Abstract
A series of previously unavailable derivatives of 2-alkyl- and 2-benzylderivatives of oxazolo[3,2-a]pyridines III were obtained via tandem ring opening and ring closure from stable mesoionic 3-acyloxazolo[3,2-a]pyridinium-2-olates I. The key intermediates of this tandem transformation are N-(b-oxoethyl)pyridones-2 II obtained by Dakin-West acylation of (pyridone-2-yl-1)acetic acid. An example of further utilization of this strategy is illustrated by preparation of unknown 2-benzylimidazo[1,2-a]pyridine from the salt I and ammonia.
Introduction
The oxazolo[3,2-a]pyridinium system has attracted our attention due to its ambident behavior toward nucleophiles and the ability to undergo opening of either the pyridine or oxazole ring forming various azolopyridines or hetaryldienes (Scheme 1, see [1,2,3,4] and review [5]):
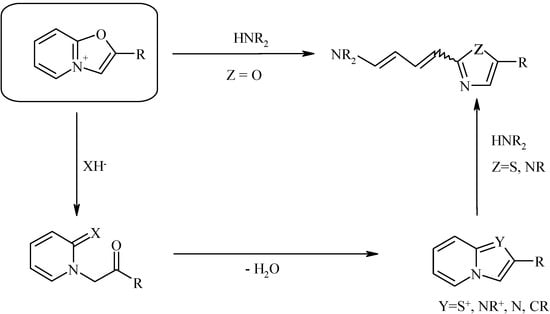
Scheme 1.
The most efficient route to the oxazolo[3,2-a]pyridinium ring system is the method of Bradsher [6] involving the cyclocondensation of 2-halopyridinium salts (A) or pyridones-2 (B) bearing a phenacyl-group at the nitrogen atom:
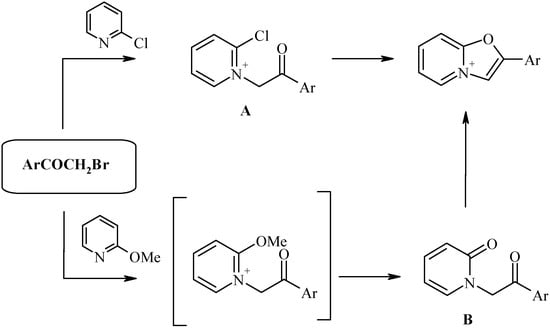
Scheme 2.
The intermediates A and B in turn could be easily prepared by alkylation of 2-substituted pyridines with α-bromoketones. This strategy gives the best results if aromatic bromoketones (phenacyl bromides) are used, leading to 2-aryl derivatives of the oxazolopyridinium salts.
2-Alkyl(aralkyl) derivatives of oxazolopyridines C would be interesting target compounds capable to further transformations. However, it is evident that the Bradsher method (e.g. cyclization of oxoalkylpyridones D) is hardly to apply for preparation of this class. Indeed, the precursors of the pyridones D would be the unsymmetrical bromoketones E, that are difficult (or even impossible) to prepare by selective bromination of methylalkylketones F.
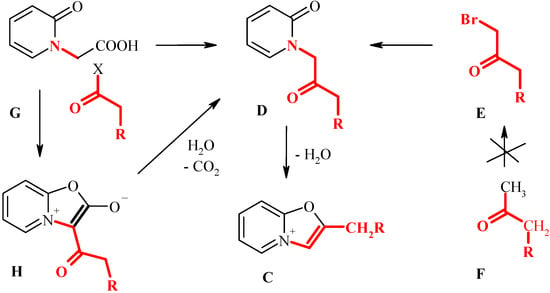
Scheme 3.
