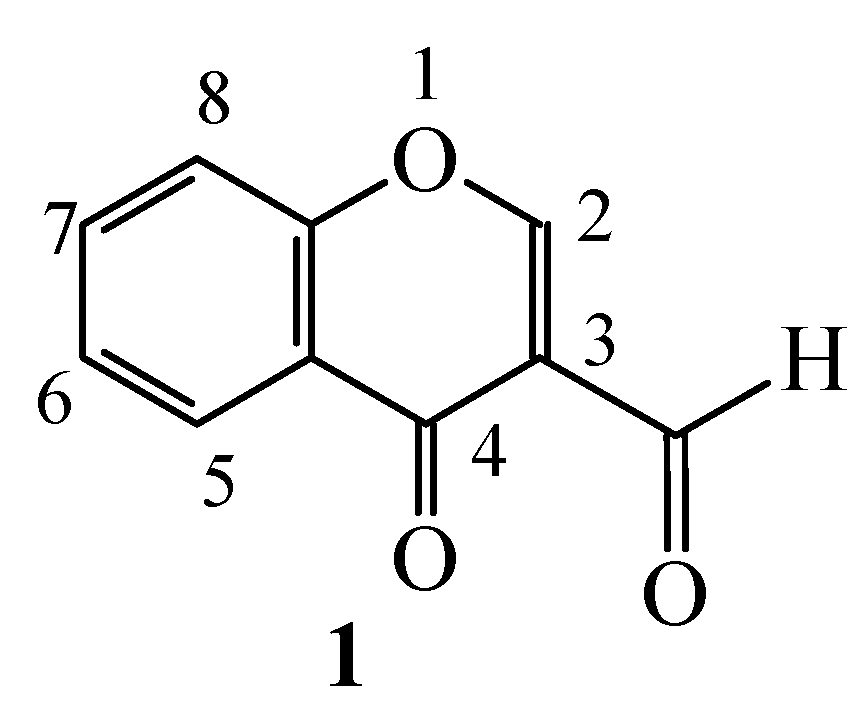
Reactions of 3-Formylchromone with Active Methylene and Methyl Compounds and Some Subsequent Reactions of the Resulting Condensation Products

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
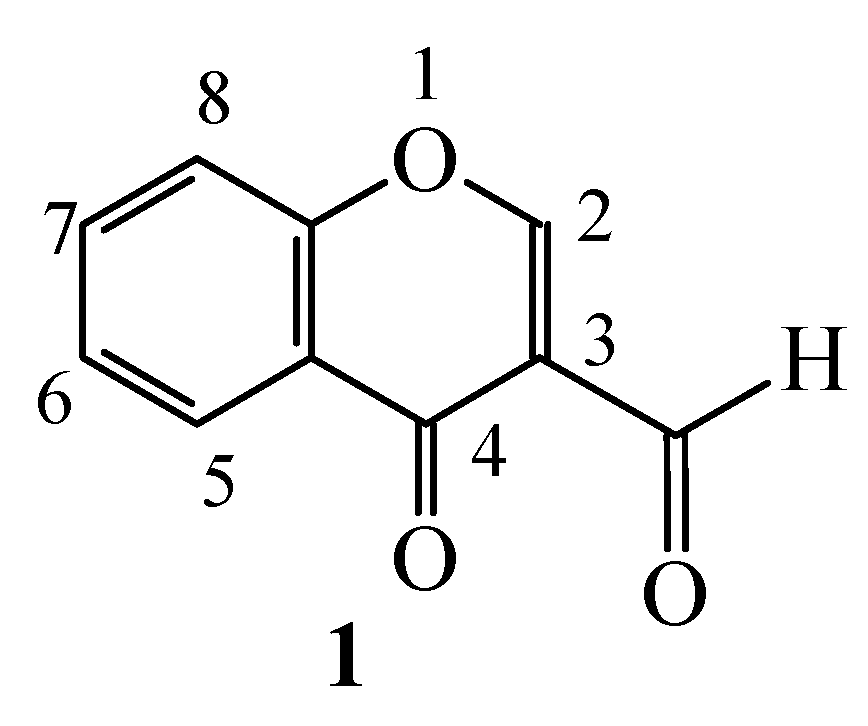
:Introduction
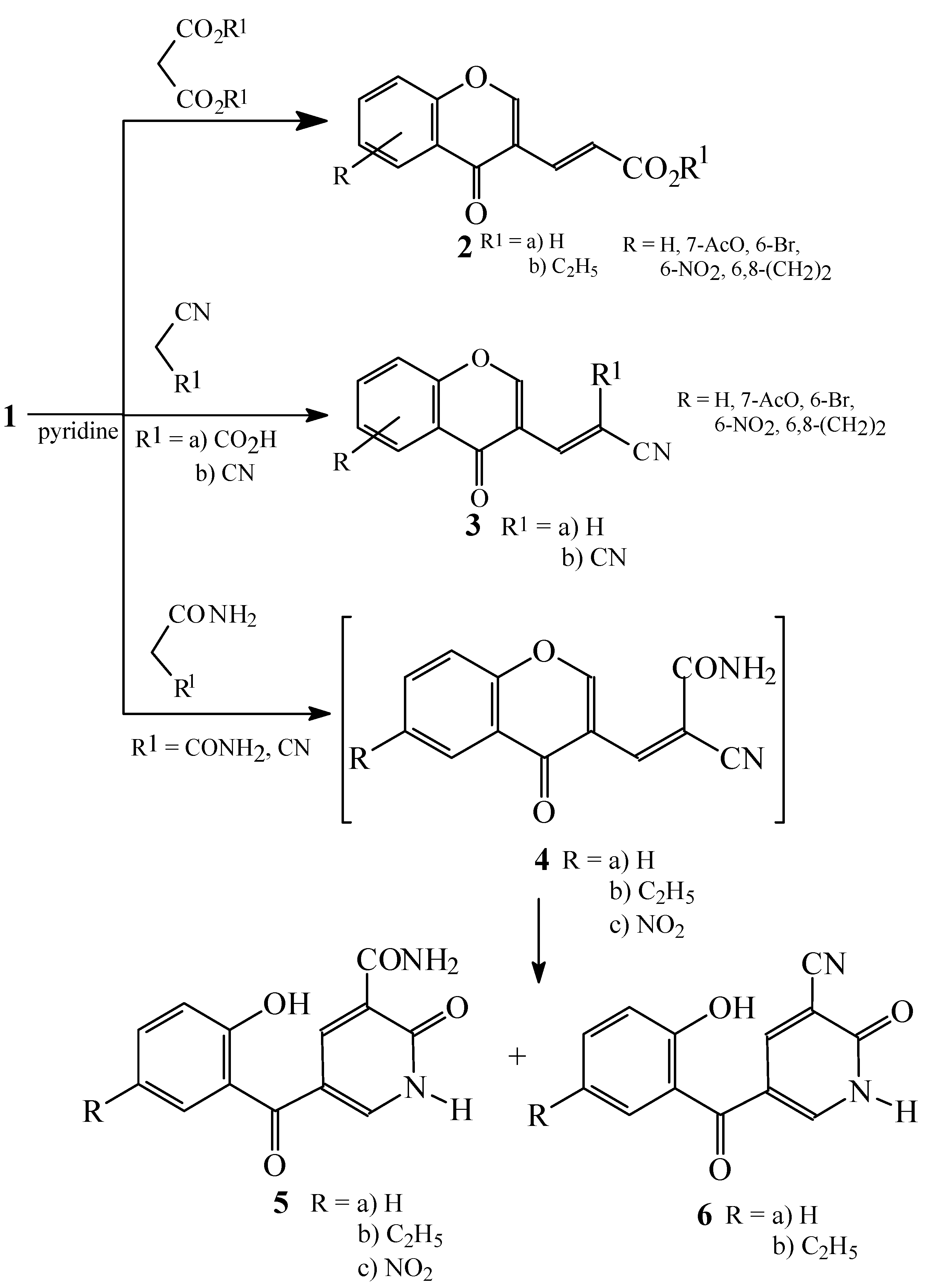
Condensations of 1 with active methylene compounds
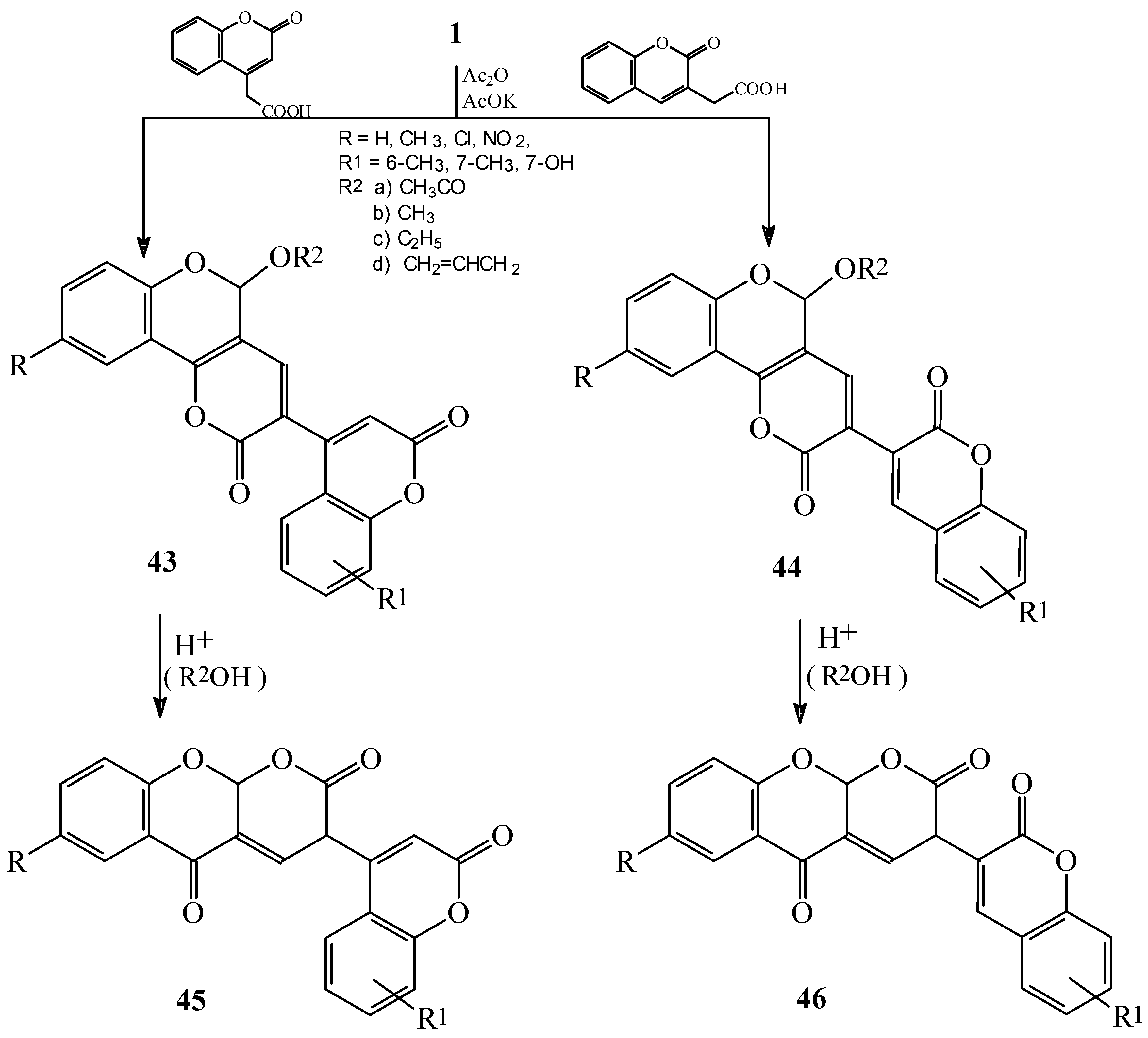
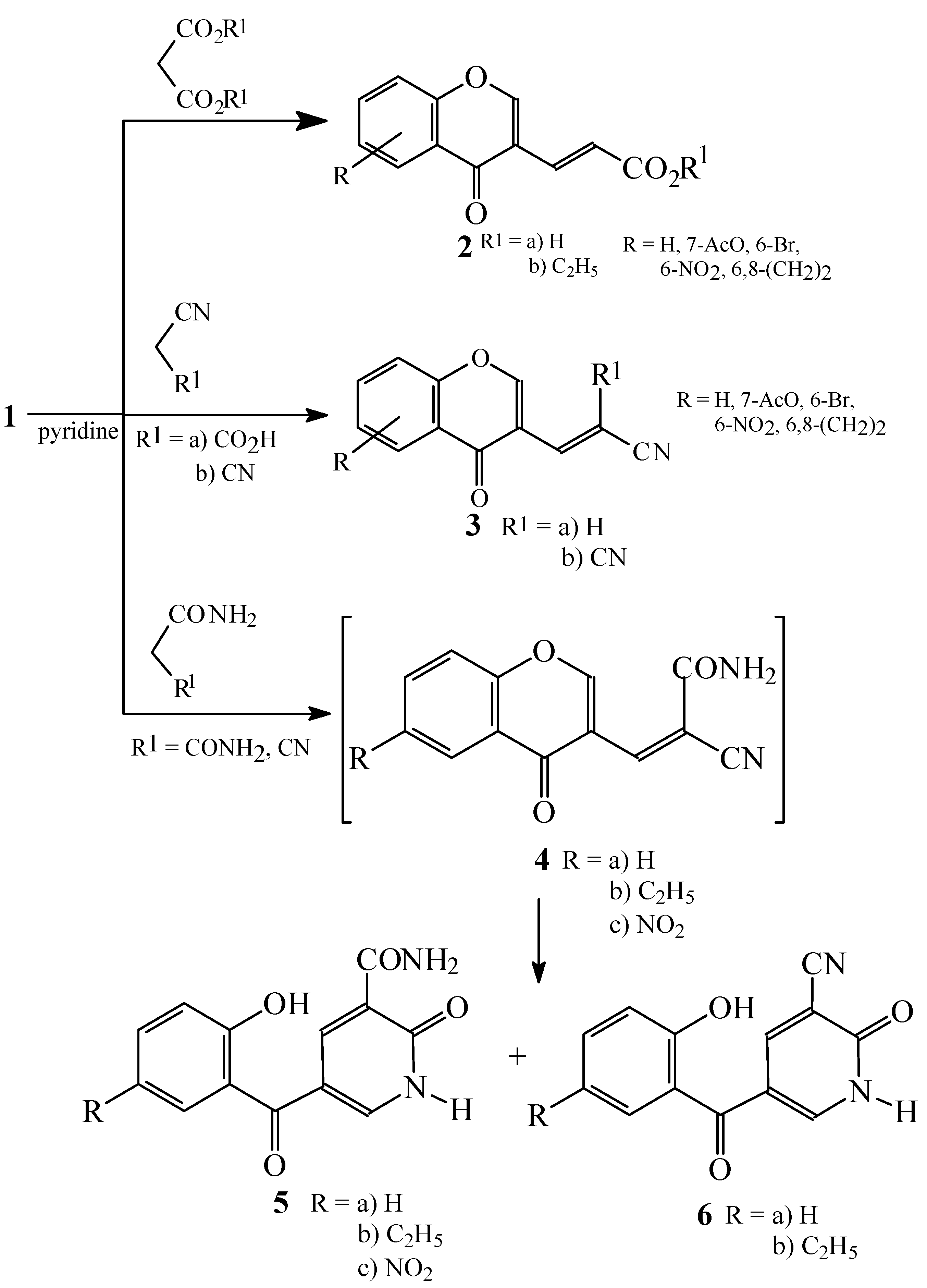
Condensations of 1 with malonic acid derivatives
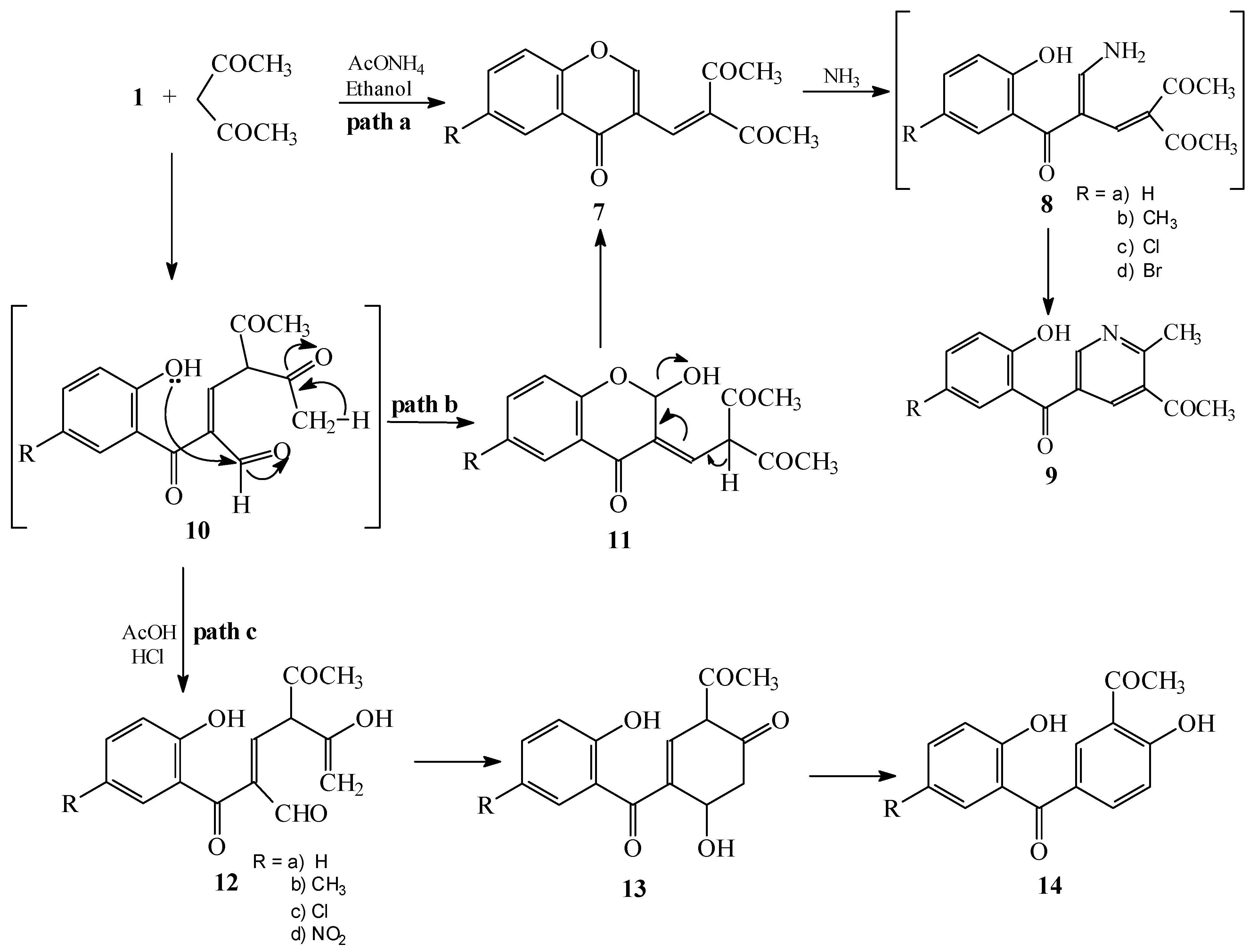
Condensations of 1 with 2,4-pentanedione
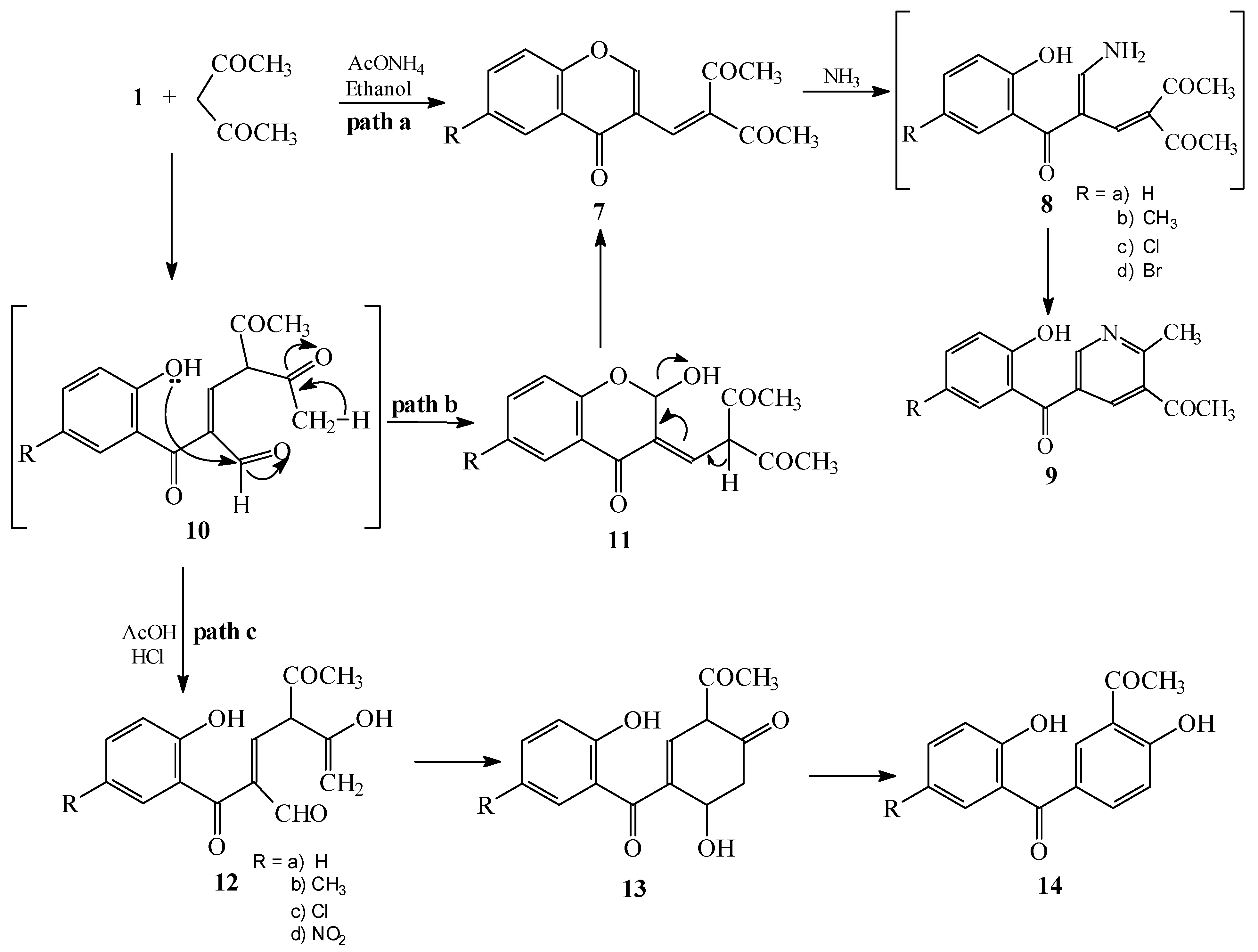
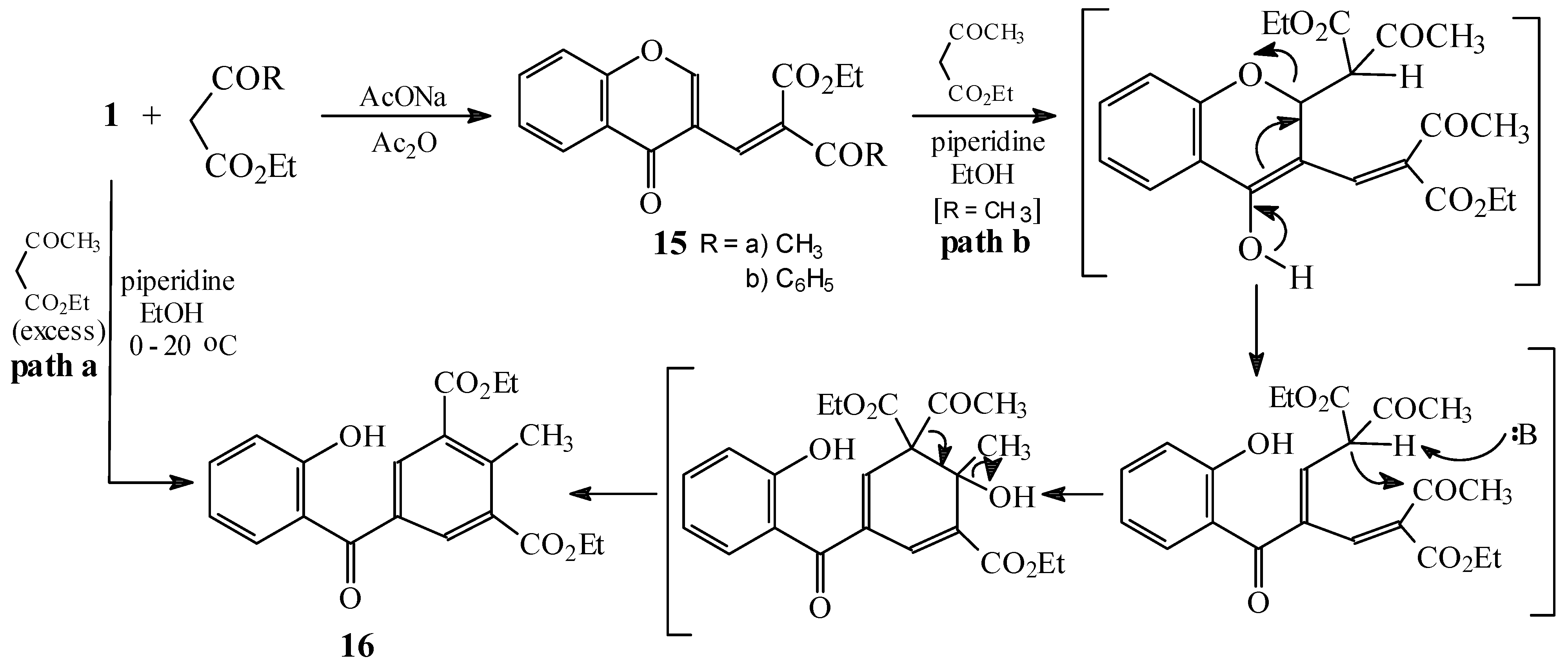
Condensations of 1 with ethyl acylacetates
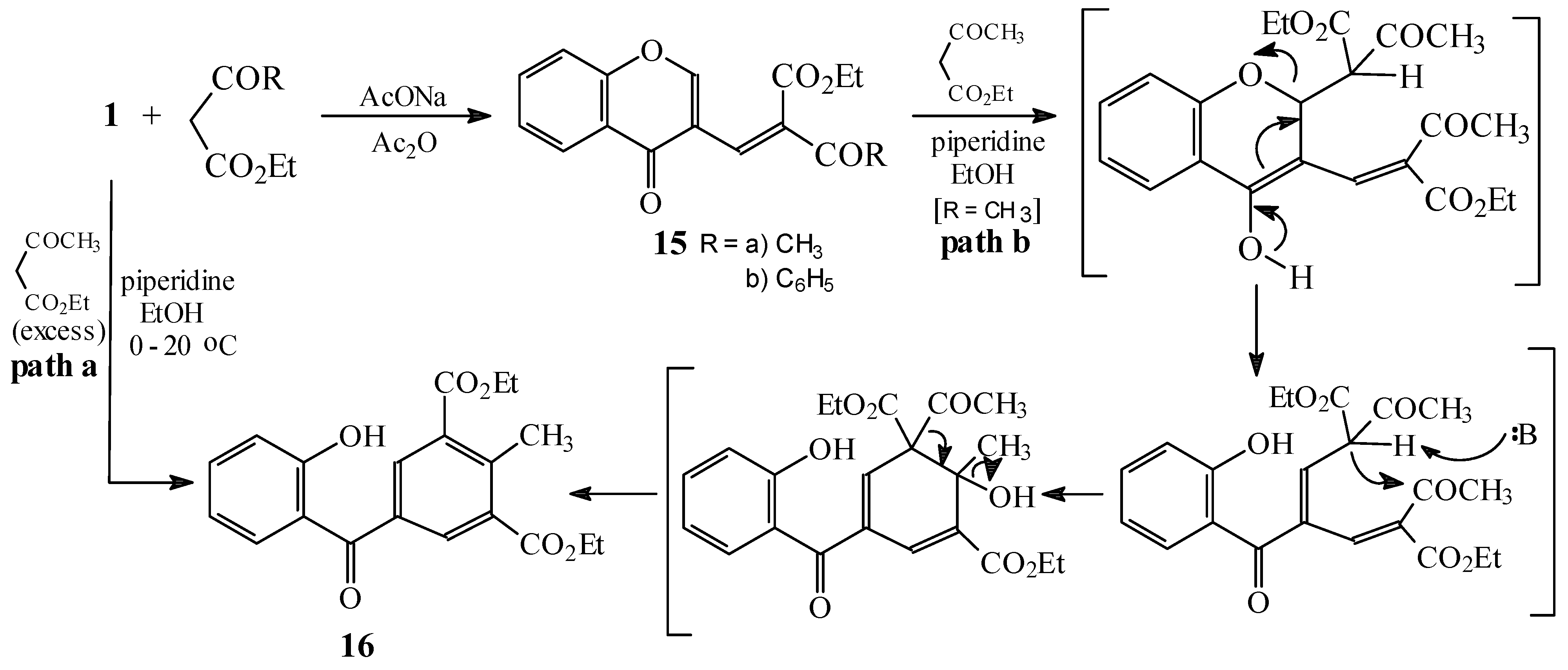
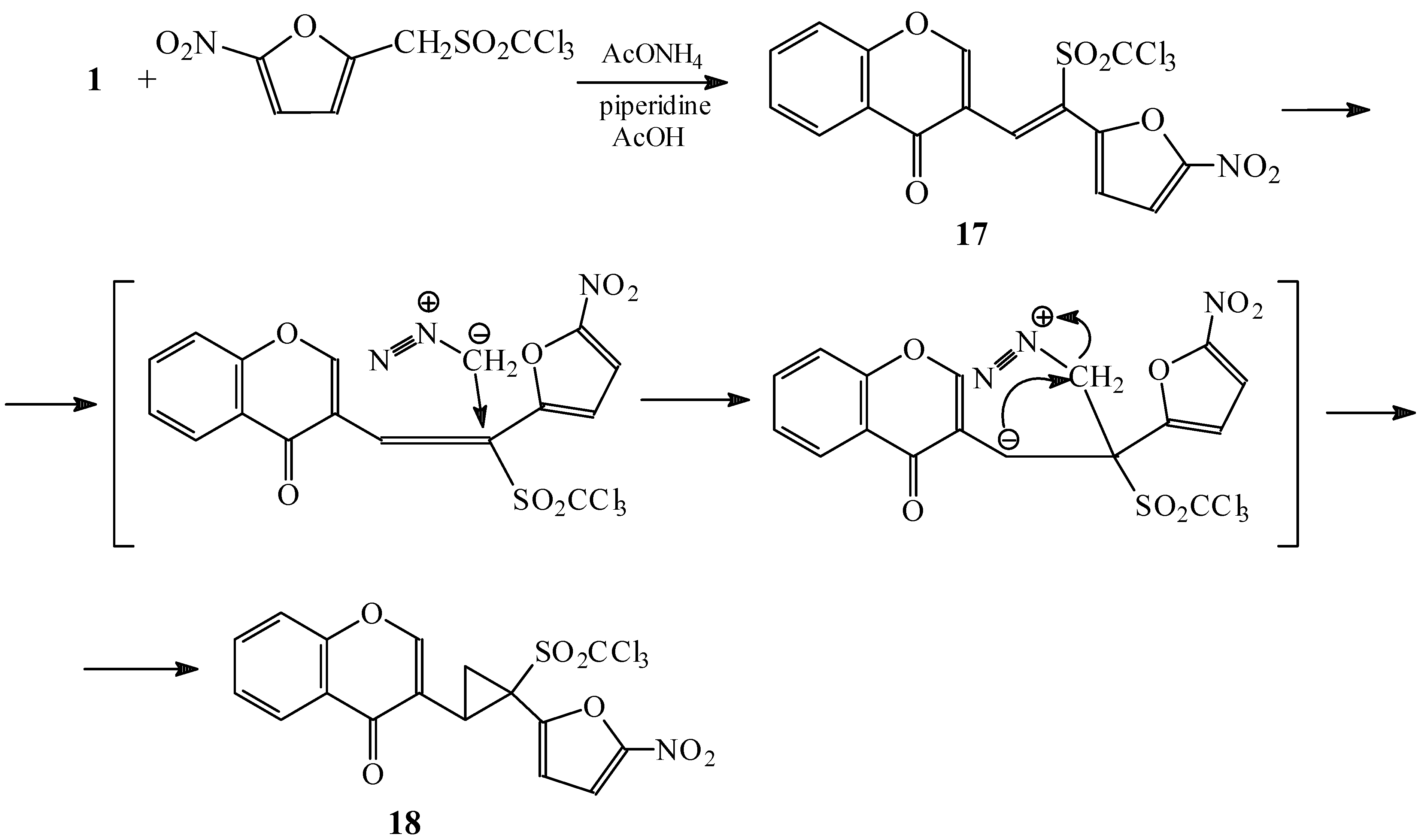
Condensations of 1 with 5-nitrofuryltrichloromethylsulphone
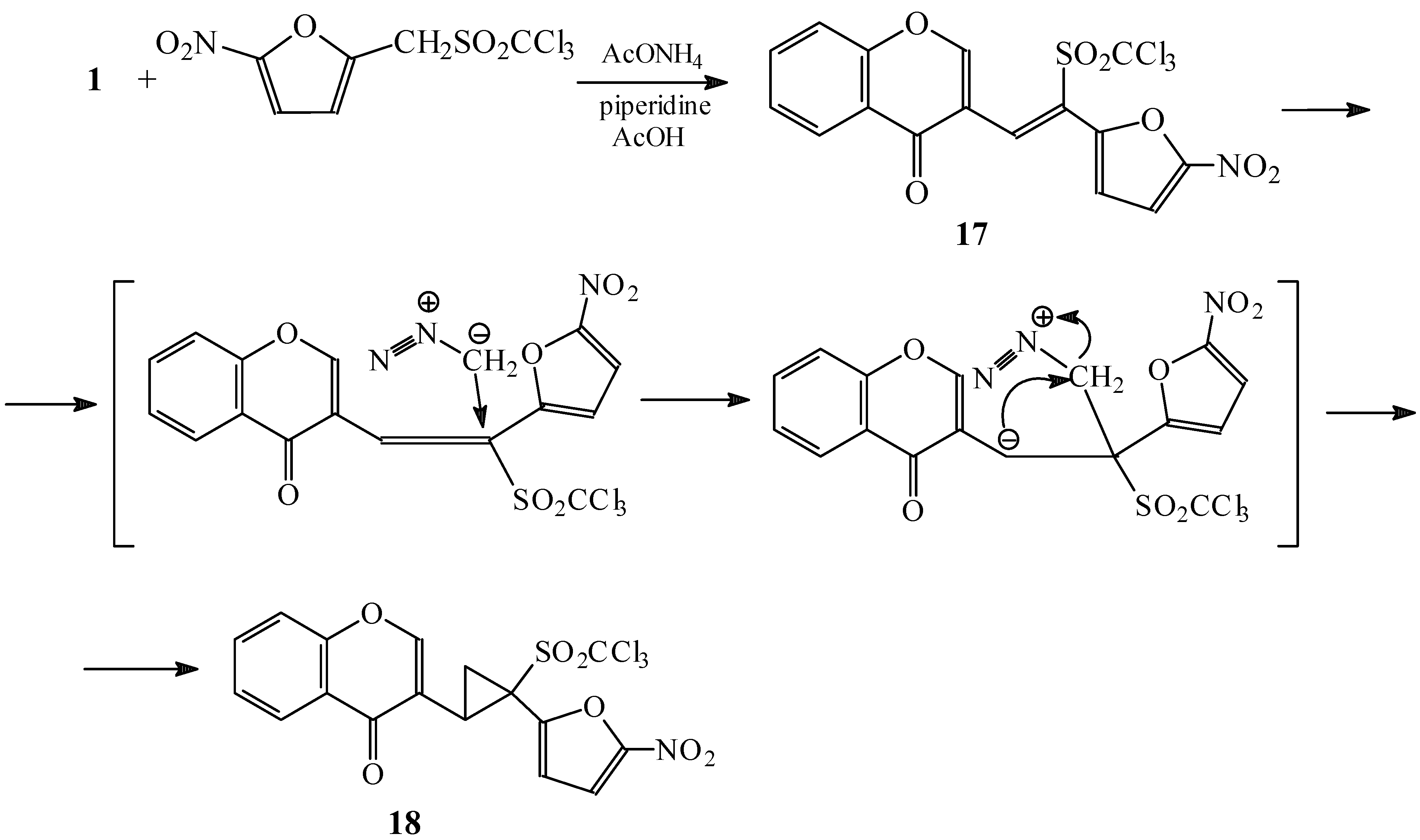
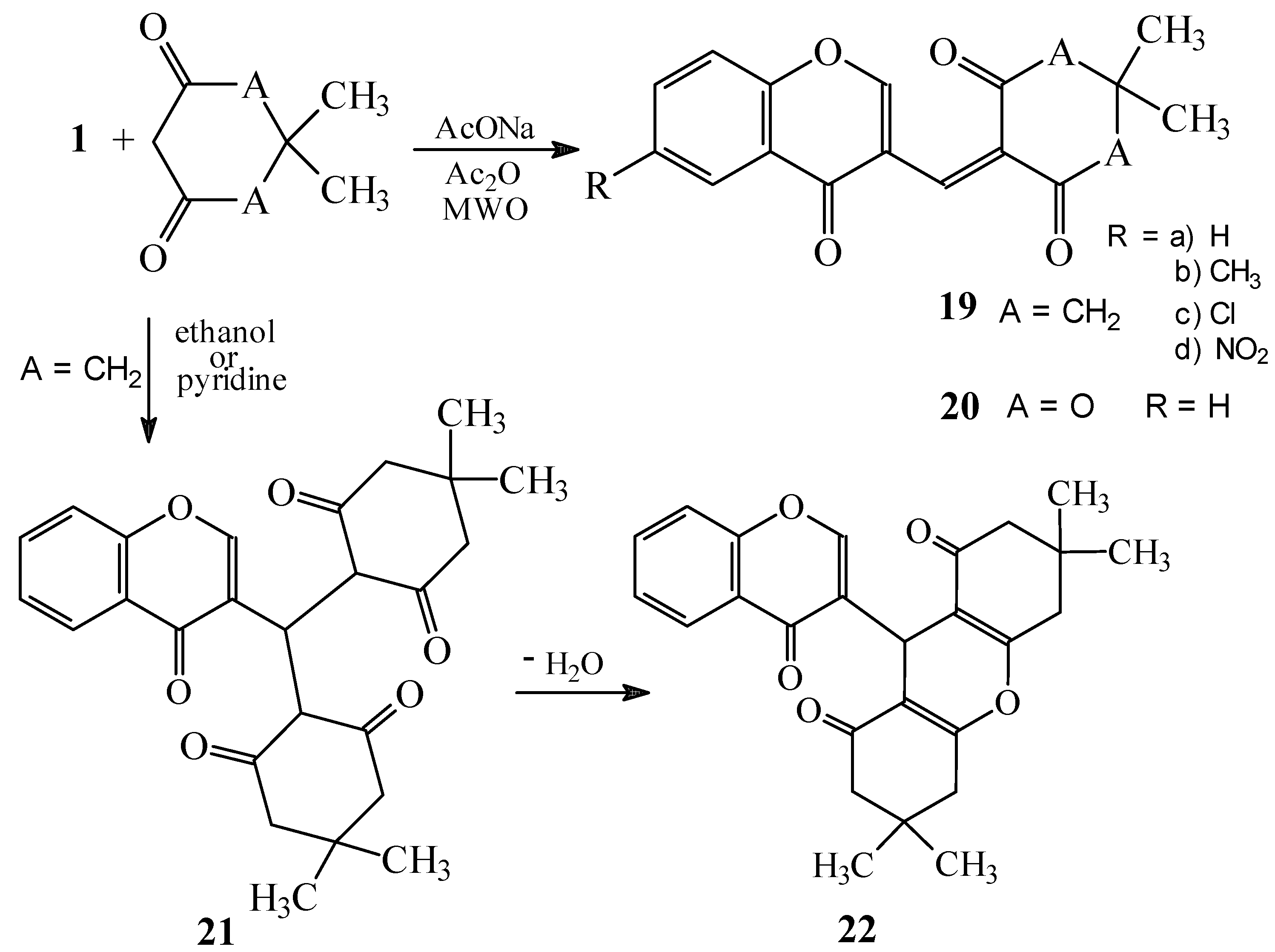
Condensations of 1 with 5,5-dimethylcyclohexane-1,3-dione and Meldrum’s acid
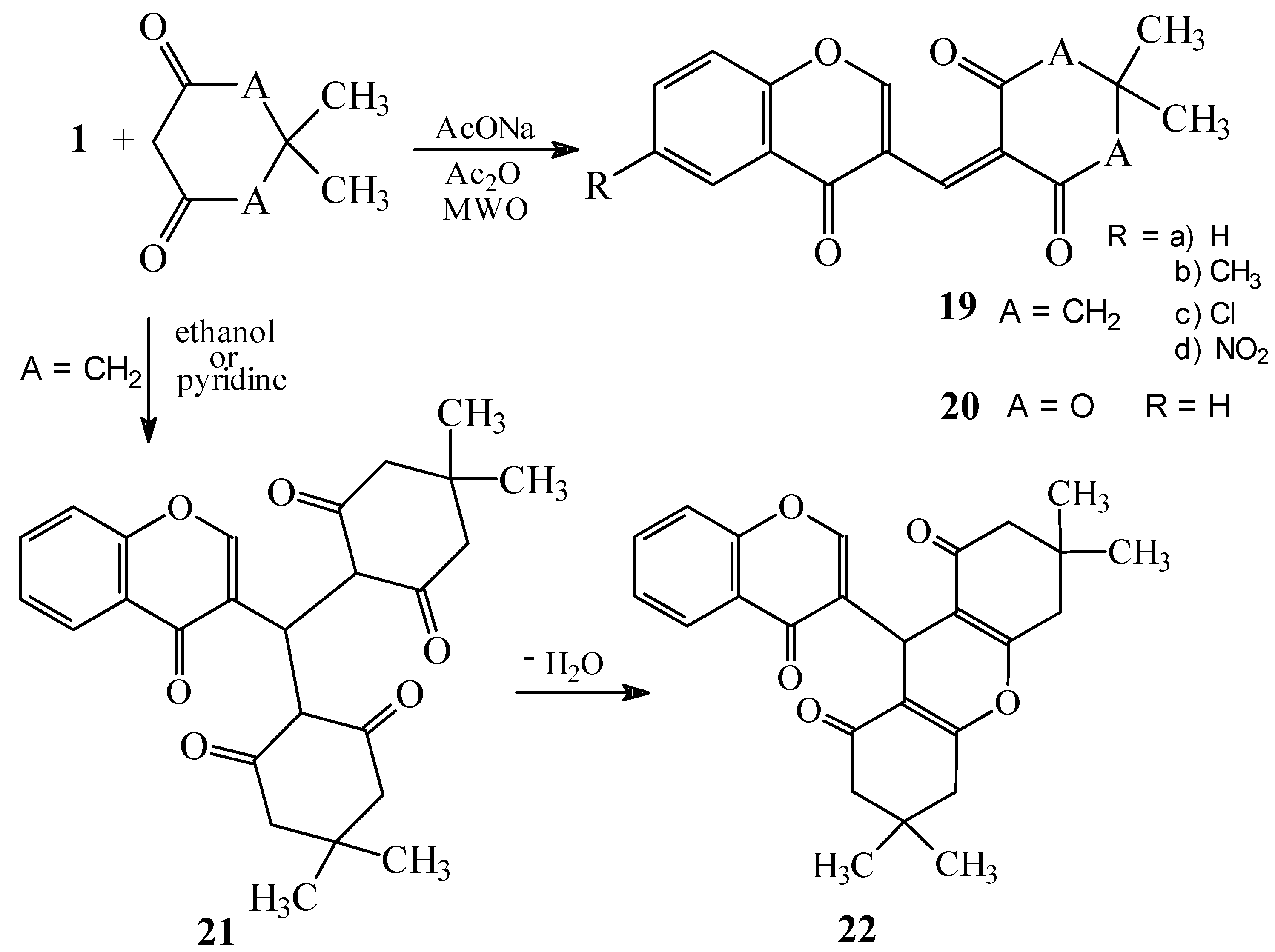
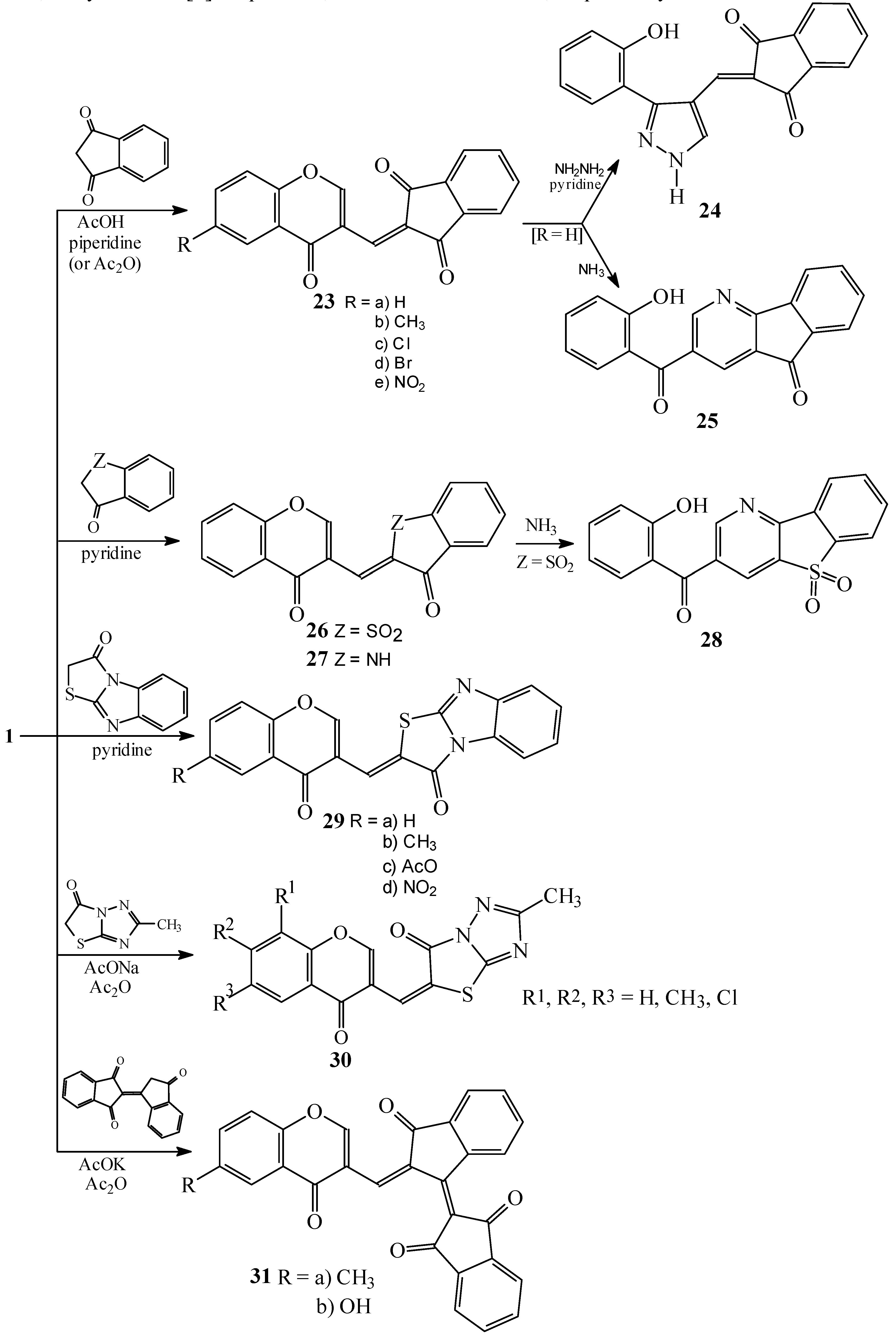
Condensations of 1 with 1,3-indandione and the other five-membered fused heterocycles
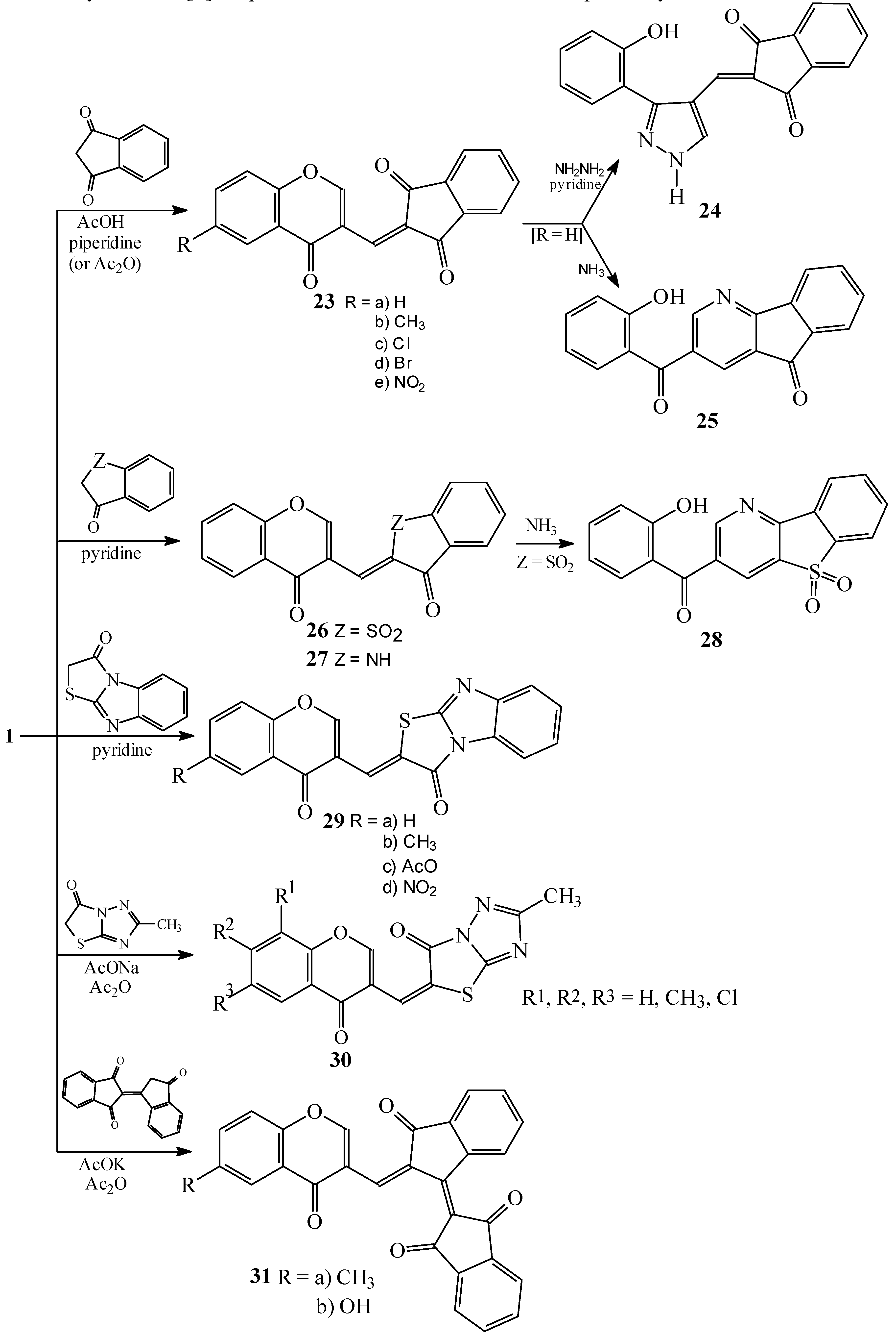
