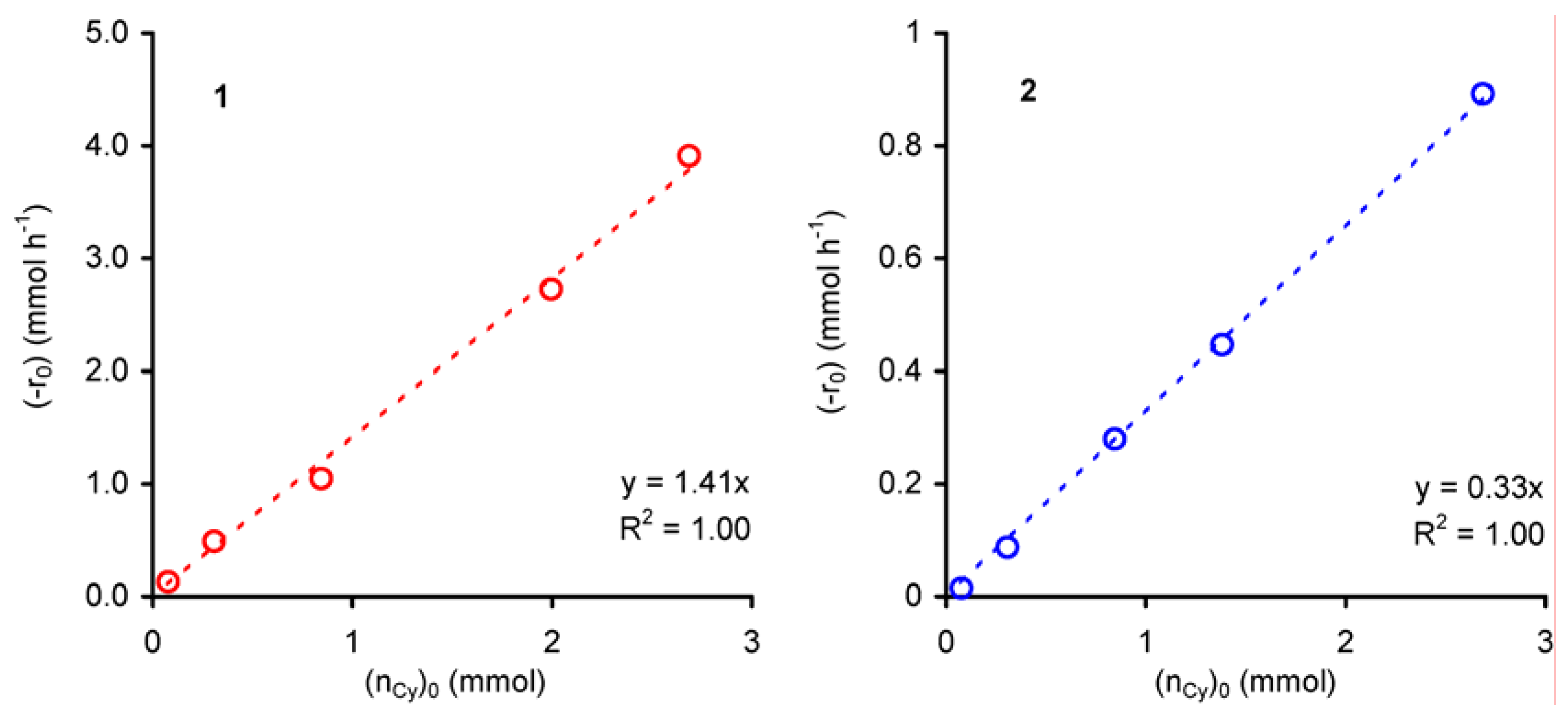
The kinetics of the liquid-phase epoxidation of cyclooctene in the presence of 1 and 2 was further investigated using the method of initial rates (-r0), and keeping the total volume of the reaction mixture constant using decane (the solvent of the purchased t-BuOOH) as solvent.