Key intermediates D can be considered as a specific type of α-aminoketones (fragment shown by red on Scheme 3). Therefore, an alternative strategy to compounds D would be Dakin-West reaction [7] i.e. acylation of a corresponding aminoacid, in our case - pyridone-2-acetic acid G. Meanwhile, the reaction of the acid G with acyl chlorides and anhydrides leads to stable mesoionic bicycles (fused munchnones) H [8,9,10]. The possibility of conversion of bicyclic munchnones H to the pyridones D has received little attention. Thus, Boyd and Wright reported that munchnones H may undergo hydrolytical cleavage under the action of hot water [9]. In our early work [11,12,13] we have observed that some bicyclic munchones can undergo cleavage of the oxazolone fragment in the acidic media.
In the present communication we report our success in applying the sequence G-H-D-C as a new strategy to prepare previously unknown class of 2-benzyl and 2-alkyl derivatives of oxazolo[3,2-a]-pyridines.
Results and Discussion
Acylation of oxazolo[3,2-a]pyridinium-2-olate.
At the initial phase we have synthesized broad series of munchnones of the type H according to Scheme 4. According to literature data the munchnones I were prepared by acylation of unstable key intermediate Ih, which in turn was generated at low temperature (0°C) [9] (Scheme 4). The known instability of the unsubstitued munchone Ih is connected with its ability to undergo self-acylation leading to undesired dimer Ii. The presence of dimer Ii (poorly soluble in most of solvents) makes difficulties in purification of mesoionic compounds Ia-g. Nevertheless, we found, that considerable decrease of the temperature (up to -45oC) depressed the dimerization process, and the desired pure munchnones I can be easily obtained by this modified protocol.
1H-NMR spectra of fused munchnones are shown in Table 1. Specific feature for the entire series of mesoionic structures I is the strong downfield shift of the signal of proton H-5 (observed at 9.8-9.9 ppm) due to the presence of magnetically anisotropic acyl group at peri-position 3.
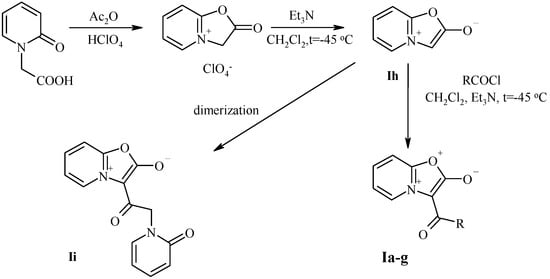
Scheme 4.

Table 1.
Characteristics of mesoionic compounds I
| № | R | Yield, % | Mp, °C | Chemical shift, p.p.m.; J, Hz | ||||
|---|---|---|---|---|---|---|---|---|
| H5 | H6 | H7 | H8 | RCO | ||||
| Ia | 4-MeOPhCH2 | 64 | 171-173 | d; 9.86; J56=6.4 | m; 7.51 | m; 7.96 | d; 7.61; J78=8.6 | m; 7.24 (2H); m; 6.77 (2H); s; 3.99 (2H); s; 3.74 (3H) |
| Ib | 4-ClPhCH2 | 60 | 194-196 | d; 9.83; J56=6.3 | m; 7.52 | m; 7.99 | d; 7.64; J78=8.4 | m; 7.34 (2H); m; 7.24 (2H); s; 4.06 (2H) |
| Ic | 4-NO2PhCH2 | 82 | 225-227 | m; 9.81 | m; 7.55 | m; 8.01 | m; 7.65 | m; 8.12 (2H); m; 7.59 (2H); s; 4.24 (2H) |
| Id | Me | 78 | 170-172* | d; 9.78; J56=6.5 | m; 7.55 | m; 8.01 | d; 7.68; J78=8.5 | s; 2.36 (2H) |
| Ie | Et | 48 | 153-155** | d; 9.90 J56=6.4 | m; 7.52 | m; 7.96 | d; 7.60; J78=8.7 | q; 2.75 (2H); J=8.0; t; 1.12 (3H); J=7.0 |
| If | Pr | 62 | 136-137 | d; 9.83; J56=6.2 | m; 7.55 | m; 8.00 | d; 7.68; J78=9.0 | t; 2.74 (2H); J=7.7; m; 1.64 (2H); t; 0.94 (3H); J=7.1 |
| Ig | Bu | 86 | 140-142 | d; 9.84; J56=6.2 | m; 7.55 | m; 8.00 | d; 7.67; J78=8.0 | t; 2.74 (2H); J=7.3; m; 1.59 (2H); m; 1.35 (2H); t; 0.91; (2H); J=7.1 |
*Lit. Mp 170-171 [8]; **lit. Mp 146-147.5 [8].
