A Java Chemical Structure Editor Supporting the Modular Chemical Descriptor Language (MCDL)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Introduction
Results and Discussion
Reconstruction of bond order from MCDL string
- Two cyclic bonds attached to a chalcogen (oxygen, sulphur) or a three-coordinated nitrogen of unknown order are replaced by single bonds. The same procedure is executed for a nitrogen atom linked with neighboring atoms with three aromatic bonds—junction of fused aromatic rings.
- The valences of positively charged heteroatoms are incremented by 1 relative to standard values: N+: 4; O+: 3.
- The order of an arbitrary selected bond is assigned to 1. The order of an adjacent bond is considered to be 2, next—1 and so on. For fusion atoms (any atom of a fused-ring system which is common to two or more rings), one bond is temporarily assigned as a double and the other two as a single. This temporary assignment is stored to allow future modification in case of an incorrect assignment.
- If the calculated number of hydrogens does not correspond to the MCDL string number, then the reconstruction process is considered to be unsuccessful. In this case, the algorithm returns to the last fused atom assignment (point 3) to reassign single and double bonds.
- If after all attempts, there is no acceptable bond assignment, the program returns to point 1, and the order of the first arbitrary selected bond is set to be 2.
- Finally, Kekule structure representation is considered to be impossible if no bond order assignment that corresponds to an MCDL string can be found.
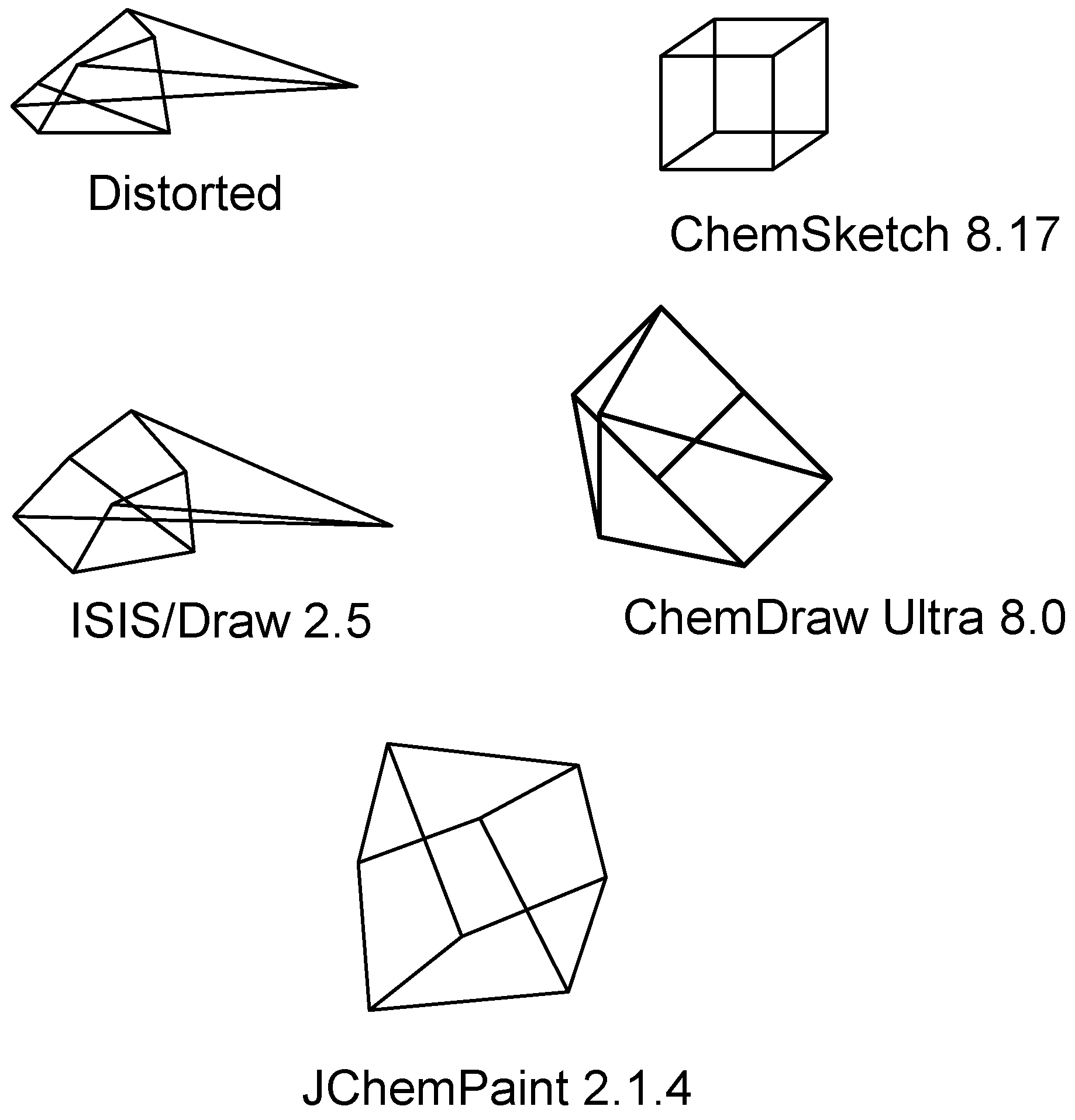
Structure diagram generation of polycyclic compounds
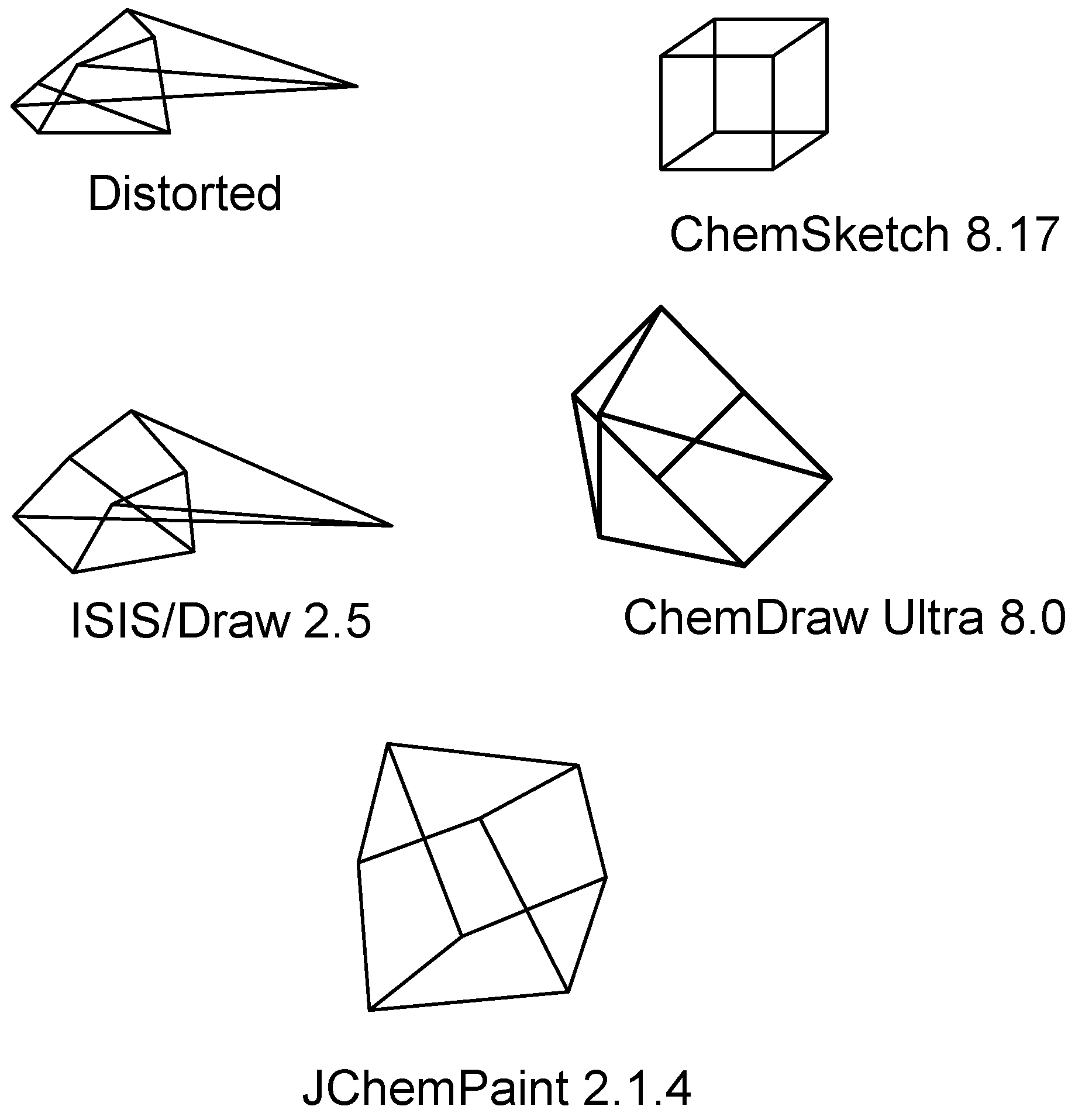
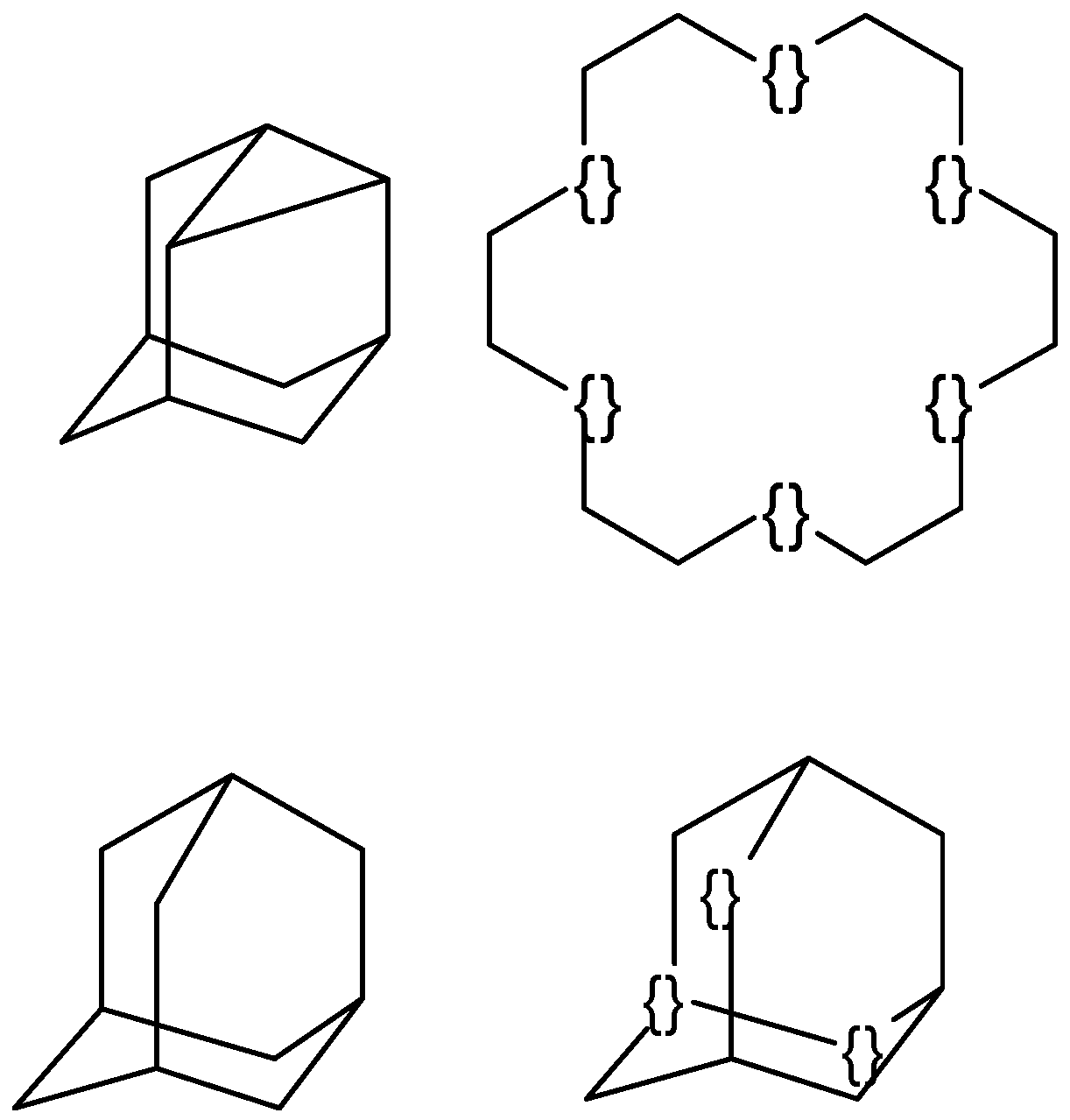
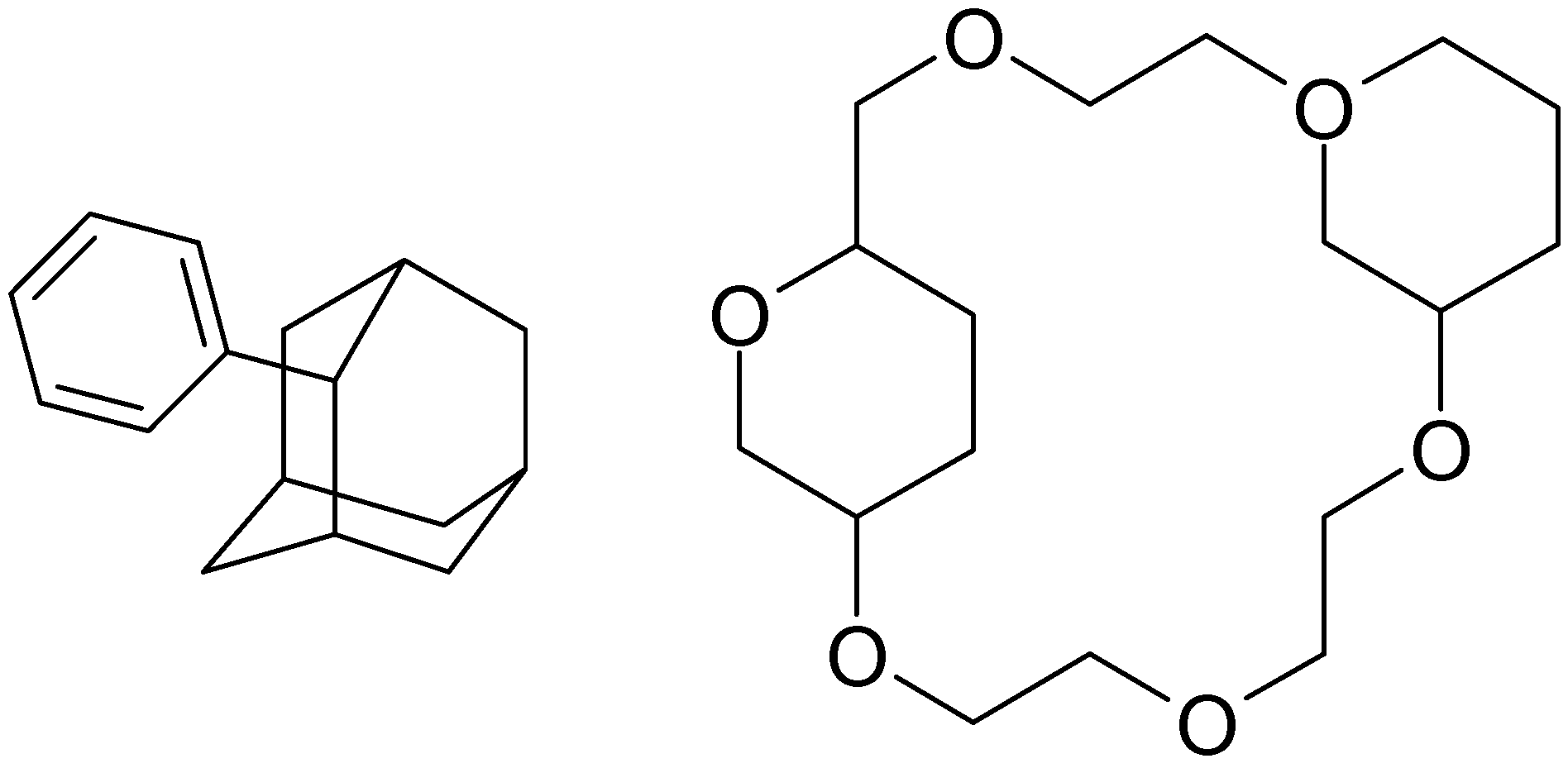
- The set of the minimum number of cycles in the compound is calculated using an algorithm [41] and stored in the LIST.