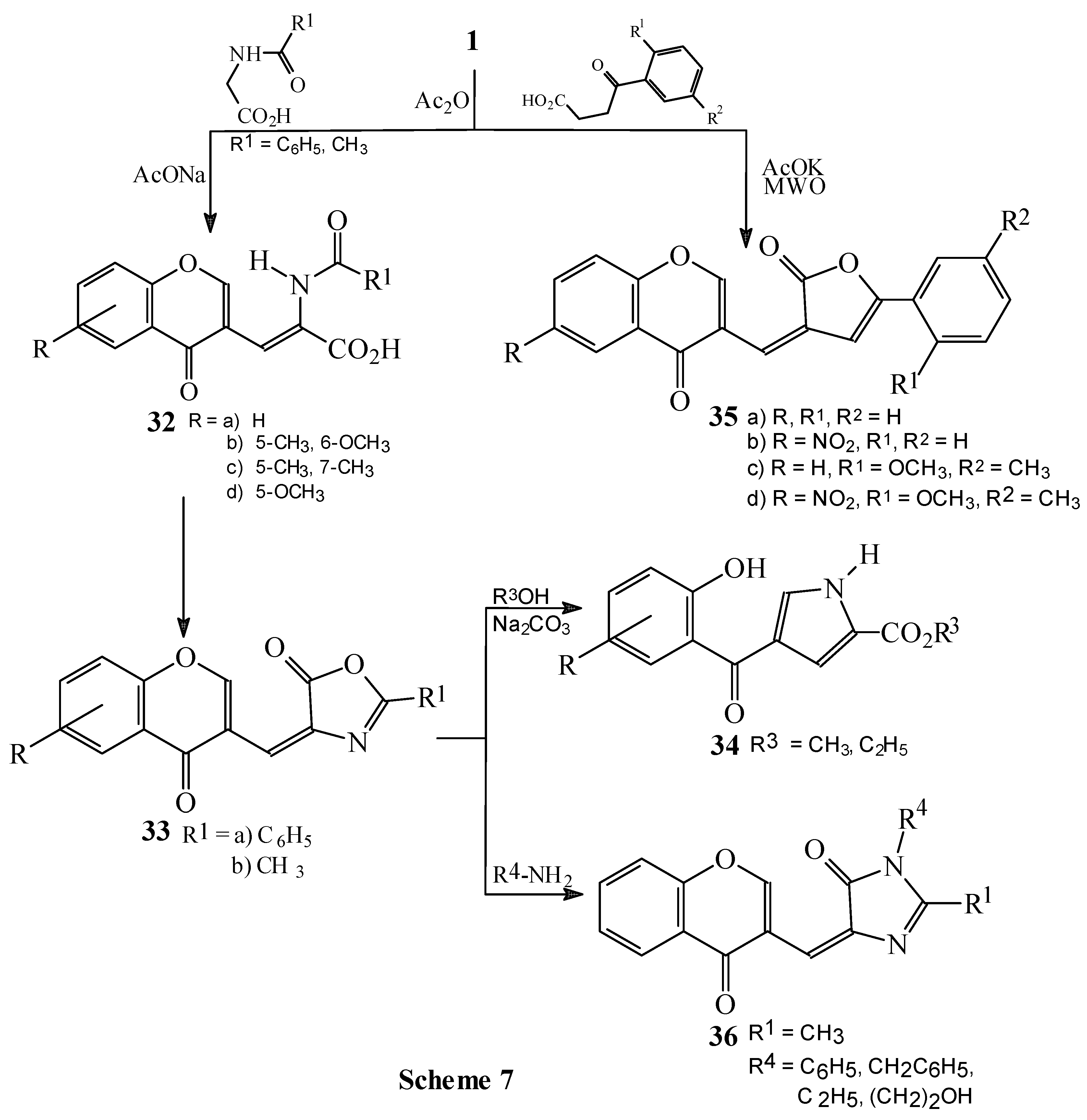
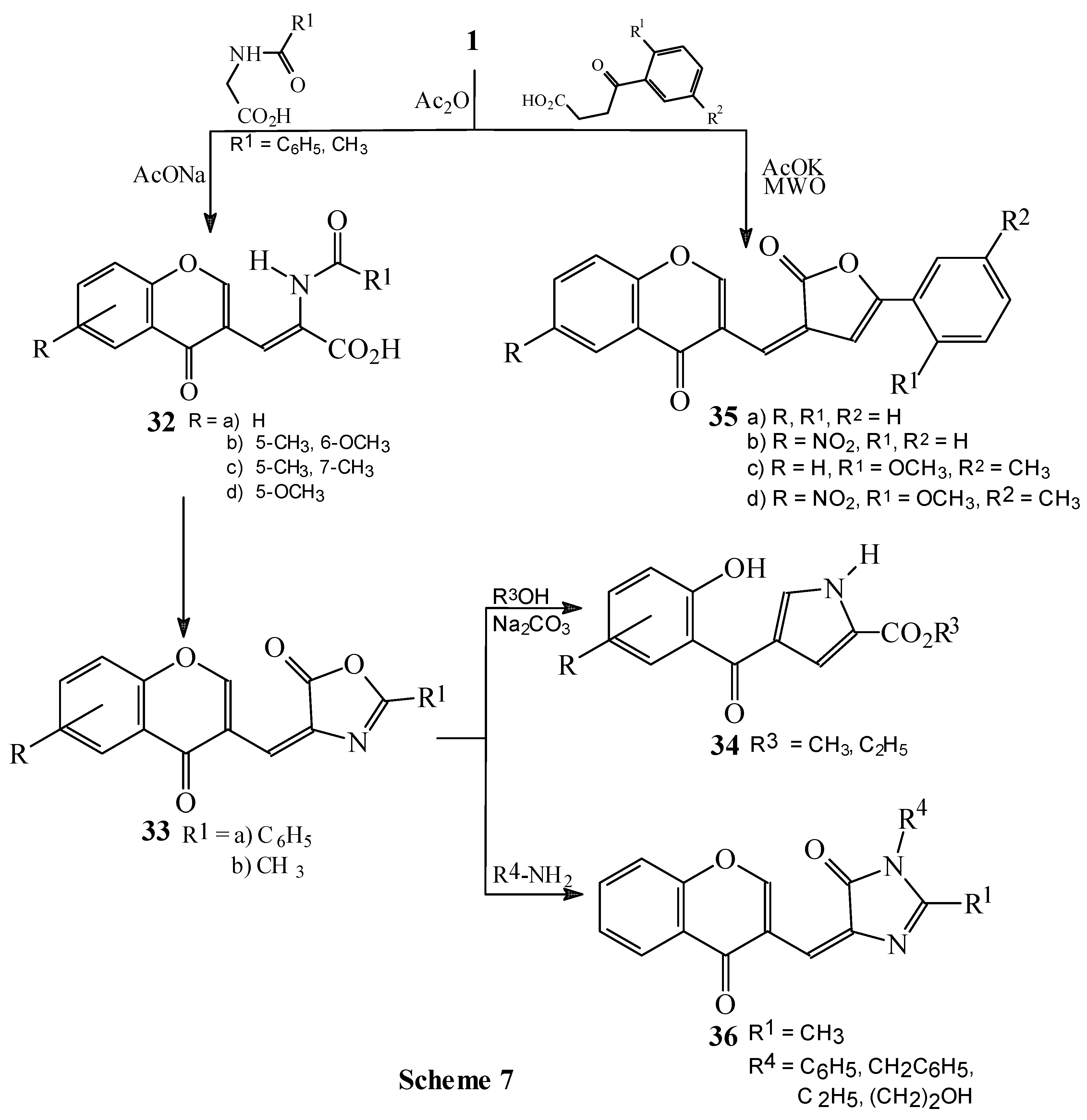
Condensations of 1 with hippuric acid, N-acetylglycine and benzoylpropionic acid
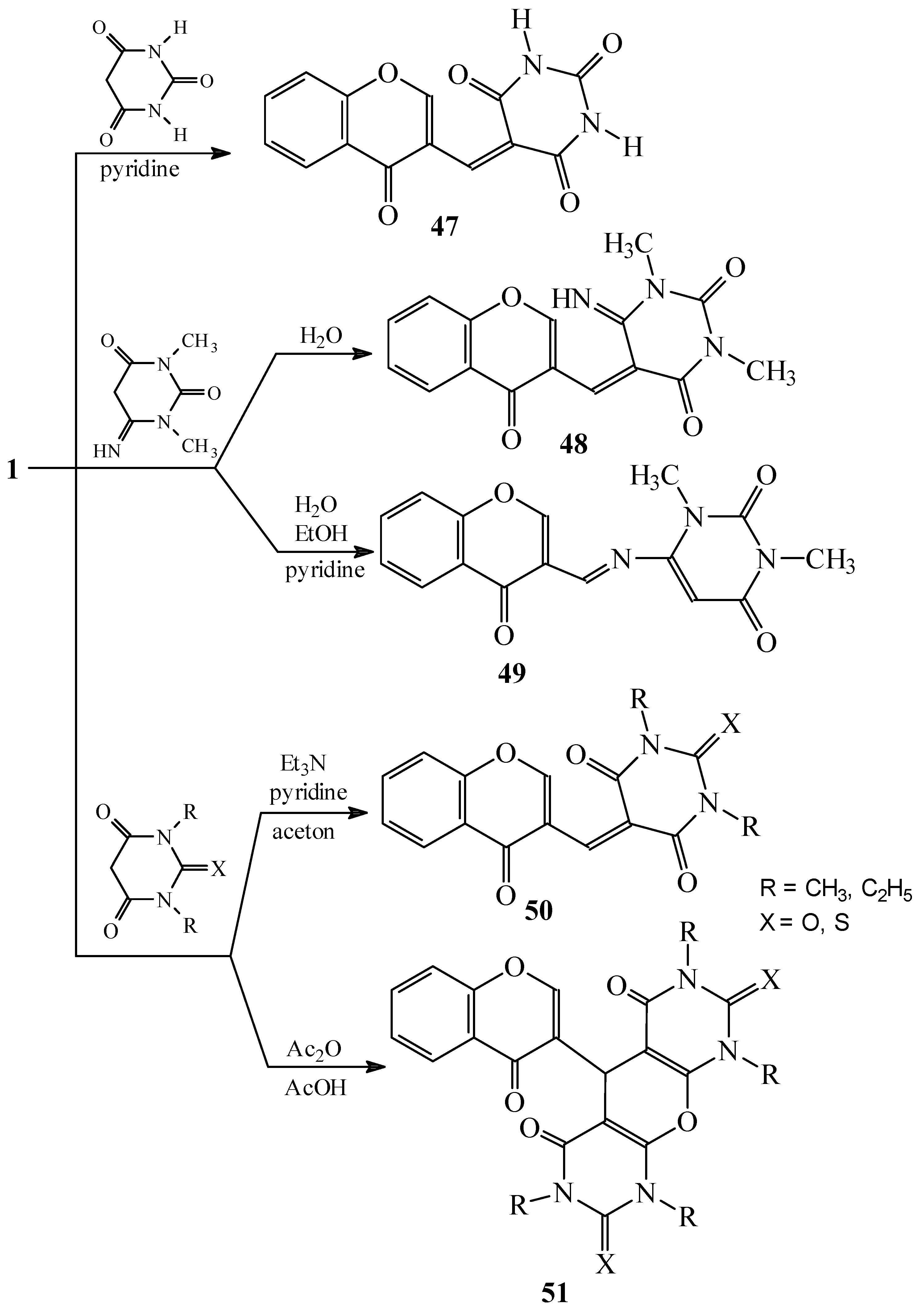
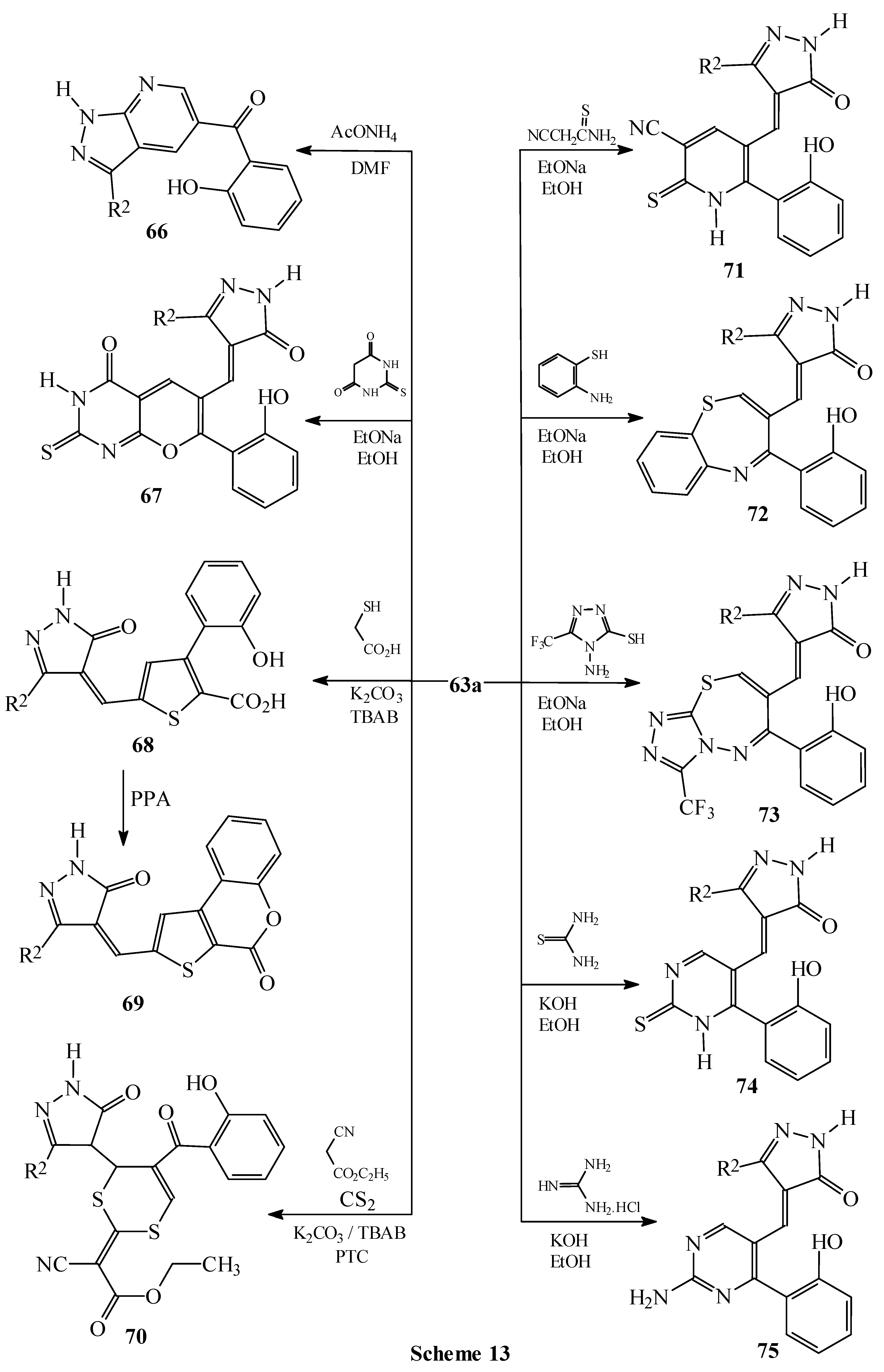
Condensations of 1 with barbituric acid derivatives
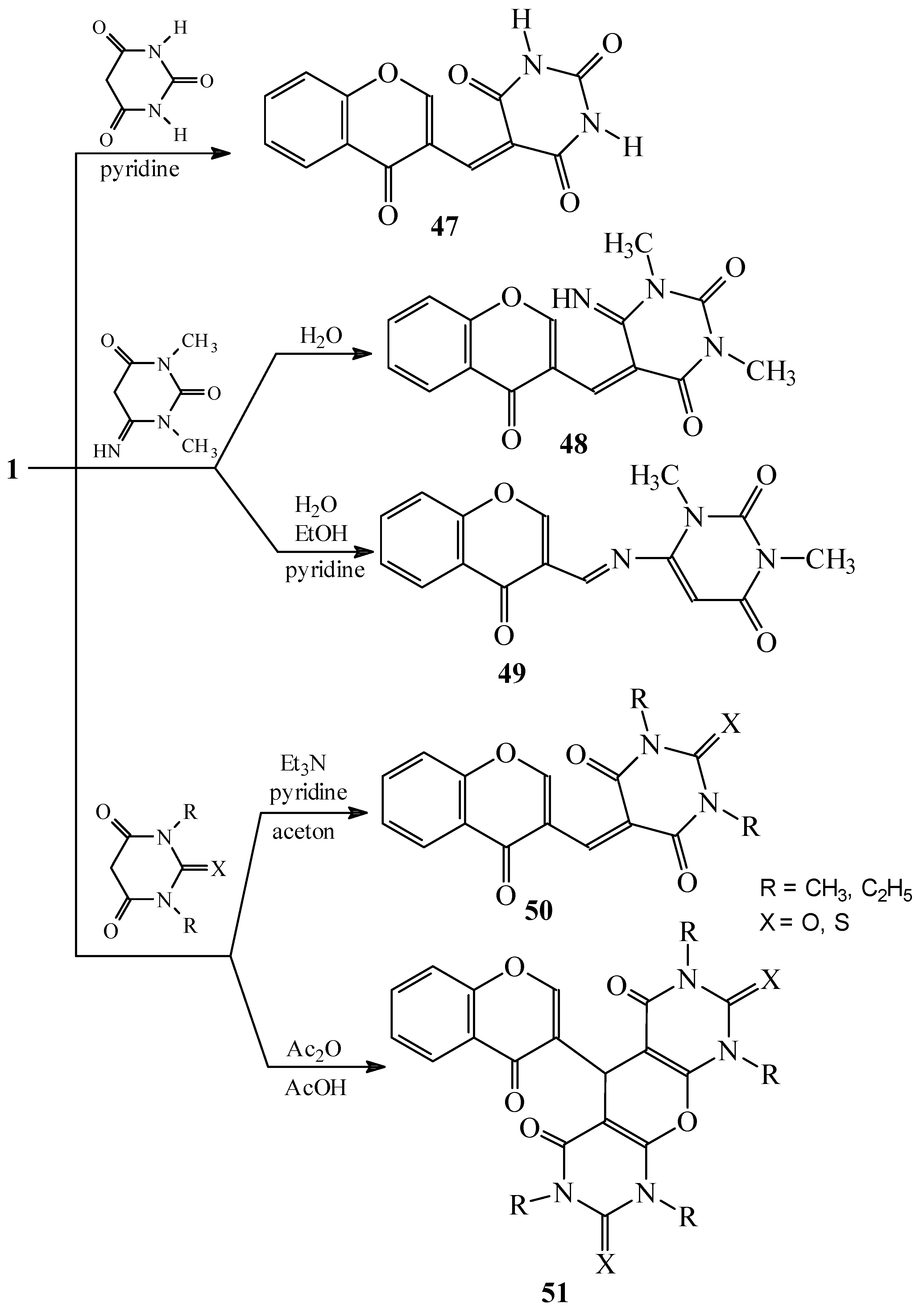
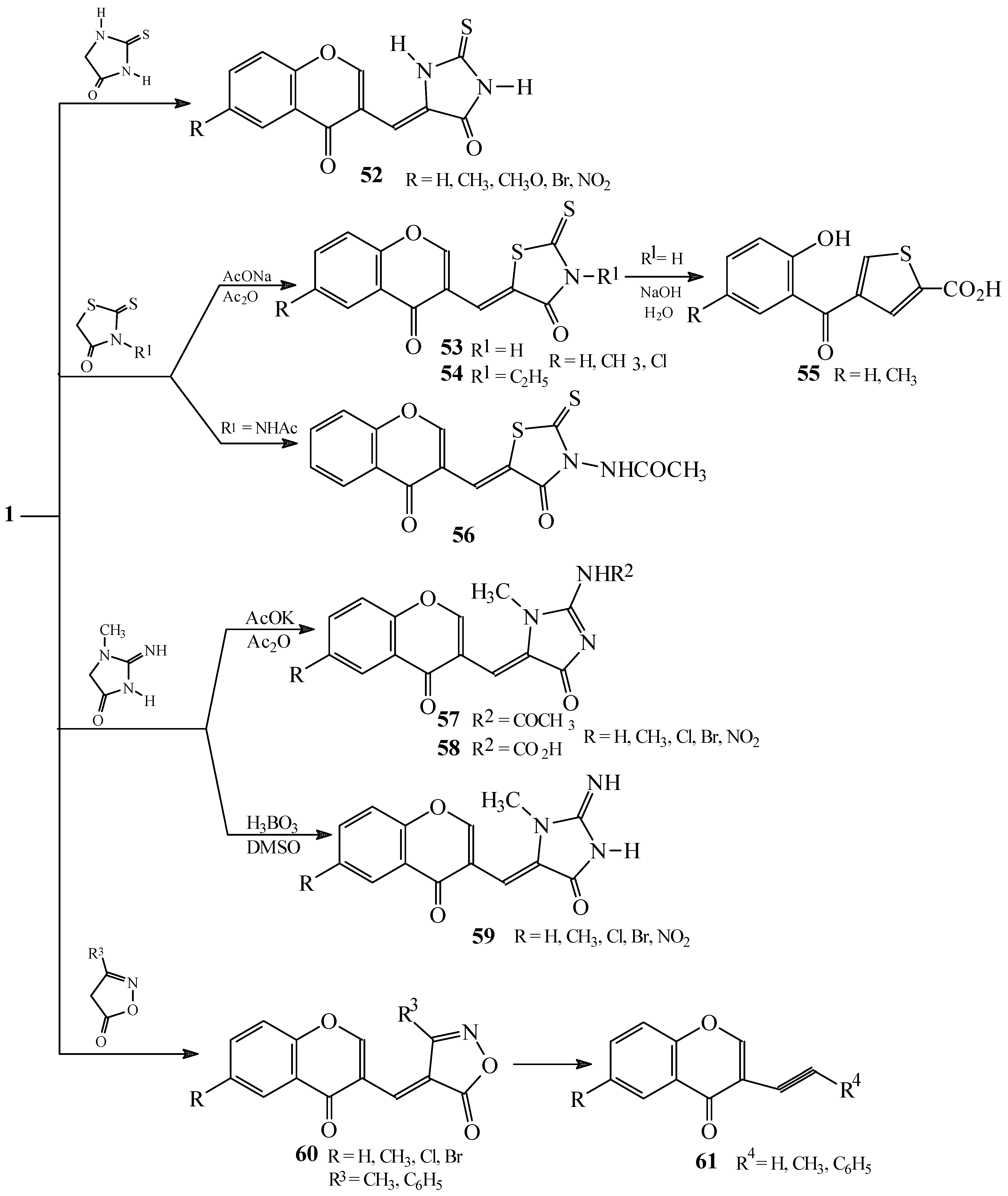
Condensations of 1 with 5-membered heterocycles
Reactions with 2-thioxoimidazolidin-4-one
Reactions with 2-thioxothiazolidin-4-one
Reactions with 2-imino-1-methylimidazolidin-4-one
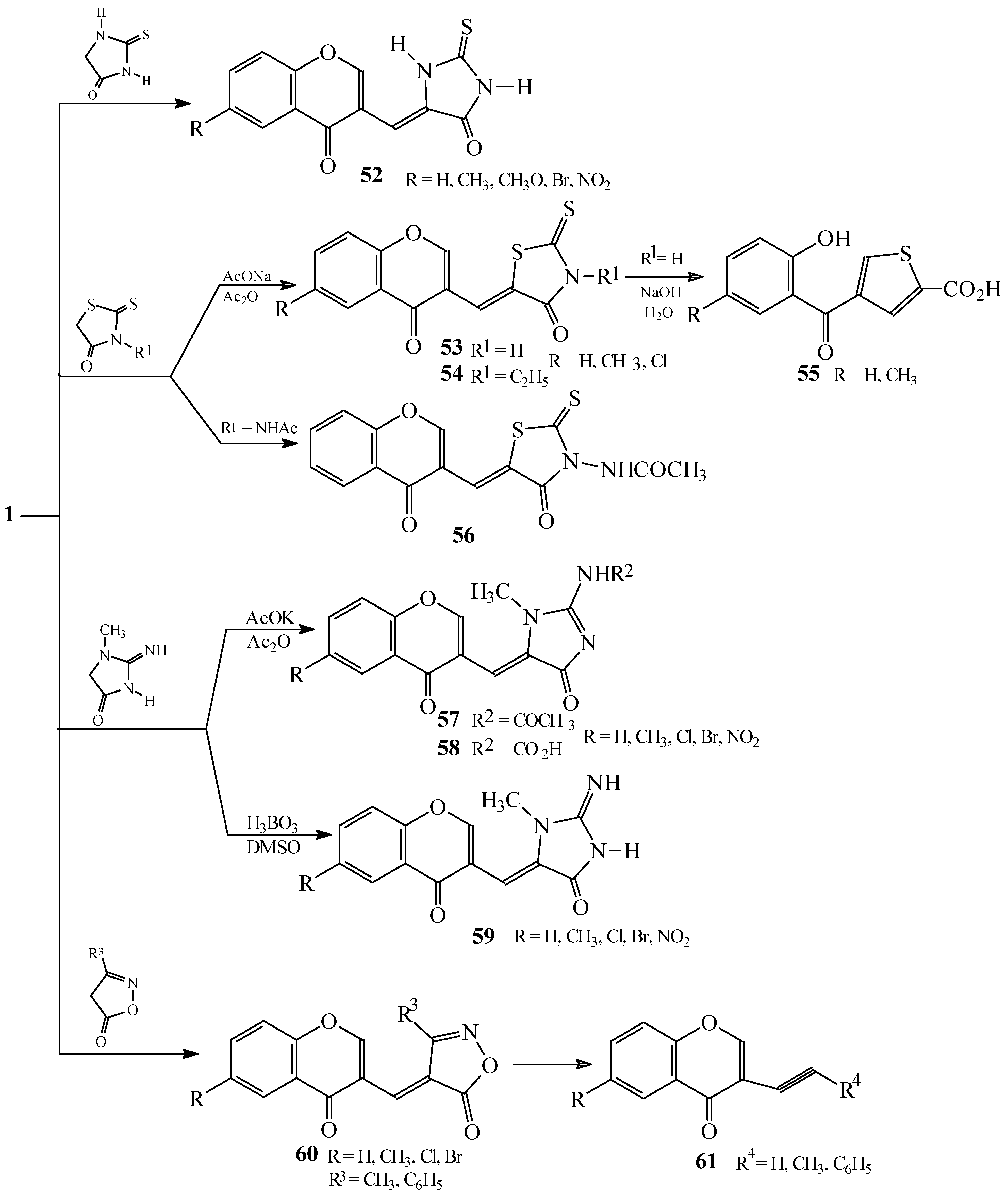
Reactions with 3-substituted isoxazol-5(4H)-ones
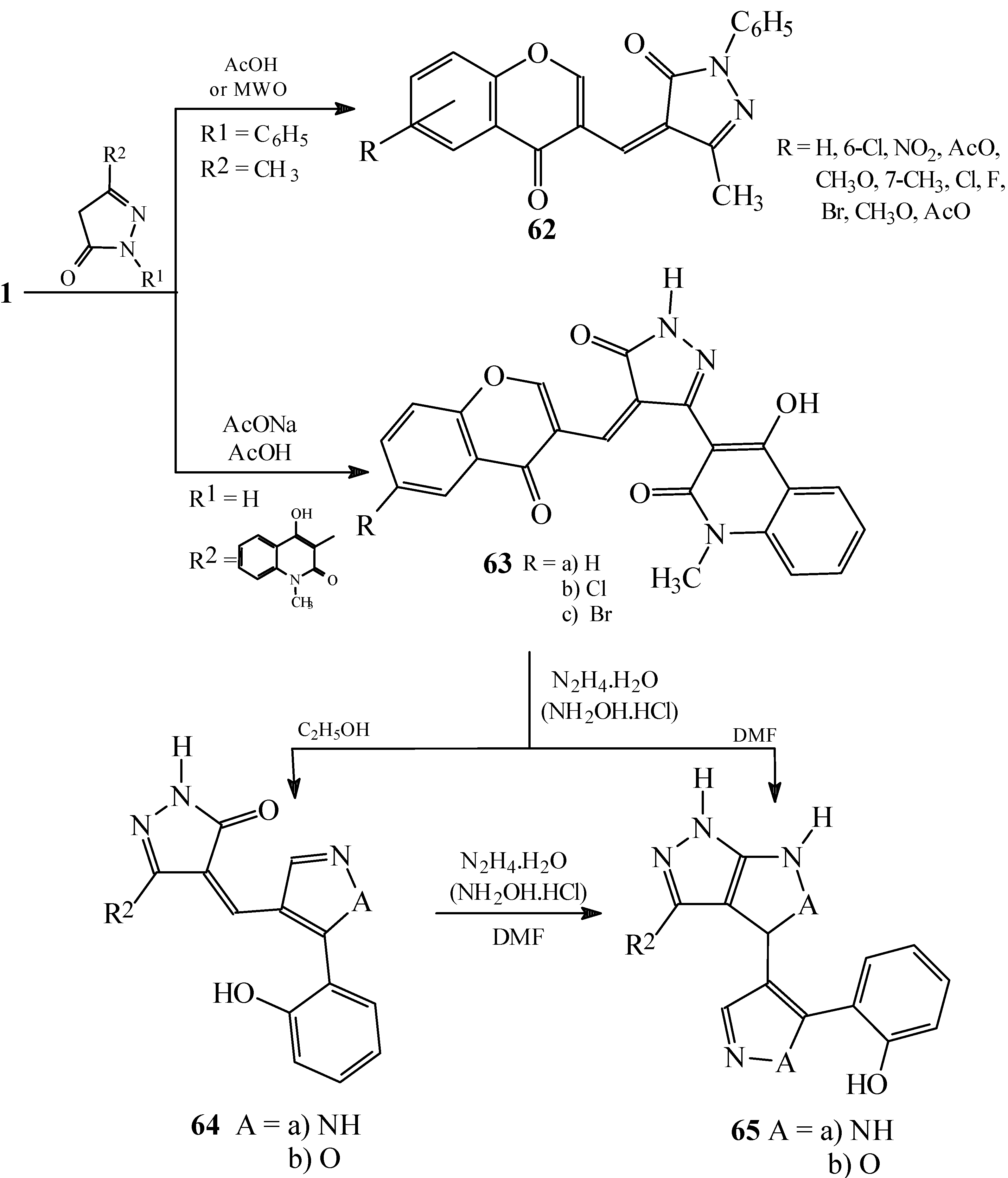
Reactions with substituted pyrazolin-5-(4H)-ones
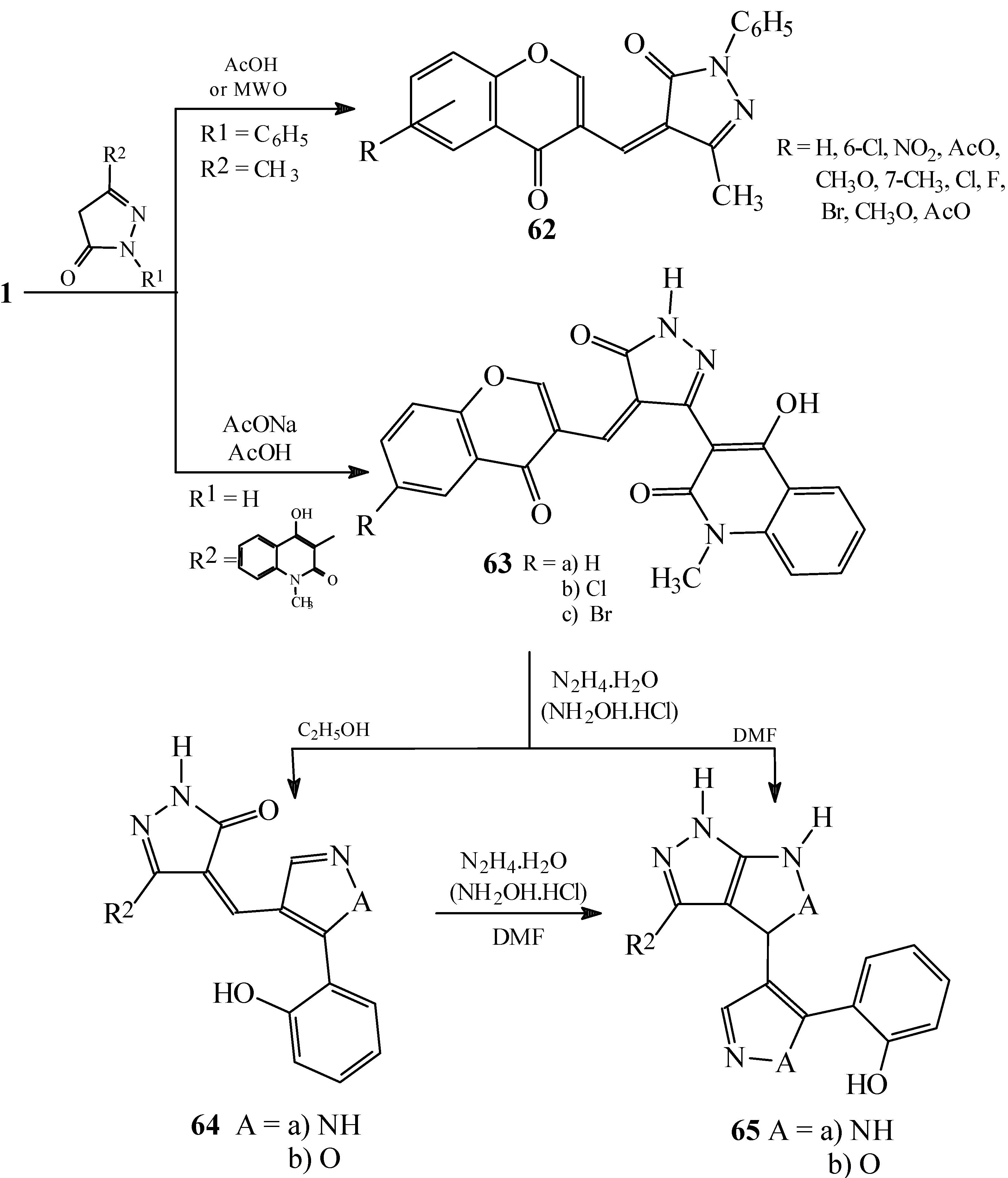
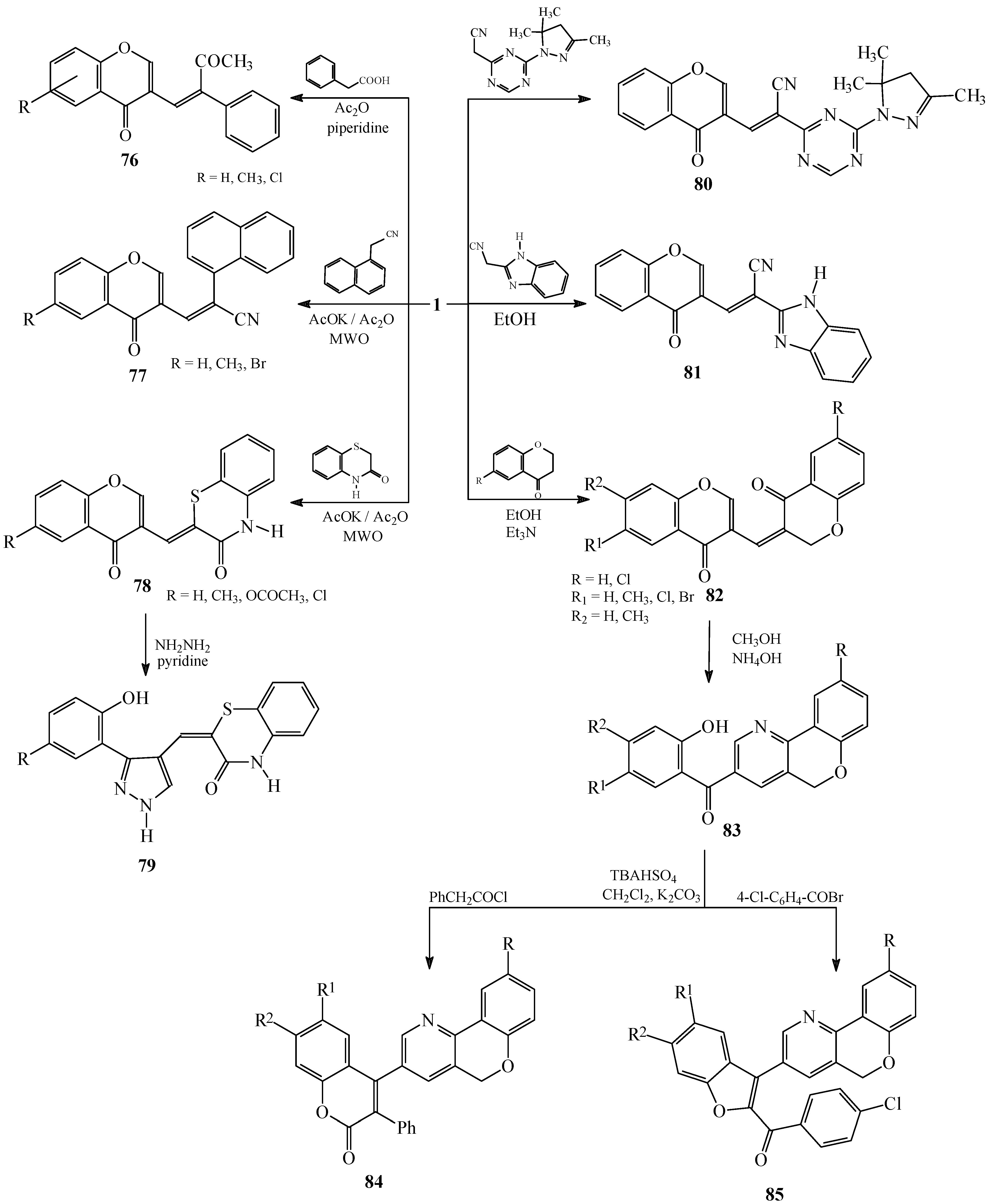
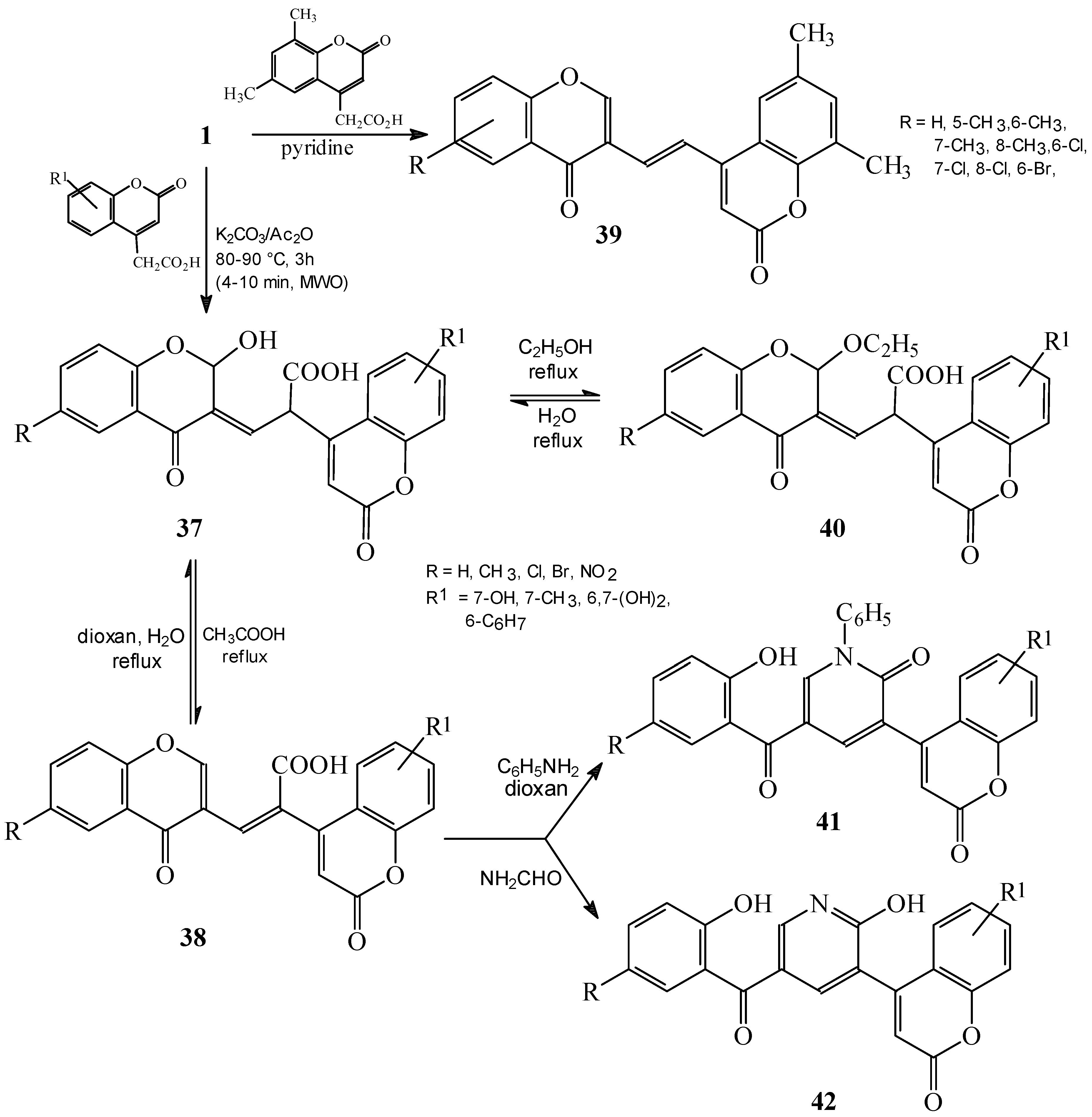
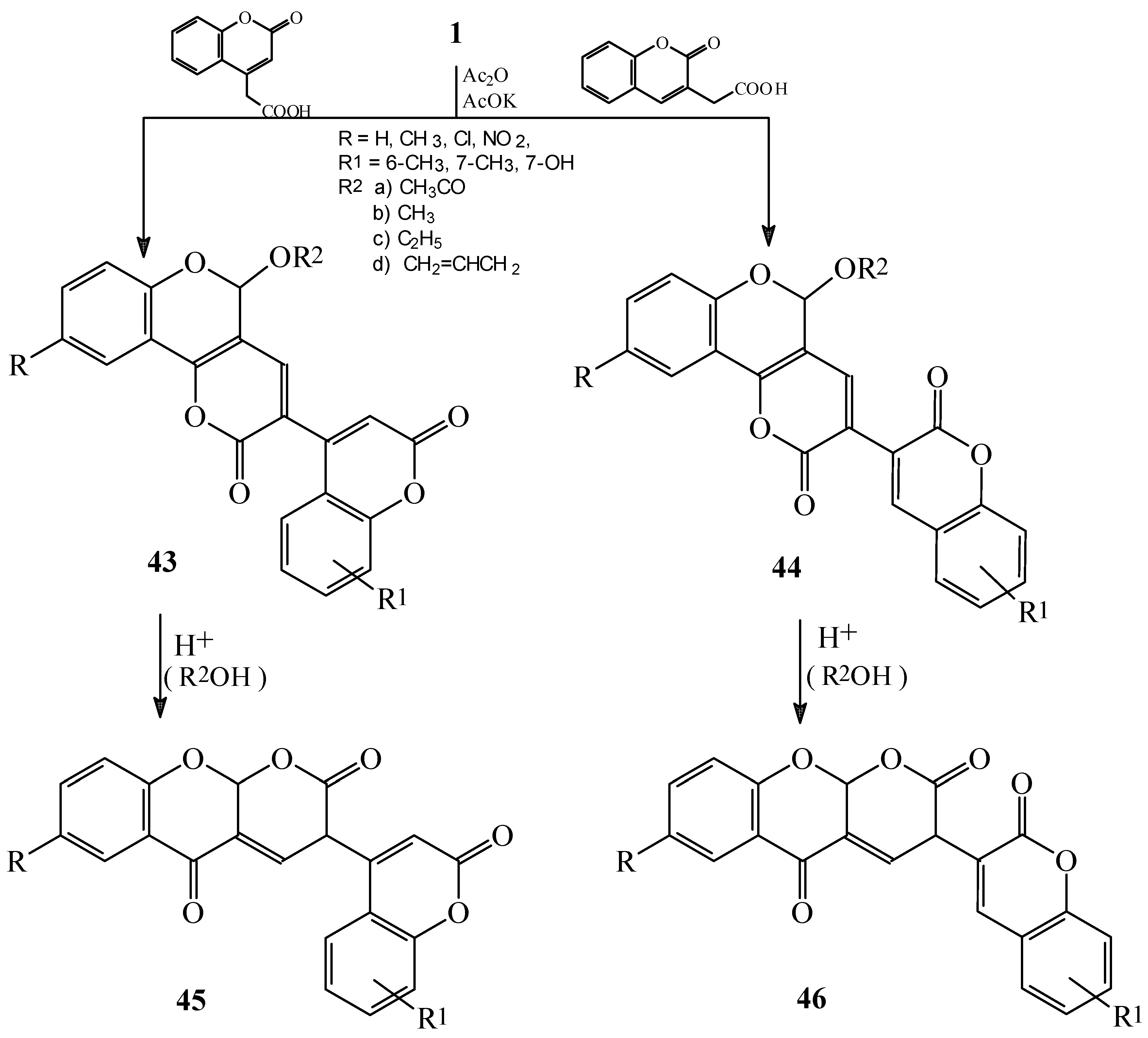
Condensations of 1 with phenylacetic acids, aryl- or heteroarylsubstituted acetonitriles and six-membered fused heterocycles
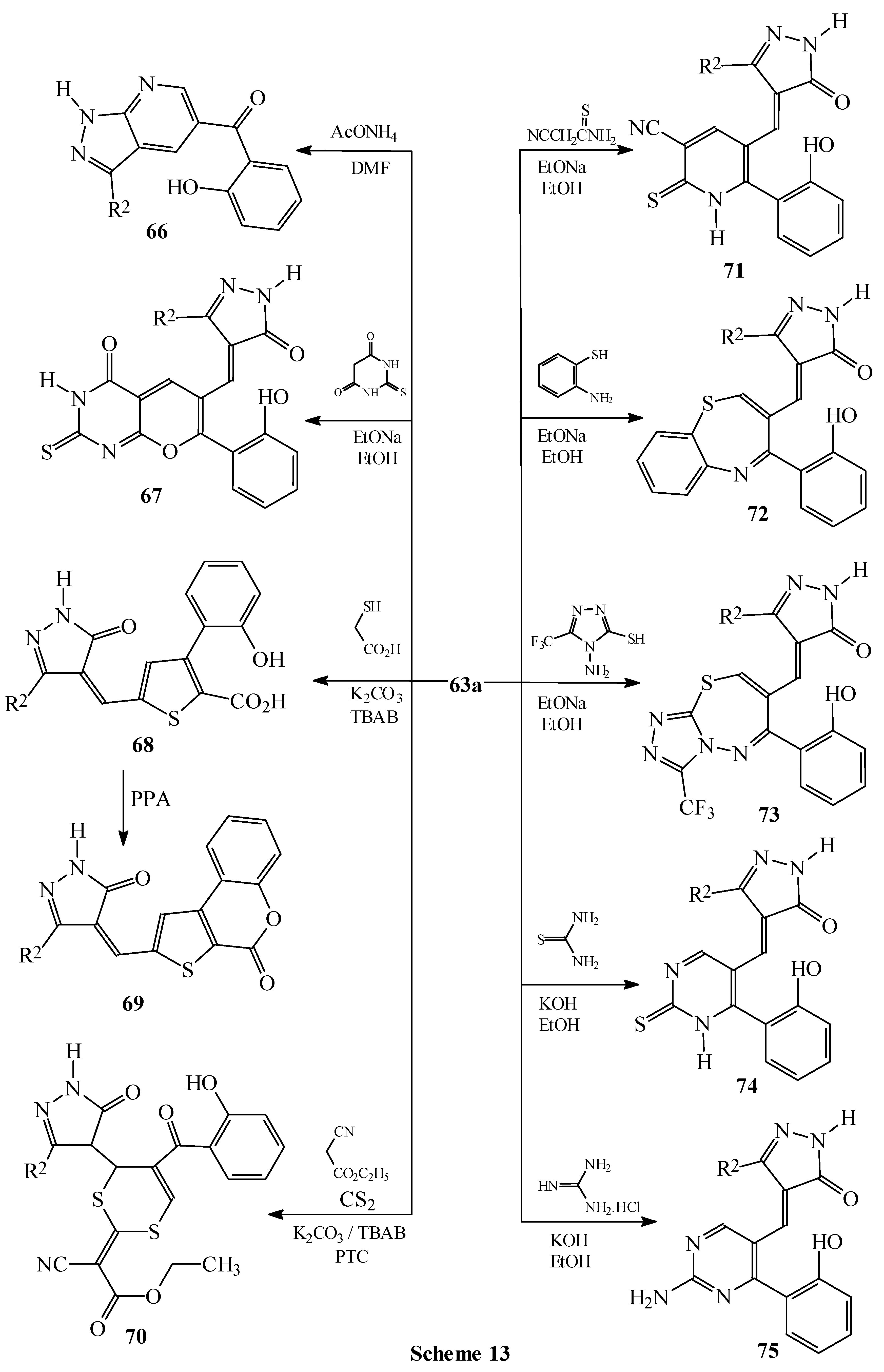
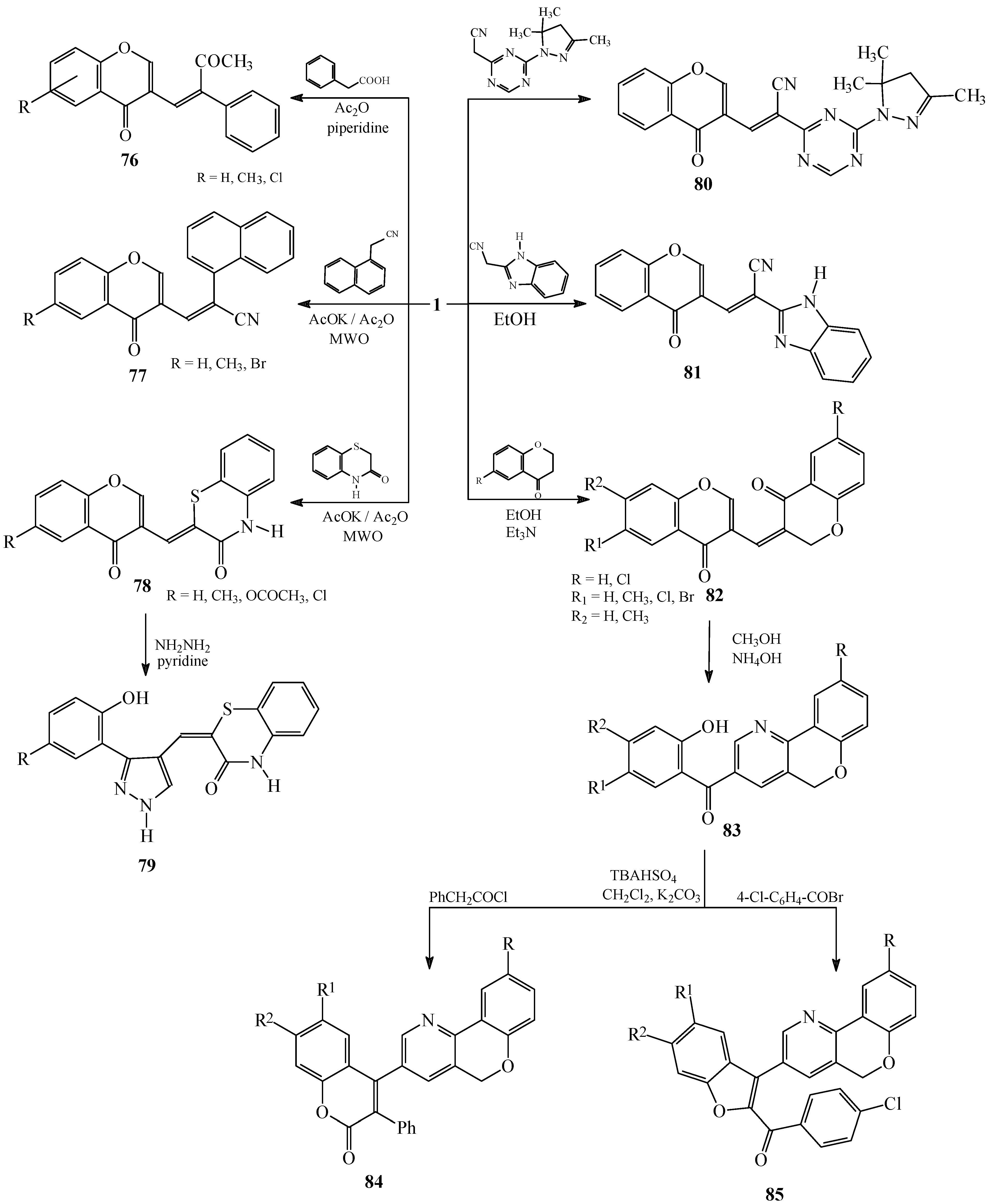
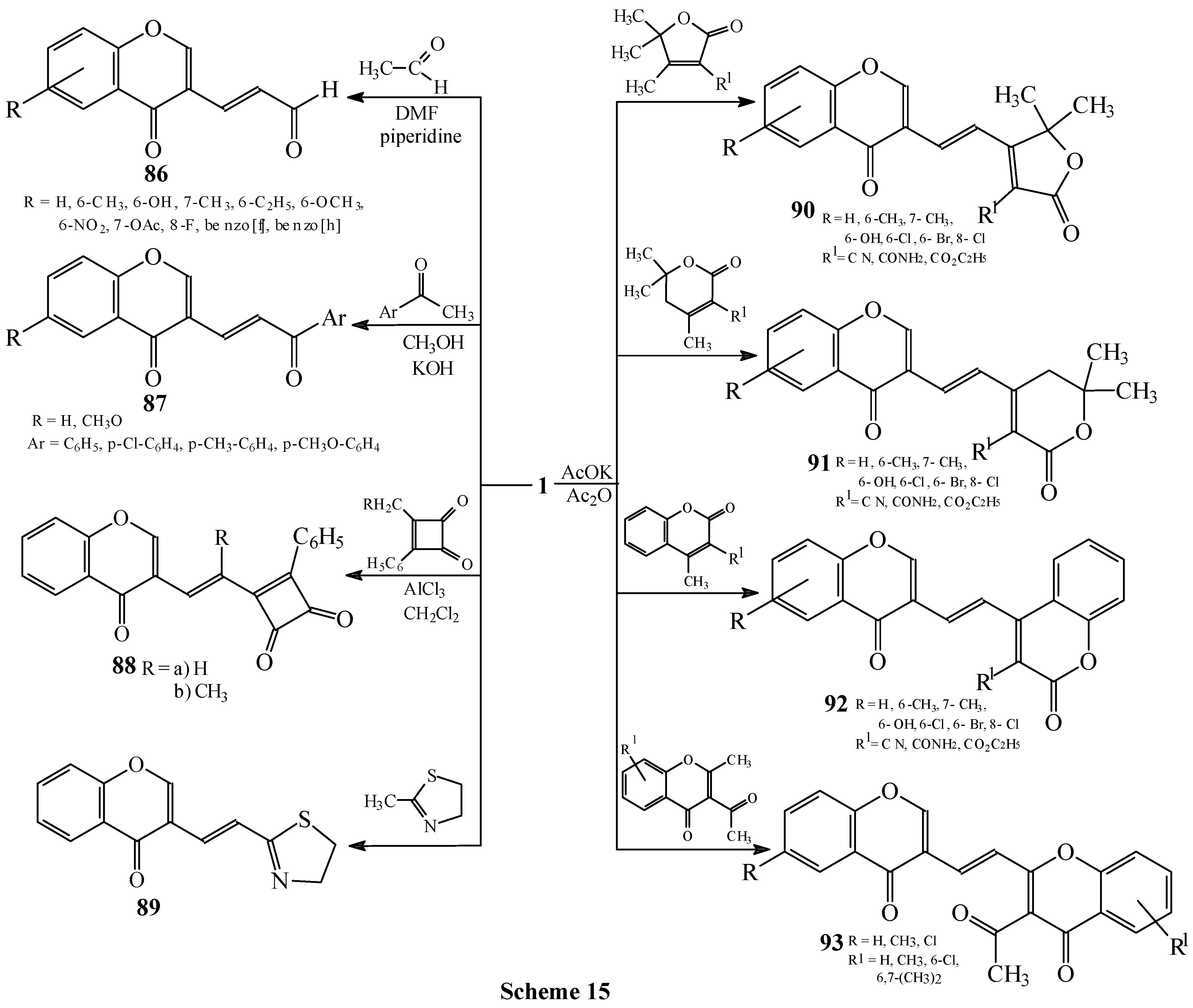
Condensations of 1 with active methyl compounds