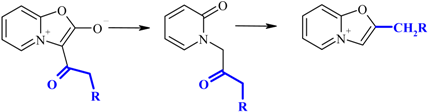
Hydrolytic cleavage of oxazolones I.
Our attempt to perform the hydrolytical cleavage of mesoionic oxazolones either in pure water or in strong sulfuric acid were unsuccessful. In the first case no indication of the reaction was observed, and in the second case undesired side reactions occurred (in particular, during the hydrolysis of munchnone Ib the strong smell of phenylacetic acid indicated that the concurrent cleavage of the acyl group might take place). After several attempts we have found that the optimal condition of hydrolysis of munchones I is the use of diluted (0.06 M) aqueous solution of hydrochloric acid leading to desired products II in good yields.
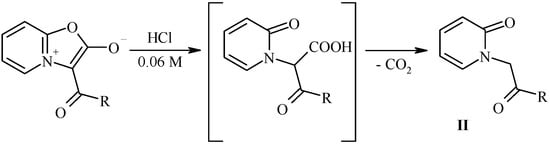
Scheme 5.
Table 2.
Characteristics of pyridones II
| № | R | Yield, % | Mp, °C | Chemical shift, p.p.m.; J, Hz | ||||
|---|---|---|---|---|---|---|---|---|
| H3 | H4 | H5 | H6 | RCOCH2 | ||||
| IIa | 4-MeOPhCH2 | 86 | 84-85 | d; 7.42; J34=7.0 | m; 7.35 | m; 6.13 | d; 6.35; J56=8.6 | m; 7.14 (2H) ;m; 6.83 (2H); s; 4.76 (2H); s; 3.76 (5H) |
| IIb | 4-ClPhCH2 | 90 | 133-135 | d; 7.45; J34=7.2 | m; 7.37 | m; 6.15 | d; 6.36; J56=9.5 | m; 7.30 (2H); m; 7.22 (2H); s; 4.81 (2H); s; 3.86 (2H) |
| IIc | 4-NO2PhCH2 | 91 | 170-172 | m; 7.50 | m; 7.37 | m; 6.16 | d; 6.36; J56=9.0 | m; 8.16 (2H); m; 7.50 (2H); s; 4.87 (2H); s; 4.06 (2H) |
| IId | Me | 67 | - | d; 7.44; J34=6.9 | m; 7.36 | m; 6.14 | d; 6.36; J56=9.2 | s; 4.72 (2H); s; 2.18 (3H) |
| IIe | Et | 52 | - | d; 7.46; J34=7.0 | m; 7.36 | m; 6.15 | d; 6.35; J56=8.7 | s; 4.71 (2H); q; 2.52 (2H); J=7.1; t; 1.05 (3H); J=7.1 |
| IIf | Pr | 59 | - | d; 7.45; J34=7.1 | m; 7.36 | m; 6.14 | d; 6.34; J56=9.2 | s; 4.70 (2H); t; 2.45 (2H); J=7.9; m; 1.61 (2H); t; 0.95 (3H); J=7.1 |
| IIg | Bu | 76 | - | d; 7.44; J34=6.6 | m; 7.36 | m; 6.14 | d; 6.35; J56=8.6 | s; 4.70 (2H); t; 2.49 (2H); J=7.8; m; 1.56 (2H); m; 1.35 (2H); t; 0.93 (3H); J=7.1 |
It should be mentioned, that the formed pyridones II are more water soluble than the parent munchones, therefore in the course of reaction the initial suspension was usually dissolved (the exception was only the case of 4-nitrobenzyl derivatives Ic, IIc, both poorly soluble in water). In the 1H-NMR spectra of pyridones II the signals of protons of N-CH2-CO group (4.7-4.8 ppm) are usually clear distinguishable from the signals of protons from another methylene group RCH2CO belonging to aliphatic or aromatic fragment (Table 2).