For all experiments cyclooctene epoxide was the only product. The dependence of the initial rate of cyclooctene conversion (-r
0) on initial amount of cyclooctene (n
Cy)
0 was studied for initial concentrations of cyclooctene ≤ 2 mol·dm
–3 at 55 ºC. For both compounds
1 and
2, the plot of (-r
0) versus (n
Cy)
0 is linear (R
2 = 0.99), suggesting an apparent first order dependence with respect to cyclooctene concentration (
Figure 4).
Figure 4.
Dependence of the initial rate of cyclooctene epoxidation (with t-BuOOH in decane), catalysed by compounds 1 or 2, as a function of the initial charge of cyclooctene.
Figure 4.
Dependence of the initial rate of cyclooctene epoxidation (with t-BuOOH in decane), catalysed by compounds 1 or 2, as a function of the initial charge of cyclooctene.
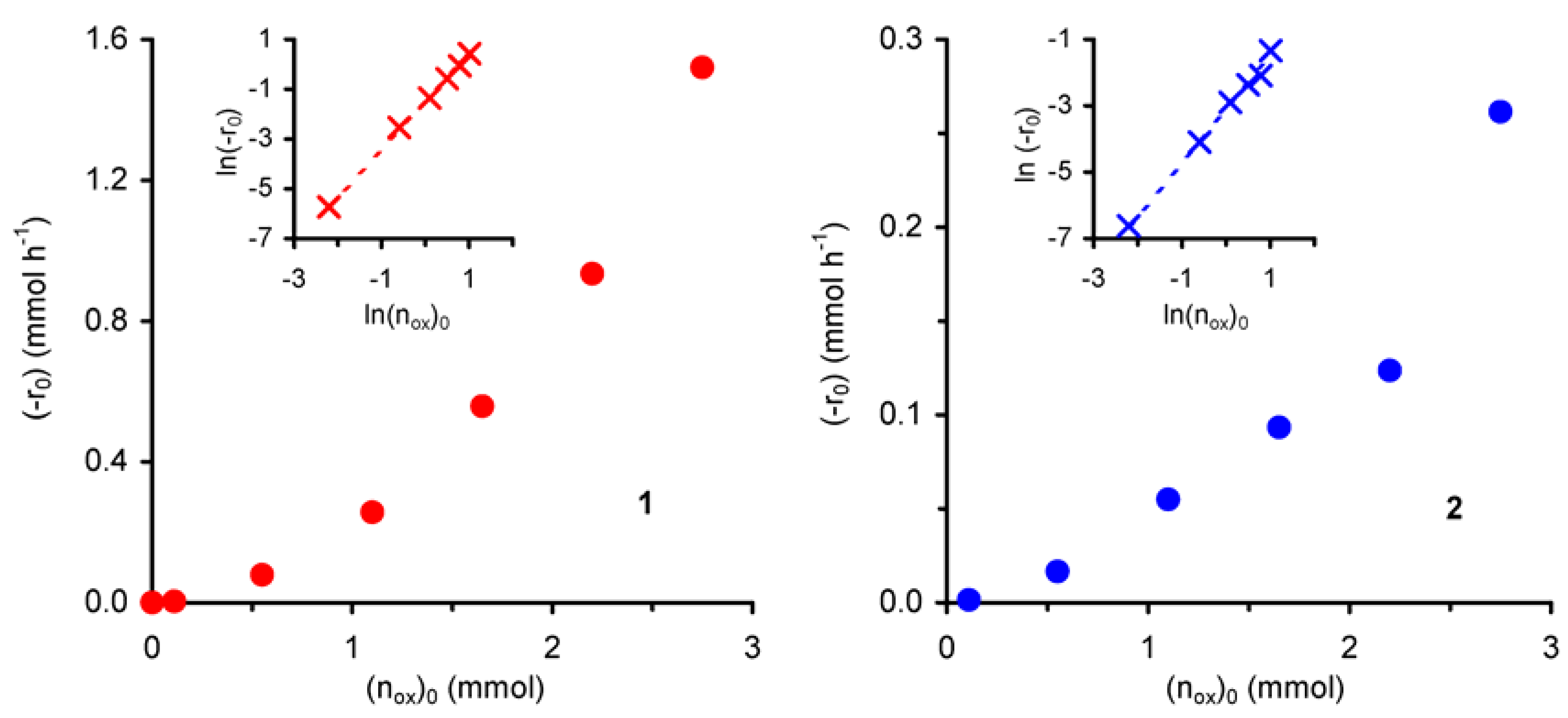
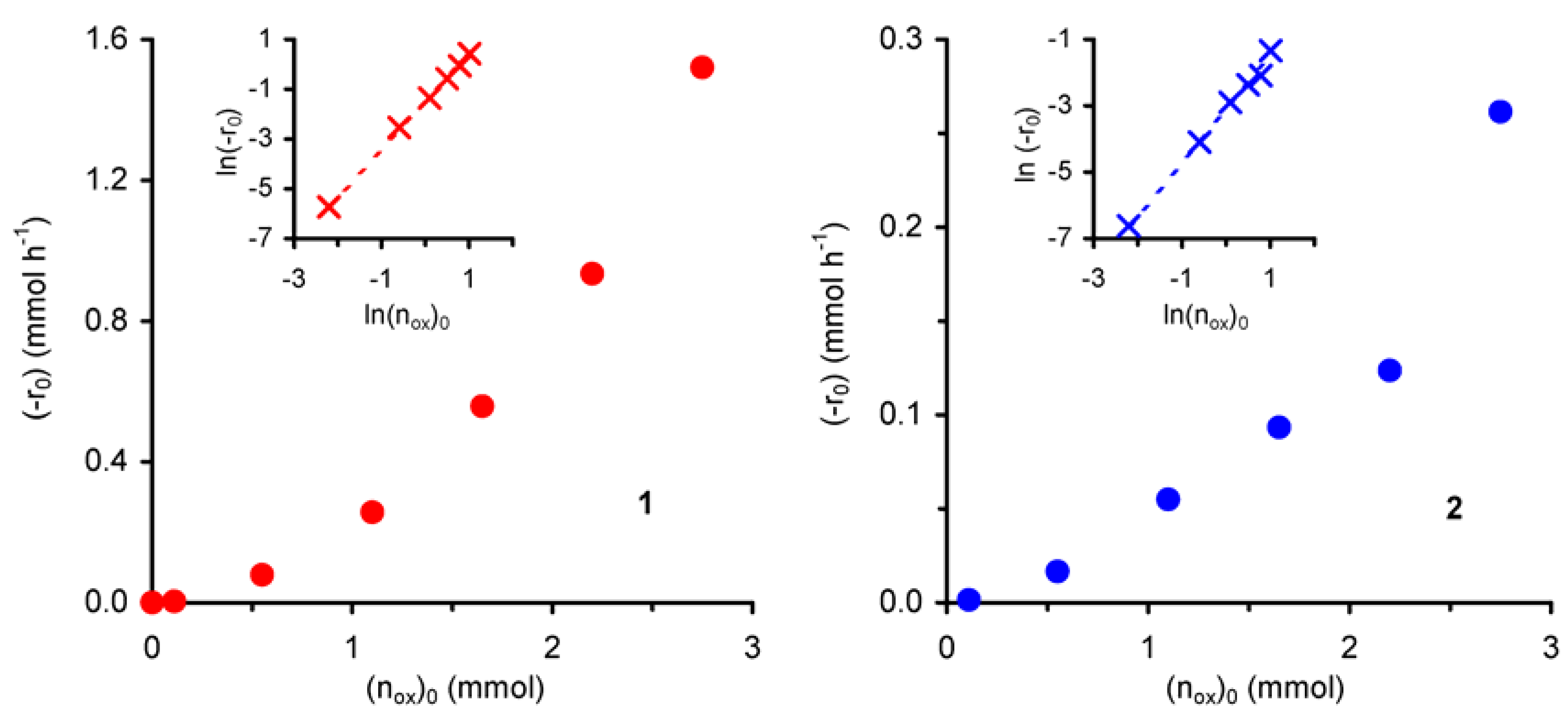
The effect of the initial amount of oxidant (n
ox)
0 on the initial reaction rate (-r
0) was studied for initial concentrations of
t-BuOOH ≤ 1.9 mol·dm
–3 at 55 ºC. Under the applied operating conditions, the reaction does not take place in the absence of
t-BuOOH. The observed initial reaction rate increases with
t-BuOOH concentration (
Figure 5).