Condensations with acetaldehyde and arylmethylketones
Condensations of 1 with 3-alkyl-4-phenyl-3-cyclobuten-1,2-diones and 3-methylthiazine
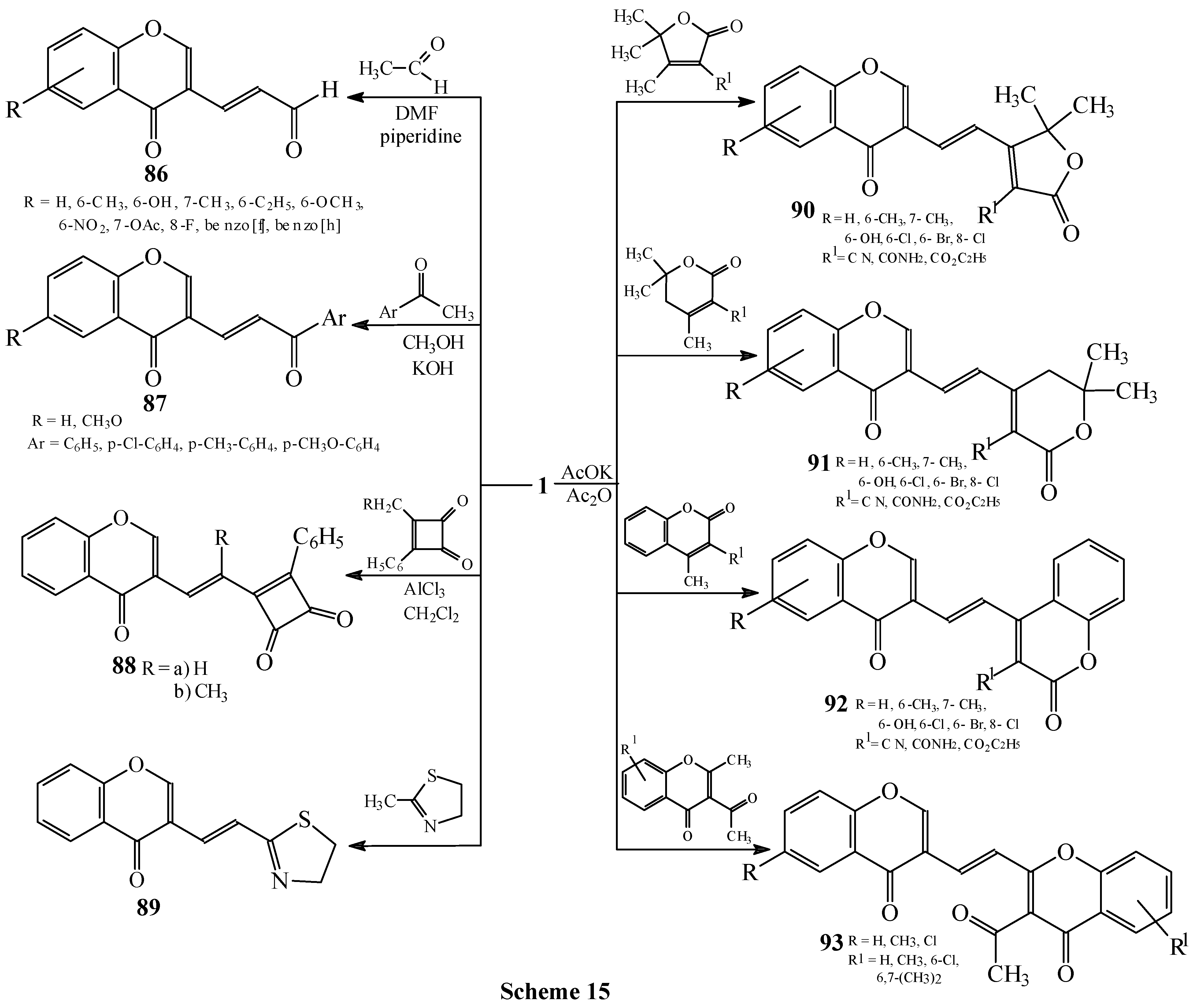
Condensations of 1 with methyl-substituted oxygen heterocycles
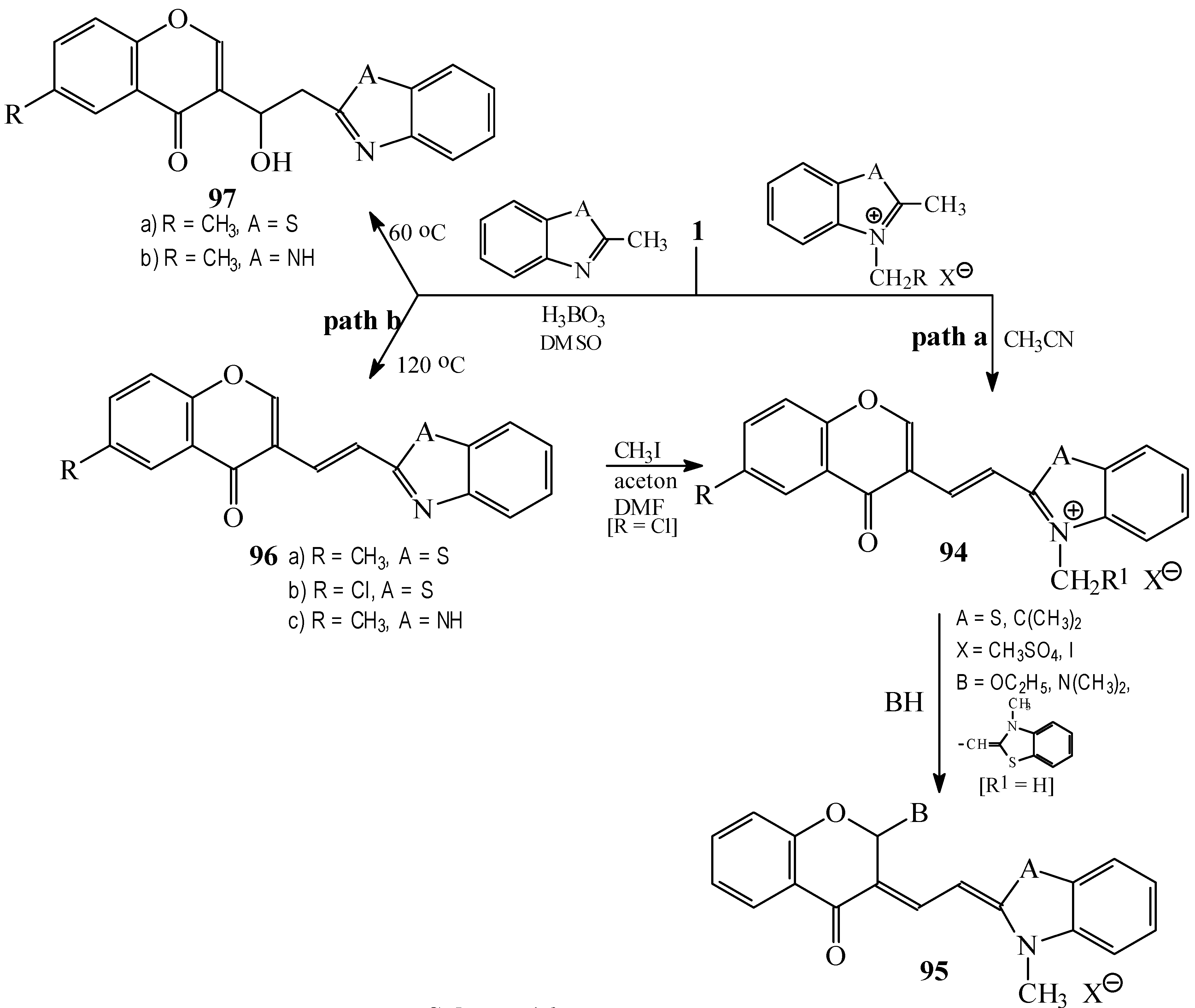
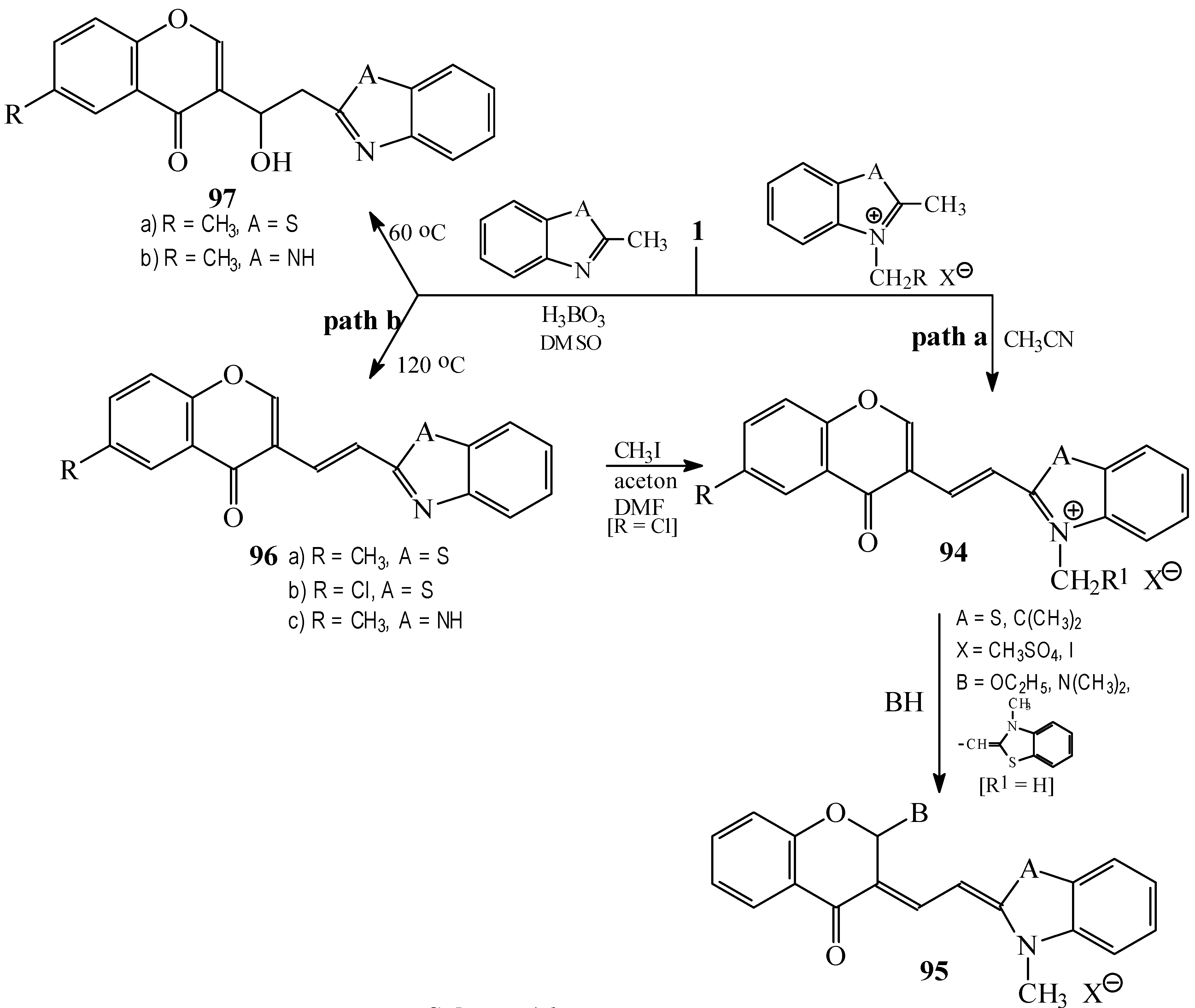
Condensations of 1 with 2-methylbenzothiazole and 2-methylbenzimidazole derivatives
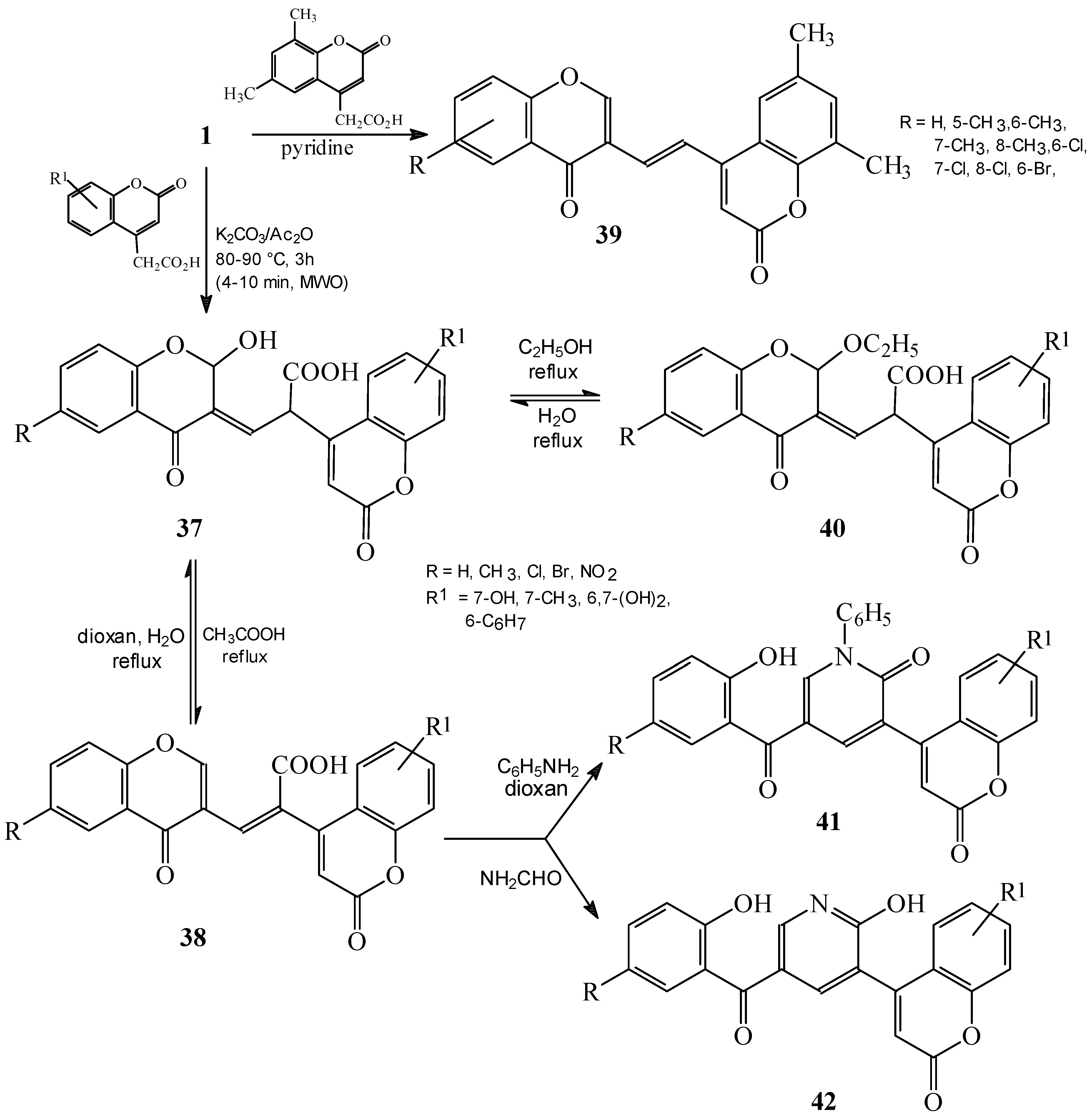
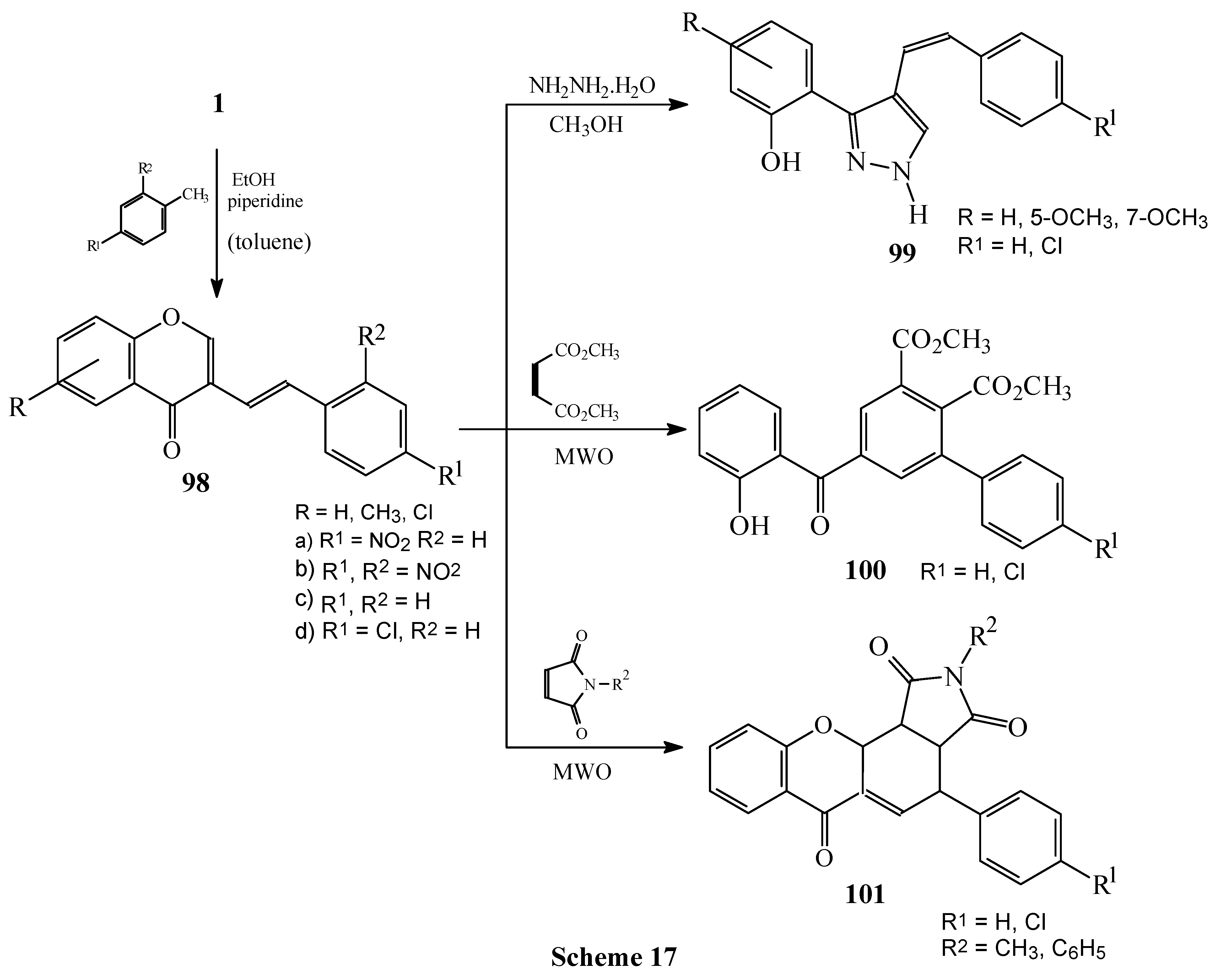
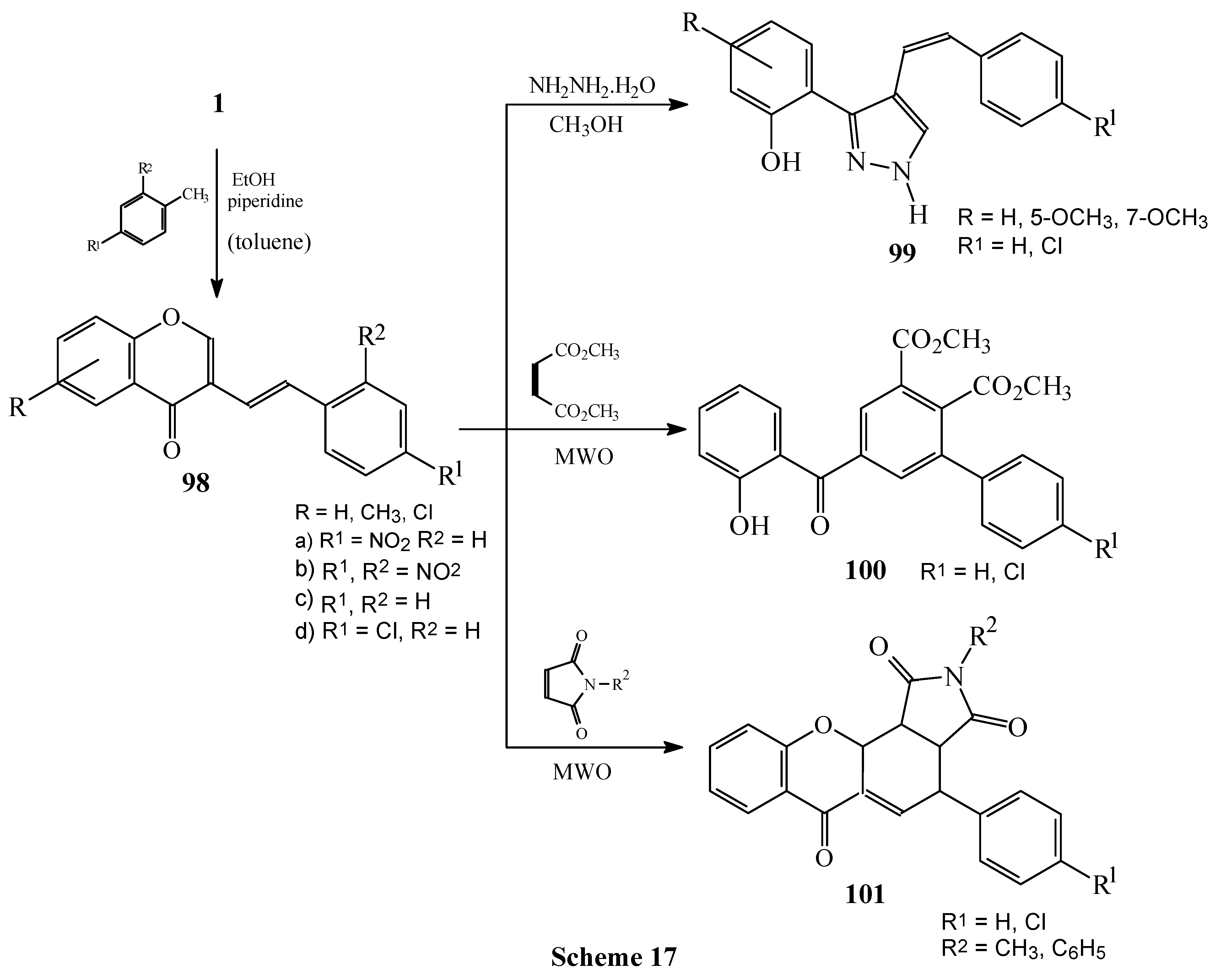
Condensations of 1 with 4-nitro and 2,4-dinitrotoluene. Subsequent reactions of 3-styrylchromones.
Conclusions
References
- Nohara, A.; Umetani, T.; Sanno, Y. Tetrahedron Lett. 1973, 1995.
- Ghosh, C. K. J. Heterocycl. Chem. 1983, 20, 1437. [CrossRef]
- Sabitha, G. Aldrichimica Acta 1996, 29, 15.
- Bram, G.; Loupy, A.; Majdoub, M.; Gutierrez, E.; Ruiz – Hitzky, E. Tetrahedron 1990, 46, 5167.
- Mingos, D. M. P.; Baghurst, D. R. Chem. Soc. Rev. 1991, 20, 1. [CrossRef]
- Abramovitch, R. A. Org. Prep. Proceed. Int. 1991, 23, 683. [CrossRef]
- Toma, S. Chem. Listy 1993, 87, 627.
- Ku, K. C.; Koh, S. Y.; Tang, T. F.; Chang, H. L. Hua Hsueh Hsueh Pao 1976, 34, 123, [Chem. Abstr. 1977, 87, 184 319m].