Synthesis of 2-substitued oxazolo[3,2-a]pyridinium perchlorates.
According to Bradsher [6] the cyclodehydratation of N-phenacylpyridones B to 2-aryloxazolo-pyridinium salts can be achieved by the action of concentrated sulfuric acid. However, all our attempts to perform conversion of N-(b-oxoethyl)pyridones-2 II to the desired 2-alkyloxazolopyridinium salts III failed. In all cases the degree of conversion has never exceeded 10-15%. Probably in these cases the completeness of the reaction was connected with the reversibility of the ring closure, and complete removal of water would shift the equilibrium. Indeed, the use of 8% oleum (completely capturing all the water) led to desired products III in good yields.
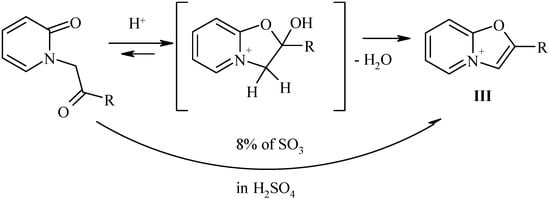
Scheme 6.
The only exception was the case of pyridone IIa (with an anisyl group) that in reaction with oleum or concentrated sulfuric acid gave a red oil, probably due to sulfonation of the benzene ring [6]. In the 1H-NMR spectra of the aromatic bicycles III novel aromatic singlet of the oxazole ring was observed at 8.5-8.7 ppm, and the most downfield doublet (at 9.2 ppm) was the signal of a-pyridinium proton H-5.
Reaction of 2-(4’-chlorobenzyl)oxazolo[3,2-a]pyridinium perchlorate (IIIb) with ammonia.
In order to investigate the reactivity of the salts III we performed simple test experiment with ammonia. Reaction of salt IIIb with solution of anhydrous ammonia in DMF resulted in formation of corresponding imidazo[1,2-a]pyridine IV (Scheme 7, Table 3), the first representative of a previously unknown class of 2-benzylimidazopyridines. Clearly, this structural type would be difficult to obtain by standard Chichibabin reaction from the practically unavailable bromoketone E (R = p-ClPh). Our attempts to perform this reaction in ethanolic or aqueous ammonia resulted in a number of byproducts.
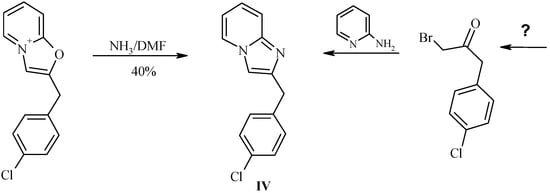
Scheme 7.
Table 3.
