An Aromatic Iodination Method, with Iodic Acid Used as the Only Iodinating Reagent

Abstract
:Introduction

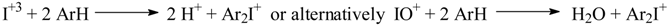

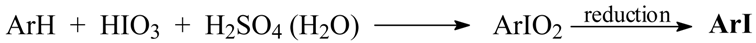
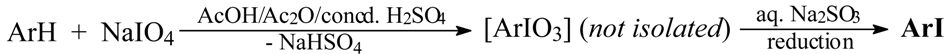

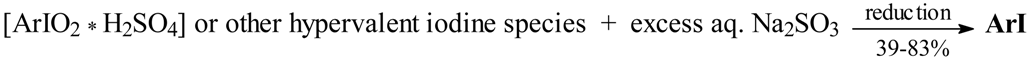

| Substrate | Product | Yield, (%) | Analysis, % I calcd (found) | Mp (°C)/Sa) | Lit, mp (°C) [10] |
|---|---|---|---|---|---|
| C6H6 | 1,4-I2C6H4 | 60 | 76.95 (76.5) | 127-128/E | 129 |
| PhI | 1,4-I2C6H4 | 58 | 76.95 (77.2) | 127.5-129/E | 129 |
| PhBr | 4-BrC6H4I | 71 | 44.86 (44.8) | 95-97/E | 92 |
| PhCl | 4-ClC6H4I | 39 | 53.22 (52.7) | 54-56/E | 57 |
| PhNO2 | 3-IC6H4NO2 | 82 | 50.96 (50.4) | 35-37/P | 35-36 |
| PhCOOH | 3-IC6H4COOH | 67 | 51.17 (51.2) | 189-190/C | 188-189 |
| PhCOOMe | 3-IC6H4COOMe | 59 | 48.43 (48.5) | 55-56/EW | 54-55 |
| PhCOOEt | 3-IC6H4COOEt | 51 | 45.97 (45.8) | bp 170-172/29 | bp 150/15 |
| 4-MeC6H4COOH | 3-I-4-MeC6H3COOH | 78 | 48.43 (48.2) | 211-212/C | 210-212 |
| 4-MeC6H4NO2 | 3-I-4-MeC6H3NO2 | 40 | 48.25 (47.9) | 52-54/E | 54-56 |
| 4-MeOC6H4NO2 | 3-I-4-MeOC6H3NO2 | 83 | 45.51 (45.1) | 94-96/E | 97 |
| 2-O2NC6H4NHCOCH3 | 4-I-2-O2NC6H3NHCOCH3 | 52 | 41.50 (41.5) | 110-112/EW | 112 |
Results and Discussion
Experimental
General
Optimized Preparations of Iodoarenes from Arenes
Possibly Optimized Preparation of 1-Bromo-4-iodylbenzene from Bromobenzene
References and Notes
- Meyer, V.; Wachter, W. Ueber Jodosobenzoësäure. Ber. Dtsch. Chem. Ges. 1892, 25, 2632–2635. [Google Scholar] [CrossRef] The German passage in the text can be translated as follows: “Next, it has to be checked out whether the reaction of aromatic compounds with sulfuric acid and iodic acid, IO2OH, corresponding to the reaction with nitric acid, NO2OH, can afford the respective compounds substitued by the –IO2 group. We can only preliminarily report that benzene reacts violently with iodic acid and sulfuric acid, and after addition of water, a solid compound precipitates out, which was not closer investigated”. Mason and Race (Ref. 3, footnote in p. 1723) commented the first German attempt to prepare PhIO2 from benzene as follows: “It does not appear that the experiment was followed up either then or after 1894, when the iodoxy- and the iodonium group were discovered”.
- Masson, I.; Race, E.; Pounder, F. E. The Iodoxy-group and its Relations. J. Chem. Soc. 1935, 1669–1679. [Google Scholar]
- Masson, I.; Race, E. The Direct Conversion of Iodic Acid and Aromatic Hydrocarbons into Iodonium Compounds. J. Chem. Soc. 1937, 1718–1723. [Google Scholar] [CrossRef]
- Beringer, F. M.; Drexler, M.; Gindler, E. M.; Lumpkin, C. C. Diaryliodonium Salts. I. Synthesis. J. Am. Chem. Soc. 1953, 75, 2705–2708. [Google Scholar] Krowczyński, A.; Skulski, L. p-Iodo-N-methylacetanilide and its Polyvalent Iodine Derivatives. Polish J. Chem. 1993, 67, 67–70. [Google Scholar]
- Beringer, F. M.; Falk, R. A.; Karniol, M.; Lillien, I.; Masullo, G.; Mausner, M.; Sommer, E. Diaryliodonium Salts. IX. The Synthesis of Substituted Diphenyliodonium Salts. J. Am. Chem. Soc. 1959, 81, 342–351. [Google Scholar]