Figure 5.
Dependence of the initial rate of cyclooctene epoxidation (with t-BuOOH in decane), catalysed by compounds 1 or 2, as a function of the initial amount of peroxide.
Figure 5.
Dependence of the initial rate of cyclooctene epoxidation (with t-BuOOH in decane), catalysed by compounds 1 or 2, as a function of the initial amount of peroxide.
For both compounds the plot of ln(-r
0) versus ln(n
ox)
0 gives a straight line (R
2 = 0.99) with slopes of 1.9 for
1 and 1.6 for
2, corresponding to the apparent reaction orders with respect to
t-BuOOH in the studied concentration range (insets of
Figure 5). These values suggest that the catalytic epoxidation process includes a number of simultaneous and consecutive elementary reactions, in agreement with the above mechanistic assumptions. Accordingly, a higher initial concentration of
t-BuOOH should increase the reaction rate by enhancing the
t-BuOOH/
t-BuOH molar ratio in the reaction medium, and thus the molar ratio between active (formed with
t-BuOOH) and inactive (formed with
t-BuOH) species.
The dependence of the initial conversion rate of cyclooctene on the reaction temperature (T) was studied in the temperature range of 35–70 ºC. For all experiments cyclooctene oxide was the only observed product. For both compounds the observed initial reaction rate (-r0) increases with temperature (plot not shown), following a linear Arrhenius plot of ln(-r0) vs 1/T (R2 = 0.99, at constant reagents and catalyst concentrations). The apparent activation energy of 1 (11 kcal·mol–1) is lower than that of 2 (20 kcal·mol–1), as would be expected considering the higher activity of 1.