Characteristics of the salts III and imidazopyridine IV
| № | R | Yield, % | Mp, °C | Chemical shift, p.p.m.; J, Hz | |||||
|---|---|---|---|---|---|---|---|---|---|
| H3 | H5 | H6 | H7 | H8 | R | ||||
| IIIb | 4-ClPhCH2 | 87 | 162-164 | s; 8.64 | d; 9.15; J56=6.1 | m; 8.45 | m; 7.90 | d; 8.27; J78=9.0 | m; 7.44 (2H); m; 7.35 (2H); s; 4.40 (2H) |
| IIIc | 4-NO2PhCH2 | 84 | 132-134 | s; 8.71 | d; 9.18; J56=6.7 | m; 8.46 | m; 7.91 | d; 8.30; J78=9.0 | m; 8.21 (2H); m; 7.72 (2H); s; 4.60 (2H) |
| IIId | Me | 42 | 127-128* | s; 8.56 | d; 9.13; J56=6.3 | m; 8.44 | m; 7.90 | d; 8.29; J78=8.9 | s; 2.68 (3H) |
| IIIe | Et | 67 | 118-120 | s; 8.61 | d; 9.13; J56=6.1 | m; 8.45 | m; 7.91 | d; 8.29; J78=8.9 | q; 3.03 (2H); J=7.8; t; 1.40 (3H); J=7.7 |
| IIIf | Pr | 74 | 98-100 | s; 8.65 | d; 9.16; J56=5.9 | m; 8.46 | m; 7.91 | d; 8.30; J78=8.9 | t; 2.98 (2H); J=7.1;m; 1.85; (2H); t; 1.09; (3H); J=8.1 |
| IIIg | Bu | 48 | - | s; 8.68 | d; 9.19; J56=6.3 | m; 8.46 | m; 7.92 | d; 8.28; J78=8.9 | t; 2.99 (2H); J=7.3; m; 1.78 (2H); m; 1.48 (2H); t; 0.98 (3H); J=7.3 |
| IV | 4-ClPhCH2 | 40 | 162-164 | s; 7.49 | d, 8.32, J56=7.1 | m, 7.11 | m, 6.73 | d, 7.38, J78=9.3 | m, 7.30 (2H); m, 7.24 (2H); s, 4.01 (2H) |
*Lit. Mp 128 [6]
Conclusions
We have found new simple route to 2-substituted oxazolo[3,2-a]pyridinium salts which, in turn could be precursors of previously unavailable structures, e.g. 2-benzylimidazo[1,2-a]pyridines. The group CH2R at the imidazopyridine and oxazolopyridines originates from the residue of an organic acid.
Experimental
General
1H- and 13C-NMR spectra were obtained using a Bruker AC 400 NMR spectrometer and were recorded at 360 MHz. All reagents and chemicals were obtained from Acros or Merck and were used as received unless otherwise noted. Ether was purified by refluxing over sodium with benzophenone for 2-2.5 hours followed by distillation. Triethylamine was distilled over sodium hydroxide, sodium and finally phthalic anhydride under an argon atmosphere. Acetic anhydride was purified by distillation over sodium acetate. DMF was stirred with calcium hydride powder for 2-3 hours at 70-80ºC, decanted from the precipitate and distilled under reduced pressure. Acyl chlorides were prepared according to a standard procedure [14].
Preparation of 2,3-dihydro-2-oxooxazolo[3,2-a]pyridinium perchlorate.
N-(Pyridone-2-yl-1)acetic acid (6 g, 39 mmol) was suspended in acetic anhydride (40 mL, freshly distilled over sodium acetate) and 70% HClO4 (6 mL, 59 mmol) was carefully added dropwise at 30-40°C. After addition of all the HClO4 the reaction was stirred for 30 min and diluted with dry Et2O (50 mL). Product was filtered off, washed with dry Et2O (12×15 mL) and dried in vacuo. Yield 8.95 g (97%). m.p. 180-182 °C (Lit. [9] m.p. 178-180 °C).
Typical procedure for the preparation of mesoionic 3-acyloxazolo[3,2-a]pyridinium-2-olates (I).
A suspension of 2,3-dihydro-2-oxooxazolo[3,2-a]pyridinium perchlorate (7.0 g, 30 mmol) in dry CH2Cl2 (100 mL) was cooled to –60 °C and freshly distilled Et3N (14.1 mL, 101 mmol) was added dropwise at –58 to -52 °C. The thus obtained slightly yellow solution was treated with a solution of the corresponding acyl chloride (30 mmol) in dry CH2Cl2 (40 mL) at –58- to -52 °C. The suspension or solution obtained was allowed to warm to RT. The solvent was evaporated to dryness, and the residue was washed with water (2×25 mL), recrystallized from iso-propanol and dried on air. In the case of synthesis of Ic, after warming the suspension the precipitate was just filtered off and washed with iso-propanol (2×25 mL). Yields and NMR data for compounds I are given in Table 1.