- The search in the fragment database is executed, and coordinates of relevant atoms are assigned when a fragment is found. If a fragment is not found, then the maximum size cycle from the LIST is drawn. If there is no cycle, then two linked atoms with maximum substitution numbers are used as the initial fragment to generate a structure diagram.
- The fragment in the database is searched for atoms with not-yet-determined coordinates. The fragment should be linked to an atom with known 2D coordinates.
- If the fragment is found, then coordinates of corresponding atoms in the structure are considered to be assigned. Then the algorithm returns to point 3, and the next fragment is searched. If there are no more qualified fragments, then the algorithm moves to point 5.
- All cycles from the list with at least one assigned coordinate atom are added to the structure. If coordinates of only one atom are known, then a spiro-cycle is added with standard bond lengths, and angles are calculated from the size of the cycle. If coordinates of two bonded atoms are known, then a fused cycle is added with the bonds’ length equal to a known bond and with the angles calculated as 2π/N, where N is the size of the cycle. If the coordinates of three and more atoms are known (polycyclic structure), then the chain is locked. The positions of new atoms are assigned using a special subroutine to avoid bond intersection (if possible). If the position of at least one more atom is determined here, then the algorithm returns to point 3, otherwise it goes to point 6.
- Coordinates of acyclic atoms (connected to cyclic atoms with known coordinates) are calculated using the standard bond length and the optimal bond angle 2π/3. In the case of long chains, coordinates of only the first (connection) atom are calculated. If the position of at least one more atom is determined here then the algorithm returns to point 3, otherwise it goes to point 7.
- A determination is made whether coordinates of all atoms are defined. “Yes” means the process is finished; “No” means that compound is a disconnected graph with two or more substructures. The process is repeated beginning from point 1 for the next fragment until full completion.

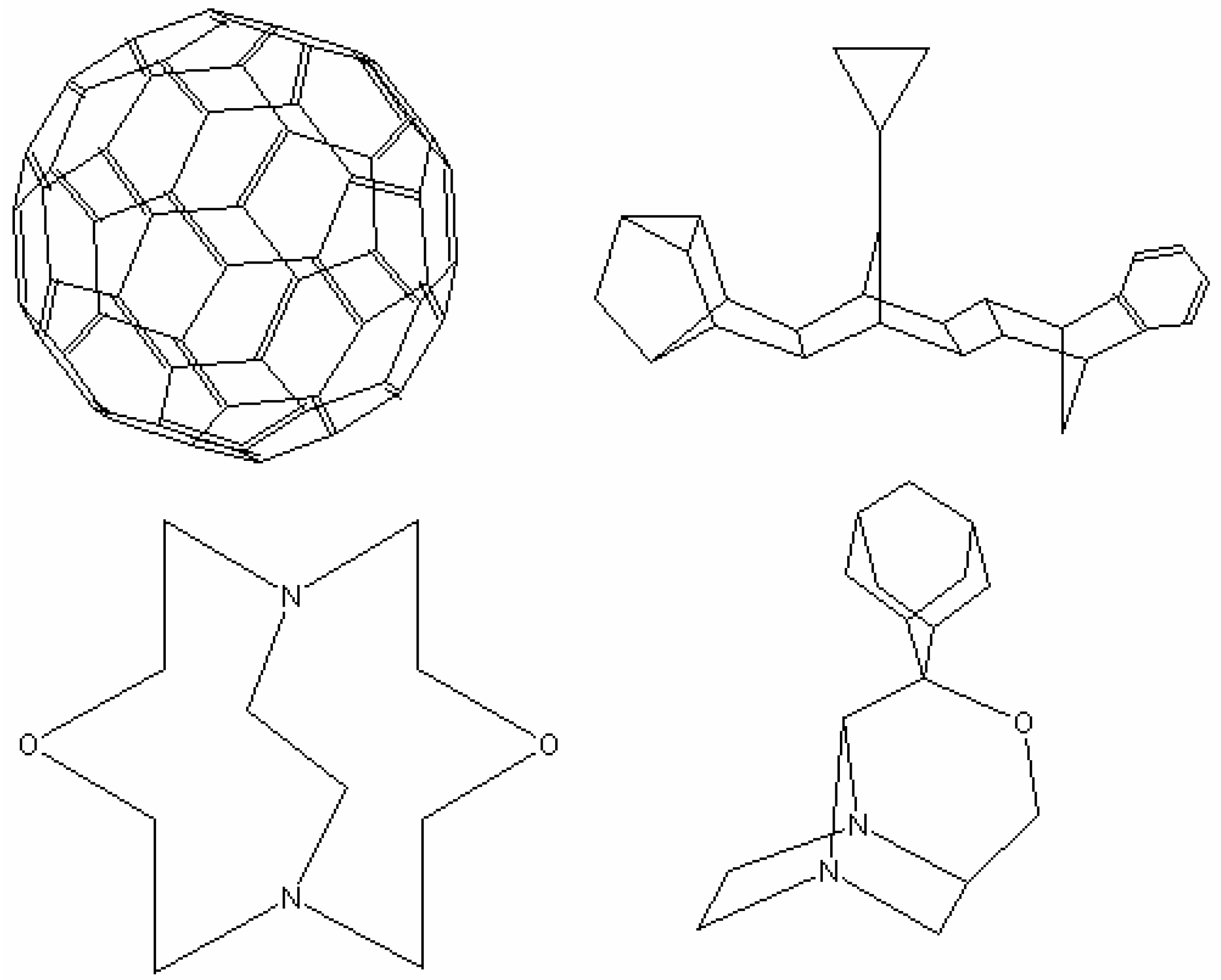
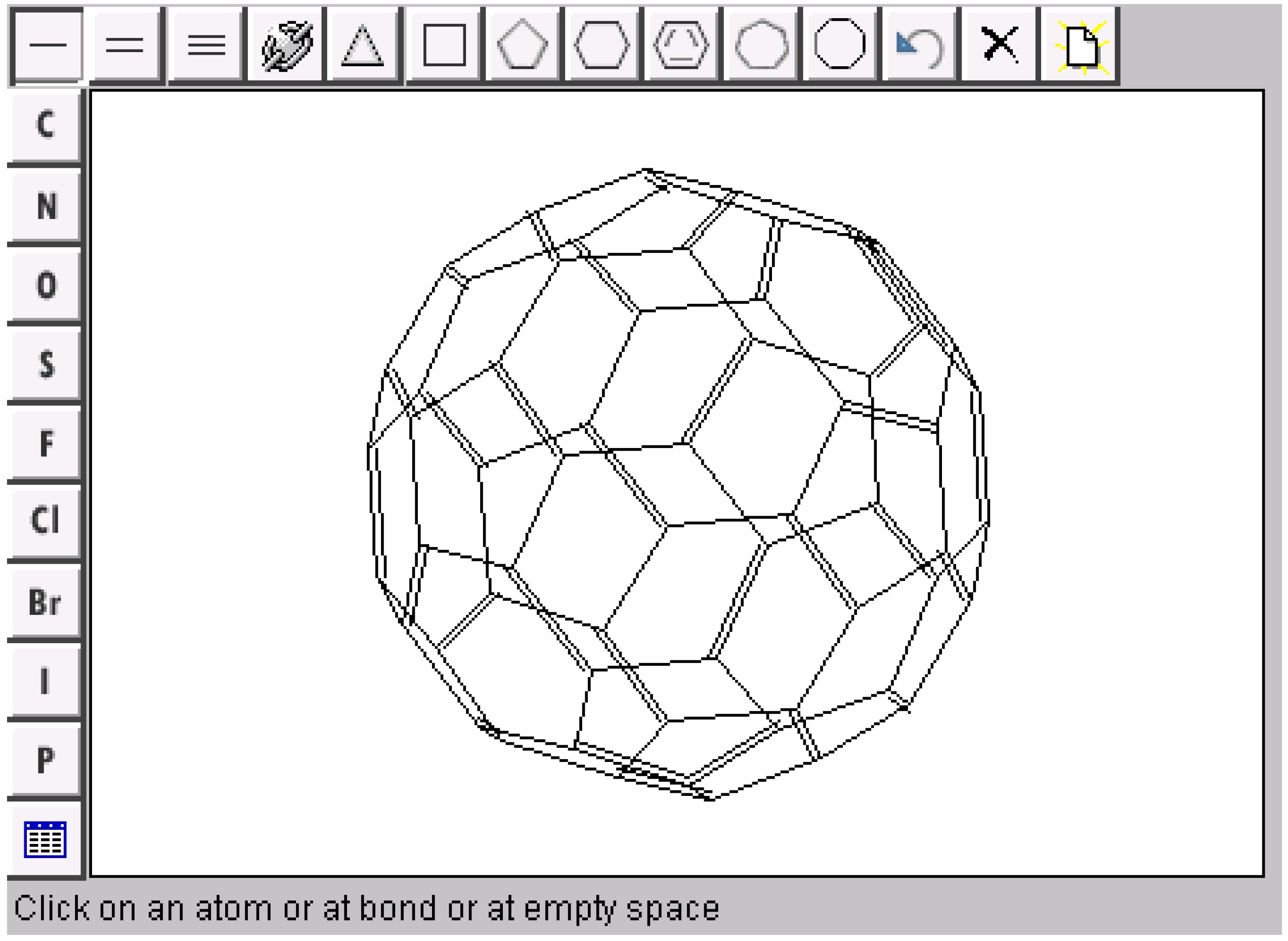
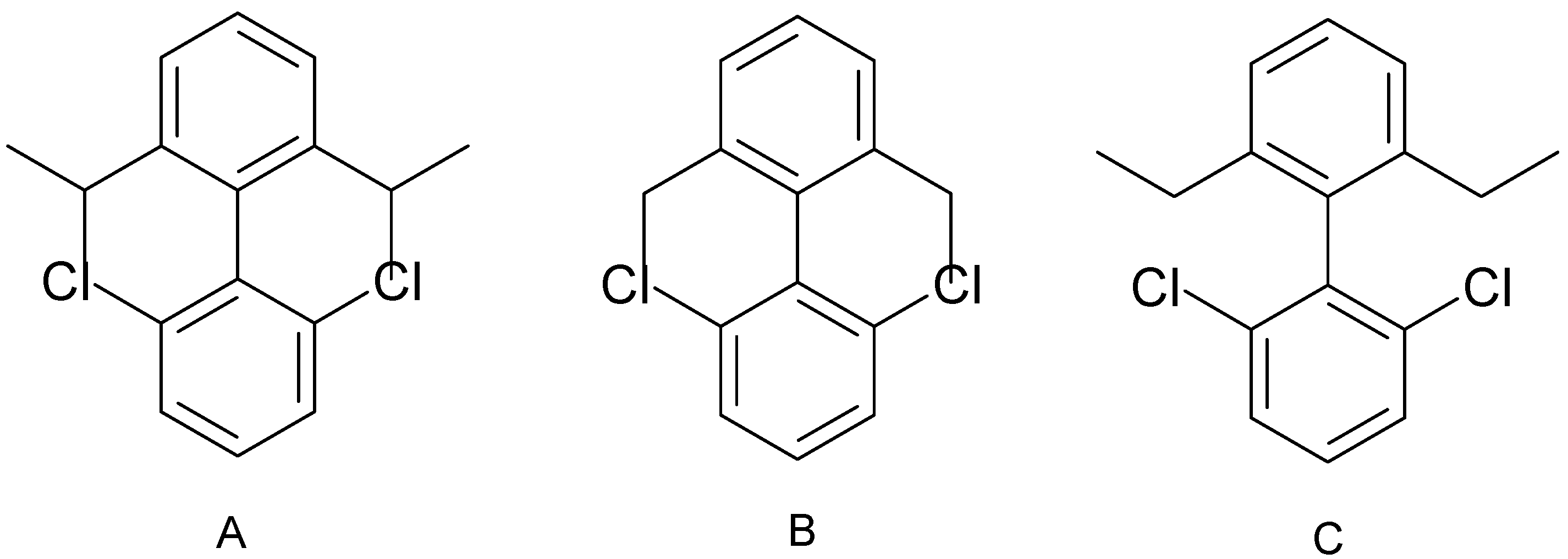
Overlapped fragments
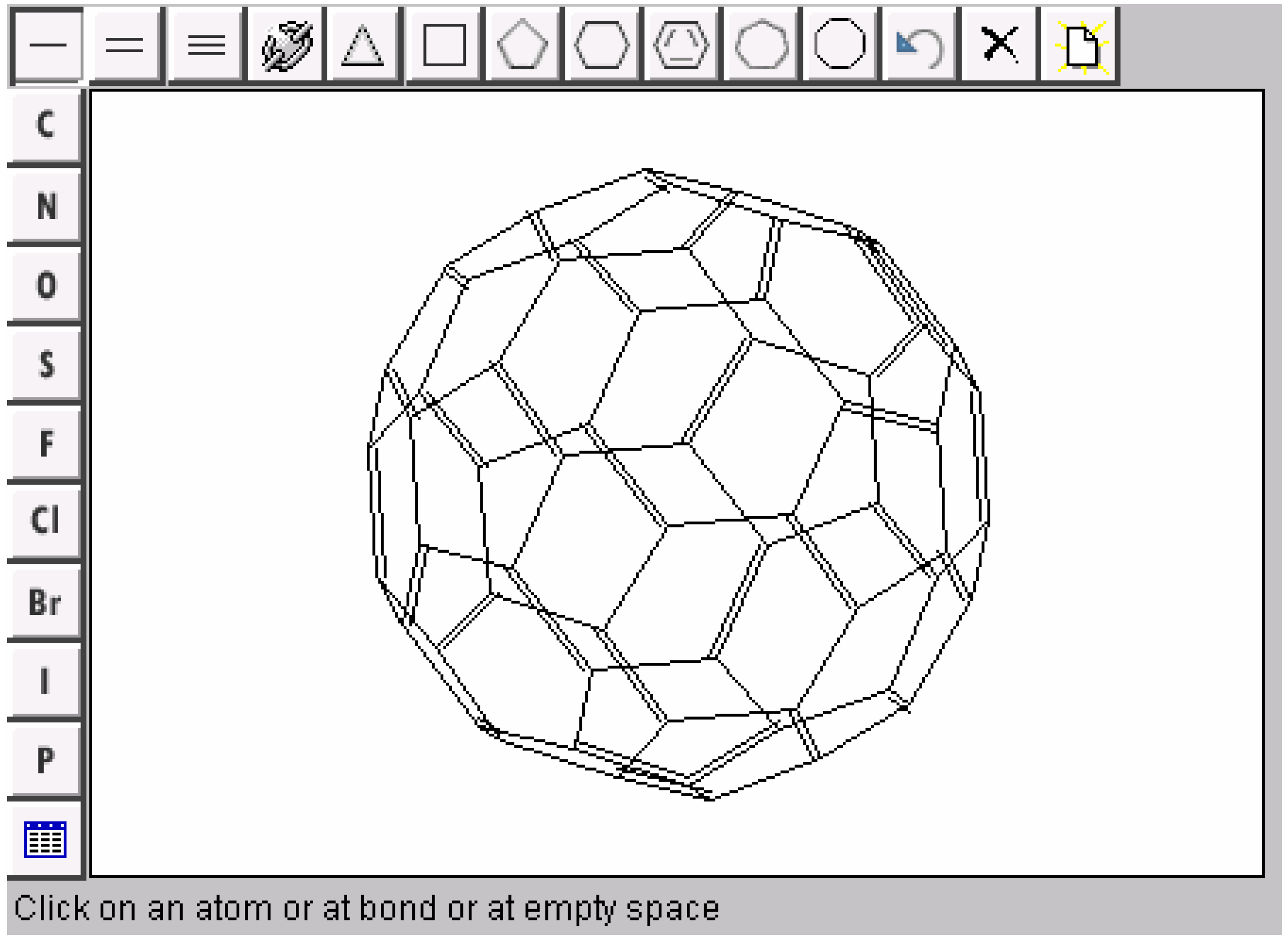
Conclusions
Supplementary materials
Acknowledgments
References and Notes
- Rzepa, H.S.; Cahser, O.; Leach, C. Recent Applications of Hyperactive Chemistry and the World-Wide-Web: Towards an Integrated Chemistry Information Environment. New Initiatives in Chemical Education: An On-line Symposium, June 3 - July 19, 1996. available at http://www.ch.ic.ac.uk/rzepa/cc96/cc96_intro.html accessed December 2005.
- Applets and visualization. http://www.morechemistry.com/links/Applets_and_Visualizations.html accessed December 2005.
- CAS. http://www.cas.org accessed December 2005.
- International Union of Pure and Applied Chemistry (IUPAC). Nomenclature of Organic Chemistry; Rigaudy, J., Klesney, S.P., Eds.; Pergamon Press: Oxford, U.K., 1979. [Google Scholar]