- Nohara, A.; Umetani, T.; Miyata, Y.; Sanno, Y. Ger Often. 1973, 2 253 914, [Chem Abstr. 1973, 79, 31 874y].
- Nohara, A.; Kuriki, H.; Saijo, T.; Ukawa, K.; Murata, T.; Sanno, Y. J. Med. Chem. 1975, 18, 34. [CrossRef]
- Nohara, A.; Umetani, T.; Sanno, Y. Japan Kokai 1975, 7 552 067, [Chem Abstr. 1976, 84, 105400t].
- Nohara, A.; Kuriki, H.; Saijo, T.; Sugihara, H.; Kanno, M.; Sanno, Y. J. Med. Chem. 1977, 20, 141. [CrossRef]
- Ellis, G. P. J. Med. Chem. 1978, 21, 1120. [CrossRef]
- Nohara, A.; Ishiguto, T.; Sanno, Y. Tetrahedron Lett. 1974, 13, 1183.
- Ghosh, C. K.; Khan, S. Synthesis 1981, 903.
- Jones, W. D.; Albrecht, W. L. J. Org. Chem. 1996, 41, 706.
- Bandyophadhyay, C.; Sur, K. R.; Patra, R. J. Chem. Res. Synop. 1998, 12, 802.
- Prousek, J.; Jurasek, A.; Kovac, J. Coll. Czech. Chem. Commun. 1980, 45, 1704. [CrossRef]
- Eiden, F.; Haverland, H. Arch. Pharm. 1967, 300, 806.
- Haas, G.; Stanton, L.; Von Sprecher, A.; Wenk, P. J. Heterocyclic Chem. 1981, 18, 607. [CrossRef]
- Gasparova, R.; Lacova, M. Coll. Czech. Chem. Commun. 1995, 60, 1178.
- Prousek, J. Coll. Czech. Chem. Commun. 1991, 56, 1361. [CrossRef]
- Polyakov, V. K.; Shevtsova, R. G.; Tsukerman, S. V. Ukr. Khim. Zh. 1981, 47, 85.