Procedures for the preparation of N-alkylated pyridones (II)
N-(3-(4’-chlorophenyl)-β-oxopropyl)-2-pyridone (IIa)
Mesoionic compound Ia (8.7 g, 30 mmol) was suspended in a 0.06 M aqueous solution of HCl (500 mL) and refluxed for 8 h (TLC control). The hot aqueous phase was decanted from the brown oily mass and cooled to RT. The white needle-like crystals obtained were filtered off and washed with water (10 mL). The mother liquors were added to the oily mass and refluxed for 2 h until all the oil was dissolved and TLC shown full conversion of the starting material. The solution was cooled to RT, and the gray needles thus obtained were filtered off and recrystallized from iso-propanol.
N-(3-(4’-nitrophenyl)-β-oxopropyl)-2-pyridone (IIb)
Mesoionic compound Ib (6.1 g, 20 mmol) was suspended in 600 mL 0.06 M aqueous solution of HCl and refluxed for 8 h (TLC control). Hot solution was decanted from solid and cooled to RT. Obtained white needle crystals were filtered off and washed with water (10 mL). Mother liquor was added to residuary starting substance and refluxing was continued for 8 h. Procedure was repeated until TLC shown full conversion of starting product. Obtained precipitate was recrystallized from iso-propanol.
N-(3-(4’-methoxyphenyl)-β-oxopropyl)-2-pyridone (IIc)
Mesoionic compound Ic (7.0 g, 25 mmol) was suspended in 0.06 M aqueous solution of HCl (500 mL) and refluxed for 4 h. After all the starting substance was dissolved the solution was refluxed for 1 h (TLC control) and then cooled to RT. The obtained mixture was extracted with CHCl3 (3×100 mL), dried over Na2SO4. The extract was evaporated obtaining yellow oil. The residue was recrystallized from iso-propanol giving white needles.
Synthesis of N-β-oxoalkyl-2-pyridones IId-g
Mesoionic compound Id-g (25 mmol) was suspended in 0.06 M aqueous solution of HCl (250 mL) and refluxed for 2 h. After all the starting substance was dissolved and solution was refluxed for 1 h (TLC control) and then cooled to RT. Obtained mixture was extracted with CHCl3 (3×75 mL), dried over Na2SO4. The extract was evaporated to give a yellow oil. Yields and NMR data for compounds II are given in Table 2.
Typical procedure for the preparation of 2-substituted oxazolo[3,2-a]pyridinium perchlorates (III):
The N-β-oxoalkyl-pyridone-2 (II, 0.74 mmol) was dissolved in 8% oleum (2 mL) and kept for 20-25 hours. Then 70% HClO4 (0.15 mL, 1.5 mmol) was carefully added to the mixture and after 20 hours the solution was poured into vigorously stirred diethyl ether (200 mL). After decanting the oily residue was mixed again with a fresh portion of ether (200 mL); this procedure should be repeated until a pure white powder formed. The precipitate was filtered, washed with ether and dried in vacuum over P2O5. Yields and NMR data for compounds III are given in Table 3.
Reaction of 2-(4’-chlorobenzyl)oxazolo[3,2-a]pyridinium perchlorate with ammonia.
2-(4’-Chlorobenzyl)oxazolo[3,2-a]pyridinium perchlorate (2.4 mmol) was dissolved in anhydrous distilled DMF (3 mL) and mixed at -25°C with a solution (4 mL) prepared by saturation of DMF with gaseous ammonia (prepared at -60°C). The reaction mixture immediately turned to deep red color. This mixture was kept for 10 hours and then poured into cold water. The product precipitated as a yellow powder, which was separated, washed with water and recrystallized from petroleum ether. Yields and NMR are given in Table 3.