- ACD/Name. http://www.acdlabs.com/products/name_lab/name/ accessed December 2005.
- MDL/CrossFire. http://www.mimas.ac.uk/crossfire/autonom.html accessed December 2005.
- ACD/ChemSketch 8.17. http://www.acdlabs.com/products/chem_dsn_lab/chemsketch/ accessed December 2005.
- http://www.cheminnovation.com/products/nameexpert.asp accessed March 2006.
- Smith, E.G. Wisswesser-Line Formula Chemical Notation; McGraw-Hill: New York, 1968. [Google Scholar]
- Ash, S.; Cline, M.A.; Homer, R.W.; Hurst, T.; Smith, G.B. SYBYL line notation (SLN): A versatile language for chemical structure representation. J. Chem. Inf. Comput. Sci. 1997, 37, 71–79. [Google Scholar] [CrossRef]
- Weininger, D. SMILES a Chemical Language and Information System. 1. Introduction to Methodology and Encoding Rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Stein, S.E.; Heller, S.R.; Tchekhovskoi, D. An Open Standard for Chemical Structure Representation: The IUPAC Chemical Identifier. In Proceedings of the 2003 International Chemical Information Conference (Nimes), Infonortics, 2003; pp. 131–143.
- http://cactus.nci.nih.gov/services/translate/trans_info.html accessed December 2005.
- Weininger, D.; Weininger, A.; Weininger, J.L. SMILES 2. Algorithm for Generation of Unique SMILES Notation. J. Chem. Inf. Comput. Sci. 1989, 29, 97–101. [Google Scholar] [CrossRef]
- Trepalin, S.V.; Yarkov, A.V.; Dolmatova, L.M.; Zefirov, N.S.; Finch, S.A.E. WinDat: An NMR Database Compilation Tool, User Interface and Spectrum Libraries for Personal Computers. J. Chem. Inf. Comput. Sci. 1995, 35, 405–411. [Google Scholar] [CrossRef]