- Ciustea, G.; Panescu, A. New synthetic routes in the chemistry of the organic iodine compounds (in Roumanian). Rev. Chim. (Bucharest) 1969, 20, 216, [Chem. Abstr. 1969, 71, 123755]. [Google Scholar]
- Skulski, L. Organic Iodine(I, III, and V) Chemistry: 10 Years of Development at the Medical University of Warsaw, Poland (1990-2000). Molecules 2000, 5, 1331–1371, Avail. at URL: http://www.mdpi.org/molecules/papers/51201311.pdf. [Google Scholar]
- Rudzki, P. An attempt of the direct iodylation of arenes. M. Sc. Thesis, Faculty of Pharmacy, Medical University of Warsaw, Poland, 2003. [Google Scholar]
- Lulinski, P.; Sosnowski, M.; Skulski, L. A Novel Aromatic Iodination Method, with Sodium Periodate Used Alone as the Iodinating Reagent. Paper presented at the 7th Electronic Conference on Synthetic Organic Chemistry (ECSOC-7, http://www.mdpi.net/ecsoc-7), November 1-30, 2003 (paper A013). Submitted for publication in Molecules.
- Dictionary of Organic Compounds,, 6th ed.; Chapman & Hall: London, 1996.
- Kazmierczak, P.; Skulski, L.; Kraszkiewicz, L. Syntheses of (Diacetoxyiodo)arenes or Iodylarenes from Iodoarenes, with Sodium Periodate as the Oxidant. Molecules 2001, 6, 881–891, Avail. at http://www.mdpi.org/molecules/papers/61200881.pdf. [Google Scholar]
- Kraszkiewicz, L.; Skulski, L. Optimized syntheses of iodylarenes from iodoarenes, with sodium periodate as the oxidant. Part II. ARKIVOC. (Gainsville, FL, USA) [online computer file]. 2003, (vi), pp. 120–125, Avail. at http://www.arkat-usa.org/ark/journal/2003/Varvoglis/AV-657A/AV-657A.htm.
- Groweiss, A. Use of Sodium Bromate for Aromatic Bromination: Research and Development. Org. Process. Res. Dev. 2000, 4, 30–33. [Google Scholar] See also: Harrison, J. J.; Pellegrini, J. P.; Selwitz, C. M. Bromination of Deactivated Aromatics Using Potassium Bromate. J. Org. Chem. 1981, 46, 2169–2171. [Google Scholar] , and references therein; Yamaoka, Y. Preparation of 1-bromo-3-nitrobenzene. Jpn. Kokai Tokkyo Koho JP 11 228,505 [99 217,344], 10 Aug 1999; [Chem. Abstr. 1999, 131, 144408].
- Sample Availability: Available from the authors.
© 2005 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Krassowska-Swiebocka, B.; Prokopienko, G.; Skulski, L. An Aromatic Iodination Method, with Iodic Acid Used as the Only Iodinating Reagent. Molecules 2005, 10, 394-400. https://doi.org/10.3390/10020394
Krassowska-Swiebocka B, Prokopienko G, Skulski L. An Aromatic Iodination Method, with Iodic Acid Used as the Only Iodinating Reagent. Molecules. 2005; 10(2):394-400. https://doi.org/10.3390/10020394
Chicago/Turabian StyleKrassowska-Swiebocka, Barbara, Grazyna Prokopienko, and Lech Skulski. 2005. "An Aromatic Iodination Method, with Iodic Acid Used as the Only Iodinating Reagent" Molecules 10, no. 2: 394-400. https://doi.org/10.3390/10020394
APA StyleKrassowska-Swiebocka, B., Prokopienko, G., & Skulski, L. (2005). An Aromatic Iodination Method, with Iodic Acid Used as the Only Iodinating Reagent. Molecules, 10(2), 394-400. https://doi.org/10.3390/10020394