References
- Babaev, E. V.; Pasichnichenko, K. Yu.; Maiboroda, D. A. Hetarenes with a bridge nitrogen atom. 6. Ambident properties of the oxazolo[3,2-a]pyridinium nucleus in reactions with nucleophiles: prediction and experimentm. Chem. Heterocycl. Comp. (Engl. translation of Khim. Geterotsikl. Soedin.) 1997, 33, 338–342. [Google Scholar]
- Maiboroda, D. A.; Babaev, E. V.; Goncharenko, L. V. Synthesis and study of the spectral and pharmacological properties of 1-amino-4-(5-aryl-2-oxazolyl)-1,3-butadienes. Khim.-Farm. Zh. 1998, 32, 24–28. [Google Scholar]
- Bradsher, C. K.; Brandau, R. D.; Boliek, J. E.; Hough, T. L. The reaction of amines with 1-phenacyl-2-bromopyridinium salts. A new route to imidazo[1,2-a]pyridinium, oxazolo[3,2-a]pyridinium, and dihydropyrido[2,1-c]-as-triazinium salts. J. Org. Chem. 1969, 34, 2129–2133. [Google Scholar]
- Babaev, E.V.; Bozhenko, S.V.; Maiboroda, D.A. Synthesis of 1-nitro-2-phenylindolizine by the ring transformation of oxazolo[3,2-a]pyridinium salt in the reaction with nitromethane. Russ. Chem. Bull. (Engl.Transl.) 1995, 44, 2203. [Google Scholar]
- Babaev, E. V. Fused Munchnones in Recyclization Tandems. J. Heterocycl. Chem. (Lectures in Heterocyclic Chemistry) 2000, 37, 519–526. [Google Scholar]
- Bradsher, C. K.; Zinn, M. Oxazolo[3,2-a]pyridinium salts. J. Heterocycl. Chem. 1967, 4, 66–70. [Google Scholar]
- Dakin, H. D.; West, R. A general reaction of amino acids. II. J. Biol. Chem. 1928, 78, 745–757. [Google Scholar]
- Lawson, A.; Miles, D. H. Some new mesoionic compounds. J. Chem. Soc. 1959, 2865–2871. [Google Scholar] [CrossRef]
- Boyd, G. V.; Wright, P. H. Anhydro-2-hydroxyoxazolo[3,2-a]pyridinium hydroxide, a mesoionic oxazolone. J. Chem. Soc. (C). 1970, 10, 1485–1490. [Google Scholar]
- Petride, H.; Raileanu, D. Mesoionic oxazolones. II. Unstable non-acylated compounds. Rev. Roum. Chim. 1988, 33, 729–739. [Google Scholar]
- Babaev, E. V.; Orlova, I. A. Heterocycles with a bridgehead nitrogen atom. 7. Novel synthesis of the oxazolo[3,2-a]pyridinium cation by recyclization of a mesoionic precursor: confirmation of a computer prediction. Chem. Heterocycl. Comp. 1997, 33, 489–490. [Google Scholar]
- Rybakov, V. B.; Babaev, E. V.; Pasichnichenko, K. Yu.; Sonneveld, E. J. X-ray mapping in heterocyclic design: VI. X-ray diffraction study of 3-(isonicotinoyl)-2-oxooxazolo[3,2-a]pyridine and the product of its hydrolysis. Crystallogr. Rep. 2002, 47, 69–74. [Google Scholar]
- Rybakov, V. B.; Zhukov, S. G.; Babaev, E. V. 1,3-Di(2-oxopyridyl-1) acetone and Its Hydroperchlorate: Formation in the Hydrolysis of Annelated Munchnone and X-Ray Diffraction Study. Russ. J. Coord. Chem. 2000, 26, 670–676. [Google Scholar]
- Tietze, L. F.; Eicher, T. Reaktionen und synthesen im organish-chemishen praktikum und forschungslaboratorium; Thieme Verlag: Stuttgart-New York, 1991. [Google Scholar]
© 2005 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.