- Rovner, S.L.; Washington, C. Chemical ‘Naming’ method unveiled. Chem. Eng. News 2005, 83, 39–40. [Google Scholar]
- http://www.iupac.org/inchi accessed December 2005.
- Prasanna, M. D.; Vondrasek, J.; Wlodawer, A.; Bhat, T.N. Application of InChI to Curate, Index, and Query 3-D Structures. Proteins 2005, 60, 1–4. [Google Scholar]
- Gakh, A.A.; Burnett, M.N. Modular Chemical Descriptor Language (MCDL): Composition, Connectivity, and Supplementary Modules. J. Chem. Inf. Comput. Sci. 2001, 41, 1494–1499. [Google Scholar] [CrossRef] [PubMed]
- Rzepa, H.; Tonge, A. VchemLab: A Virtual Chemistry Laboratory. The Storage, Retrieval, and a Display of Chemical Information Using Standard Internet Tools. J. Chem. Inf. Comput. Sci. 1998, 38, 1048–1053. [Google Scholar]
- Csizmadia, F.J. Chem: Java Applets and Modules Supporting Chemical Database Handling from Web Browsers. J. Chem. Inf. Comput. Sci. 2000, 40, 323–324. [Google Scholar] [CrossRef] [PubMed]
- Ertl, P.; Jacob, O. WWW-based chemical information system. THEOCHEM 1997, 419, 113–120. [Google Scholar] [CrossRef]
- Krause, S.; Willighagen, E.; Steinbeck, C. JChemPaint-Using the Collaborative Forces of the Internet to Develop a Free Editor for 2D Chemical Structures. Molecules 2000, 5, 93–98. [Google Scholar]
- Steinbeck, C.; Han, Y.; Kuhn, S.; Horlacher, O.; Luttman, E.; Willighagen, E. The Chemistry Development Kit (CDK): An Open-Source Java Library for Chemo- and Bioinformatics. J. Chem. Inf. Comput. Sci. 2003, 43, 493–500. [Google Scholar]
- http://www.chemaxon.com/products.html accessed March 2006.