- Franz, C.; Heinisch, G.; Holzer, W.; Mereiter, K.; Strobe, B.; Zheng, C. Heterocycles 1995, 41, 2527.
- Gaikwad, M. S.; Karale, B. K.; Mane, A. S.; Chavan, V. P.; Shingare, M. S. Indian J. Heterocycl. Chem. 2001, 10, 313.
- Gasparova, R.; Lacova, M. Unpublished results.
- Fitton, A. O.; Frost, J. R.; Suschitzky, H.; Houghton, P. G. Synthesis 1977, 133.
- Lacova, M.; Loos, D.; Chovancova, J.; Kralova, K. 3rd International Electronic Conference on Synthetic Organic Chemistry, ECSOC-3, September 1 – 30, 1999.
- Karale, B. K.; Chavan, V. P.; Hangarge, R. V.; Mane, A. S.; Gill, C. H.; Shingare, M. S. Indian J. Heterocycl. Chem. 2001, 11, 81.
- Bartosova, B.; Lacova, M.; Gaplovsky, A.; Chovancova, J.; Pronayova, N.; Simunek, P.; Loos, D. 6th Electronic Conference on Synthetic Organic Chemistry, ECSOC-6, September 1-30, 2002.
- Eiden, F.; Schikorr, W. Arch. Pharm. 1972, 305, 187.
- Lacova, M.; Gasparova, R.; Loos, D.; Liptay, T.; Pronayova, N. Molecules 2000, 5, 167.
- Hanefeld, W.; Schlitzer, M. J. Heterocyclic Chem. 1995, 32, 1019. [CrossRef]
- Wentrup, C.; Winter, H. W. Angew. Chem. 1978, 90, 643. [CrossRef]
- Sabitha, G.; Reddy, M. M.; Archana, B.; Yadav, J. S. Synth Commun 1998, 28, 573.