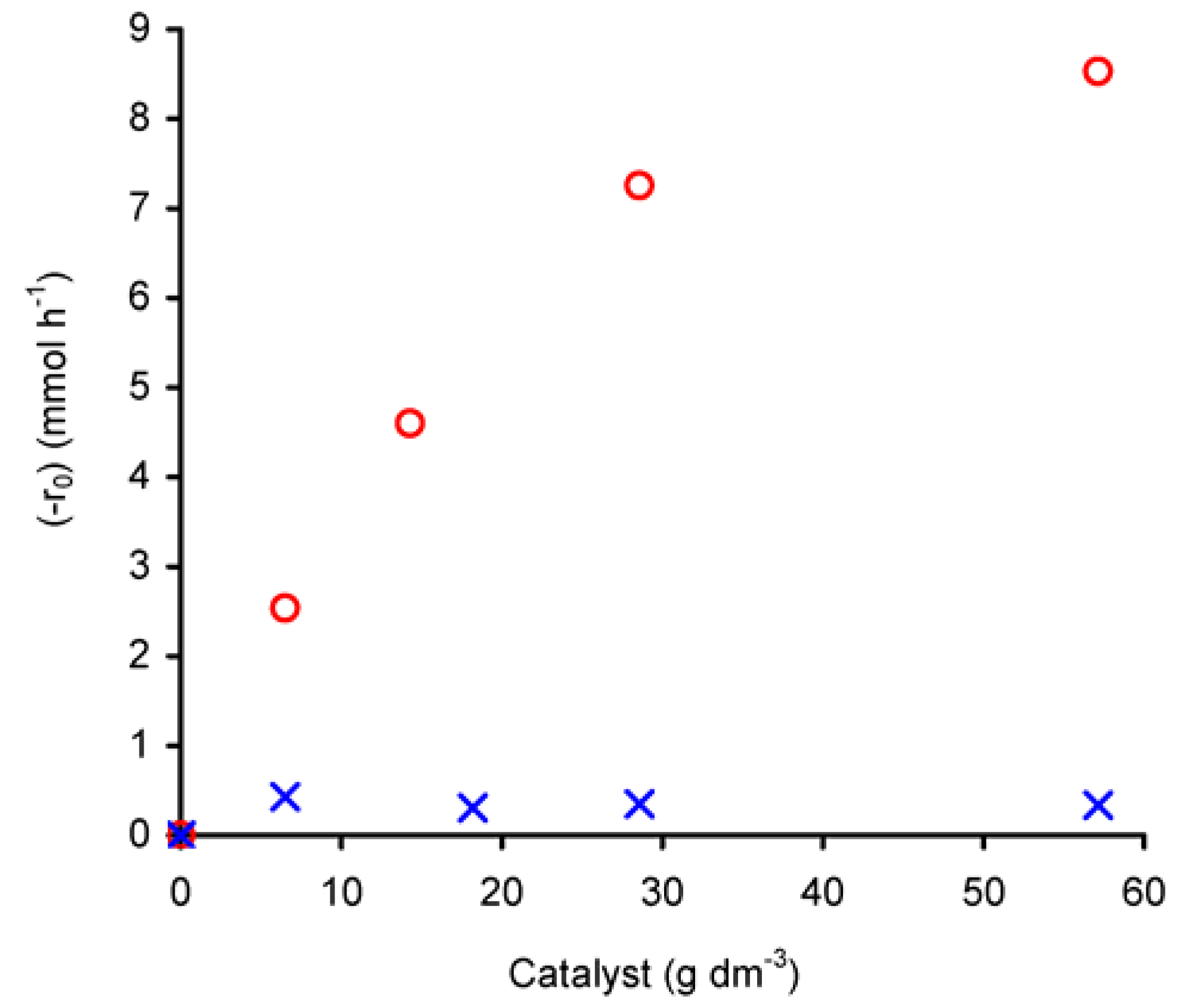
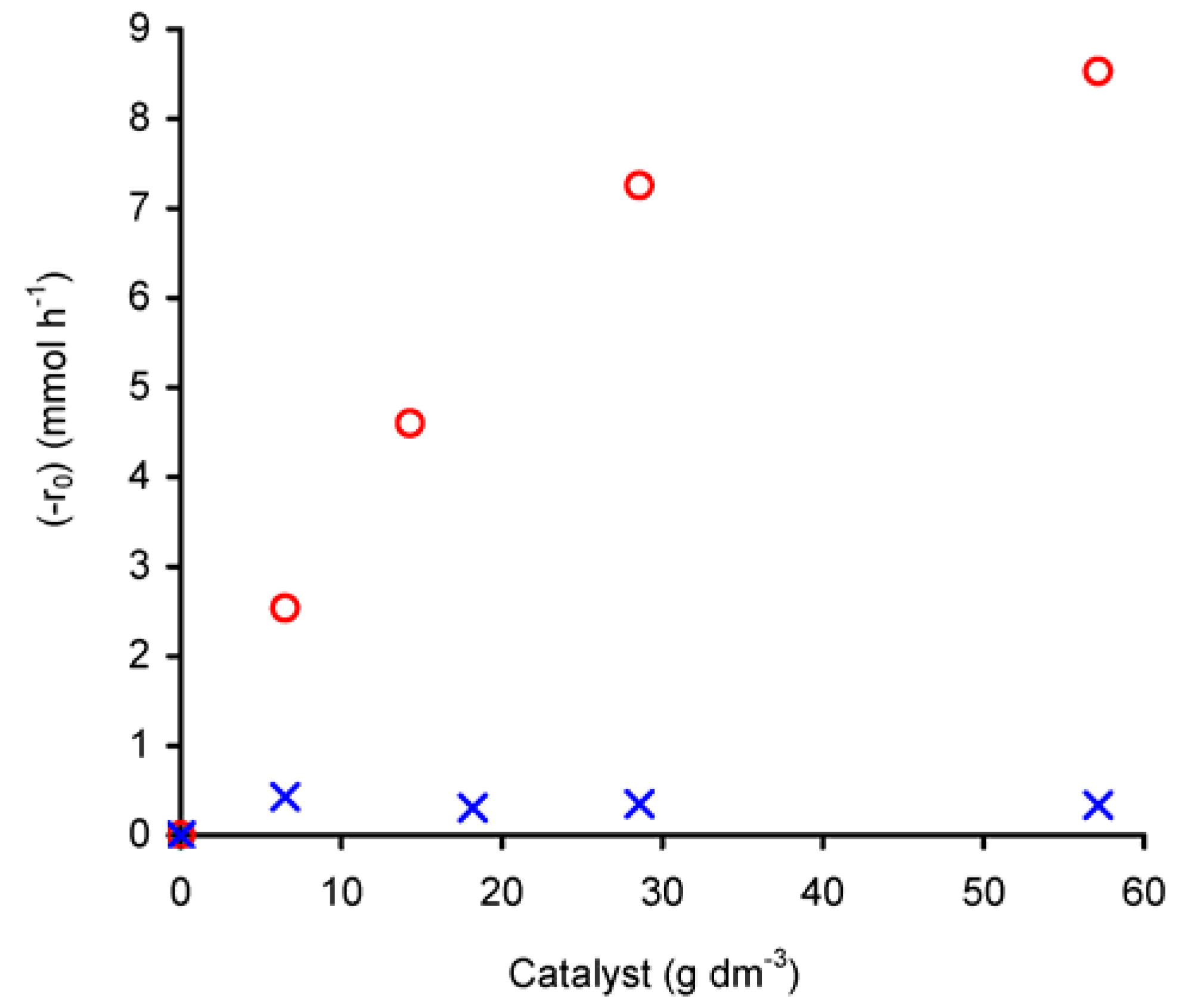
The dependence of (-r
0) on the initial amount of catalyst was studied in the range of 7–57 g
cat·dm
–3, at 55 ºC (
Figure 6). For compound
1 the reaction rate increases with catalyst charge. For W/L < 30 g
cat· dm
–3, the plot of ln(-r
0) versus ln(n
cat)
0 gives a straight line (R
2 = 0.99) with a slope of 0.7 (not shown), corresponding to the apparent reaction order with respect to initial catalyst amount ((n
cat)
0) in the studied concentration range. However, for charges greater than 30 g
cat·dm
–3, the differential rate law tends to become independent of the amount of catalyst, which may be due to a change in the controlling mechanism or catalyst solubility. On the other hand, at higher complex concentrations the productive utilisation of
t-BuOOH may be levelled off due to its decomposition into
t-BuOH and molecular oxygen.
Figure 6.
Dependence of the initial rate of cyclooctene epoxidation (with
t-BuOOH in decane), catalysed by compounds
1 (
![Molecules 11 00298 i003]()
) or
2 (
![Molecules 11 00298 i004]()
), as a function of the initial amount of charged catalyst.
Figure 6.
Dependence of the initial rate of cyclooctene epoxidation (with
t-BuOOH in decane), catalysed by compounds
1 (
![Molecules 11 00298 i003]()
) or
2 (
![Molecules 11 00298 i004]()
), as a function of the initial amount of charged catalyst.