- Dalby, A.; Hourse, J.G.; Hounshell, W.D.; Gurchurst, A.K.I.; Grier, D.L.; Leland, B.A.; Laufer, J. Description of several chemical structure file formats used by computer program developed at Molecular Design Limited. J. Chem. Inf. Comput. Sci. 1992, 32, 244–255. [Google Scholar] [CrossRef]
- Bremser, W. HOSE—a novel substructure code. Anal. Chim. Acta 1978, 103, 355–365. [Google Scholar] [CrossRef]
- JChemPaint applet demo. http://jchempaint.sourceforge.net/applet accessed December 2005.
- Sun Java Run Time Enviroment. http://java.sun.com/j2se/downloads.html accessed December 2005.
- http://directory.google.com/Top/Computers/Programming/Languages/Java/Development_Tools/Obfuscators/ accessed December 2005.
- http://java.sun.com/j2se/1.3/docs/guide/extensions/spec.html accessed December 2005.
- Mayer, I. Charge, bond order and valence in the abinitio SCF theory. Chem. Phys. Lett. 1983, 97, 270–274. [Google Scholar] [CrossRef]
- Mayer, I. Comments on the quantum-theory of valence and bonding - choosing between alternative definitions. Chem. Phys. Lett. 1984, 110, 440–444. [Google Scholar] [CrossRef]
- Cioslowski, J.; Mixon, S.T. Covalent Bond Orders in the Topological Theory of Atoms in Molecules. J. Am. Chem. Soc. 1991, 113, 4142–4145. [Google Scholar] [CrossRef]
- Baber, J.C.; Hodgkin, E.E. Automatic assignment of chemical connectivity to organic molecules in the Cambridge Structural Database. J. Chem. Inf. Comput. Sci. 1992, 32, 401–406. [Google Scholar] [CrossRef]
- Helson, H.E. Structure Diagram Generation. Rev. Comput. Chem. 1999, 13, 313–398. [Google Scholar]
- Fricker, P.C.; Gastreich, M.; Rarey, M. Automated Drawing of Structural Molecular Formulas under Constraints. J. Chem. Inf. Comput. Sci. 2004, 44, 1065–1078. [Google Scholar] [CrossRef]
- MDL™ ISIS Draw 2.5. http://www.mdl.com/products/framework/isis_draw/index.jsp accessed: December 2005.
- Cambridge ChemDraw Ultra 8.0. http://www.cambridgesoft.com/products/family.cfm?FID=2 accessed December 2005.
- http://cdk.sf.net/api/org/openscience/cdk/layout/TemplateHandler.html and http://cdk.sf.net/api/org/openscience/cdk/layout/StructureDiagramGenerator.html accessed December 2005.
- Figueras, J. Ring Perception Using Breadth-First Search. J. Chem. Inf. Comput. Sci. 1996, 36, 986–991. [Google Scholar] [CrossRef]
- MCDL applet. http://www.zelinsky.ru/mcdl/mcdl.html.
- Source codes. https://sourceforge.net/projects/mcdl.
- Schnur, D. Design and Diversity Analysis of Large Combinatorial Libraries Using Cell-Based Methods. J. Chem. Inf. Comput. Sci. 1999, 39, 36–45. [Google Scholar]
- Bajorath, J. Selected Concepts and Investigations in Compound Classification, Molecular Descriptor Analysis, and Virtual Screening. J. Chem. Inf. Comput. Sci. 2001, 41, 233–245. [Google Scholar]
© 2006 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Trepalin, S.V.; Yarkov, A.V.; Pletnev, I.V.; Gakh, A.A. A Java Chemical Structure Editor Supporting the Modular Chemical Descriptor Language (MCDL). Molecules 2006, 11, 219-231. https://doi.org/10.3390/11040219
Trepalin SV, Yarkov AV, Pletnev IV, Gakh AA. A Java Chemical Structure Editor Supporting the Modular Chemical Descriptor Language (MCDL). Molecules. 2006; 11(4):219-231. https://doi.org/10.3390/11040219
Chicago/Turabian StyleTrepalin, Sergei V., Alexander V. Yarkov, Igor V. Pletnev, and Andrei A. Gakh. 2006. "A Java Chemical Structure Editor Supporting the Modular Chemical Descriptor Language (MCDL)" Molecules 11, no. 4: 219-231. https://doi.org/10.3390/11040219
APA StyleTrepalin, S. V., Yarkov, A. V., Pletnev, I. V., & Gakh, A. A. (2006). A Java Chemical Structure Editor Supporting the Modular Chemical Descriptor Language (MCDL). Molecules, 11(4), 219-231. https://doi.org/10.3390/11040219