- Polyakov, V. K.; Babitch, J. P.; Shevtsova, R. G.; Trusevich, N. D. Izv. Vysh. Utch. Zaved. Khim., Khim. Technol. 1987, 30, 42, [Chem. Abstr. 1988, 108, 112 324p].
- Karale, B. K.; Chavan, V. P.; Mane, A. S.; Hangarge, R. V.; Shingare, M. S. Synth. Commun. 2002, 32, 497.
- Abass, M.; Hassan, A. Chem. Papers 2003, 57, 267.
- Sarma, G. V. S. R.; Reddy, V. M. Ind. J. Heterocycl. Chem. 1993, 3, 111, [Chem. Abstr. 1994, 121, 108 619q].
- Shingare, M. S.; Karale, B. K.; Gill, C. H.; Ganage, K. N.; Bachute, M. T. Ind. J. Heterocycl. Chem. 1999, 9, 153.
- Brzozowski, Z.; Saczewski, F. Eur. J. Med. Chem., Chem. Ther. 2002, 37, 709.
- von Harnish, H. Justus Lieb. Ann. Chem. 1972, 765, 14.
- Reddy, K. V.; Sabitha, G.; Rao, S. Org. Prep. Proced. Intern. 1996, 28, 325. [CrossRef]
- Ghosh, C. K.; Tewari, N.; Bhattacharyya, A. Synthesis 1984, 614.
- Skumat, A. P.; Babitch, Y. P.; Pivnenko, N. C.; Polyakov, V. K.; Lavrushkin, V. F. Zh. Obsh. Khim. 1989, 59, 1116.
- Shankar, M. S. S.; Reddy, R. B.; Chandra Mouli, G. V. P.; Reddy, Y. D. J. Ind. Chem. Soc. 1989, 66, 30.
- Polyakov, V. K.; Shevtsova, R. G.; Tsukerman, S. V. Ukr. Khim. Zh. 1982, 48, 772.
- Ried, W.; Vogl, M. Chem. Ber. 1982, 115, 403. [CrossRef]