Contrary to that observed for 1, (-r0) for compound 2 is independent of the initial amount of catalyst, that is, it exhibits an apparent zero-order rate law with respect to the catalyst amount. Given the low solubility of 2 in the reaction medium it is possible that the reaction rate is mainly governed by some soluble metal species and that increasing the concentration of catalyst does not further increase the amount of these active soluble species due to saturation of the solution.
Based on the above results, for the studied reaction temperature and intervals of initial concentrations, the initial rate (-r0) law of cyclooctene conversion in the presence of 2 may be expressed as (-r0) = k(nCy)0(nox)01.6, where k represents the reaction rate constant. In the case of 1, for W/L < 30 gcat·dm–3 and for the specified limits of initial concentrations of reagents and reaction temperature, the initial rate (-r0) law in the presence of 1 may be expressed as (-r0) = k(nCy)0(nox)01.9(ncat)00.7, where k represents the reaction rate constant.
We have previously reported that
1 can be recycled without significant loss of activity, originating the same kinetic profiles as the fresh catalyst [
9]. In this work the stability of
2 was studied in a similar fashion. After the first reaction cycle the catalyst was separated by centrifugation, washed several times with n-hexane and dried at room temperature prior to its reuse. As found for compound
1, the conversion profiles for fresh and recovered
2 were similar, suggesting that these compounds are fairly stable under the applied reaction conditions.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 ) and MoO2(OSiPh3)2(bpy) (2) (
) and MoO2(OSiPh3)2(bpy) (2) (  ).
).
 ) and 2 (
) and 2 (  ).
).