- Caujolle, R.; Baziard-Mouysset, G.; Favrot, J. D.; Payard, M.; Loiseau, P. R.; Amarouch, H.; Linas, M. D.; Seguela, J. P.; Loiseau, P. M. Eur. J. Med. Chem. 1993, 28, 29. [CrossRef]
- Melikyan, G. S.; Lacova, M.; Kralova, K.; El-Shaaer, H. M.; Henselova, M.; Avetisyan, A. A. Chem. Papers 1993, 47, 388.
- Lacova, M.; El-Shaaer, H. M.; Loos, D.; Matulova, M.; Chovancova, J.; Furdik, M. Molecules 1998, 3, 120.
- Gasparova, R.; Lacova, M.; El-Shaaer, H. M.; Odlerova, Z. Il Farmaco 1997, 52, 251.
- Kralova, K.; Sersen, F.; Gasparova, R.; Lacova, M. Chem. Papers 1998, 52, 776.
- Tolmatchev, A. I.; Shulezhko, L. M.; Briks, Y.; Katchkovski, A. D. Khim. Geterocikl. Soed. 1990, 9, 1271.
- Sonawane, S. A.; Chavan, V. P.; Shingare, M. S.; Karale, B. K. Ind. J. Heterocycl. Chem. 2002, 12, 65.
- Sandulache, A.; Silva, A. M. S.; Pinto, D. C. G.A.; Almeida, L. C. M. P. M.; Cavaleiro, J. A. S. New J. Chem. 2003, 27, 1592.
- Silva, V. L. M.; Silva, A. M. S.; Pinto, D. C. G. A.; Cavaleiro, J. A. S. 10th Blue Danube Symposium on Heterocyclic Chemistry, Vienna, September 3-6, 2003.
- Pinto, D. C. G. A.; Silva, A. M. S.; Carrillo, J. R.; Diaz-Ortiz, A.; de la Hoz, A.; Cavaleiro, J. A. S. 10th Blue Danube Symposium on Heterocyclic Chemistry, Vienna, September 3-6, 2003.
- Pinto, D. C. G. A.; Silva, A. M. S.; Almeida, L. M. P. M.; Carrillo, J. R.; Diaz-Ortiz, A.; de la Hoz, A.; Cavaleiro, J. A. S. Synlett 2003, 10, 1415.
- Sample availability: Not applicable.
© 2005 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Gasparová, R.; Lácova, M. Reactions of 3-Formylchromone with Active Methylene and Methyl Compounds and Some Subsequent Reactions of the Resulting Condensation Products. Molecules 2005, 10, 937-960. https://doi.org/10.3390/10080937
Gasparová R, Lácova M. Reactions of 3-Formylchromone with Active Methylene and Methyl Compounds and Some Subsequent Reactions of the Resulting Condensation Products. Molecules. 2005; 10(8):937-960. https://doi.org/10.3390/10080937
Chicago/Turabian StyleGasparová, R., and M. Lácova. 2005. "Reactions of 3-Formylchromone with Active Methylene and Methyl Compounds and Some Subsequent Reactions of the Resulting Condensation Products" Molecules 10, no. 8: 937-960. https://doi.org/10.3390/10080937
APA StyleGasparová, R., & Lácova, M. (2005). Reactions of 3-Formylchromone with Active Methylene and Methyl Compounds and Some Subsequent Reactions of the Resulting Condensation Products. Molecules, 10(8), 937-960. https://doi.org/10.3390/10080937