Synthesis and X-ray Structure of a New Pyrrolo[1,2-b]-pyridazine Derivative

Abstract
:Introduction
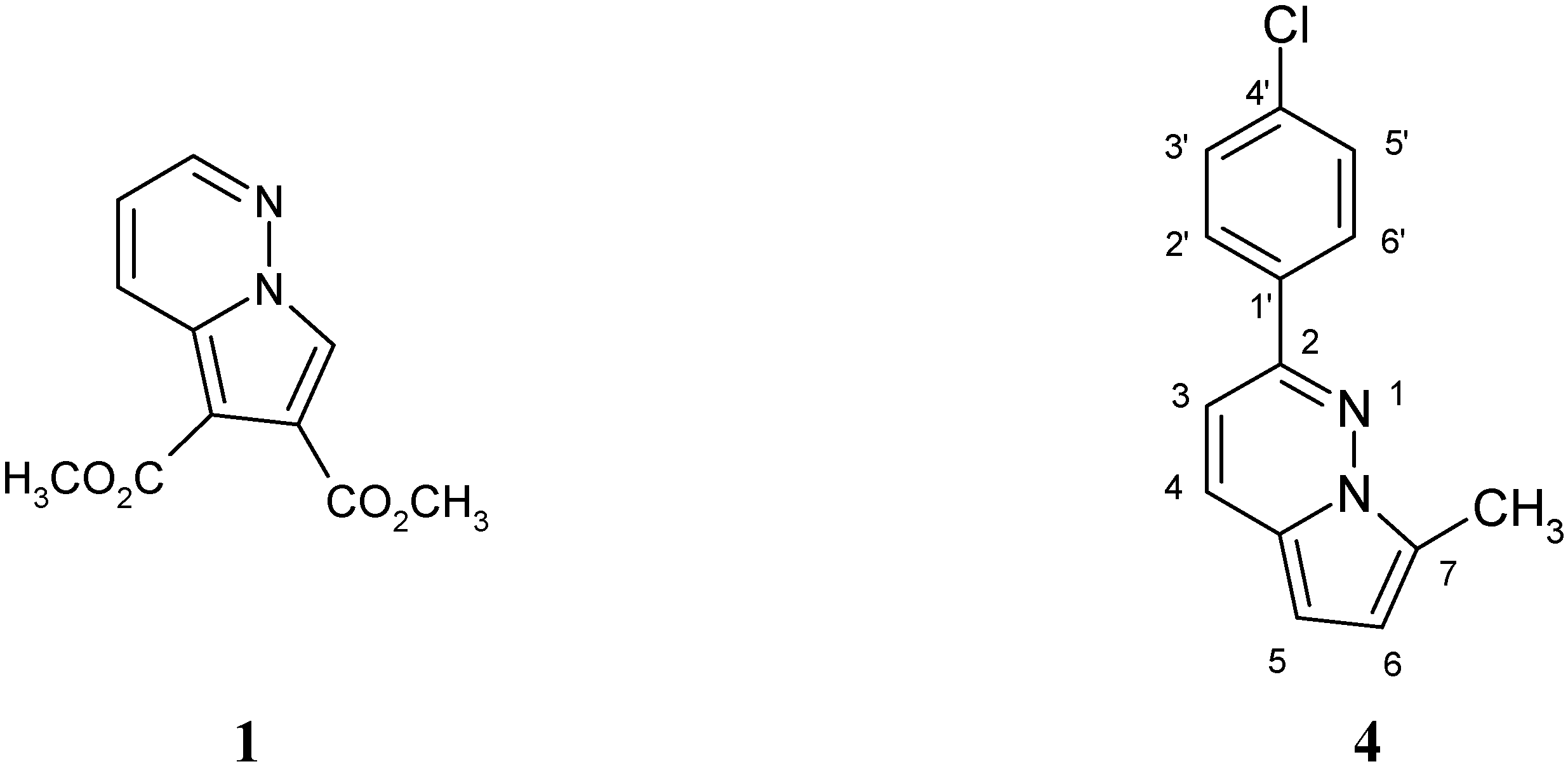
Results and Discussion
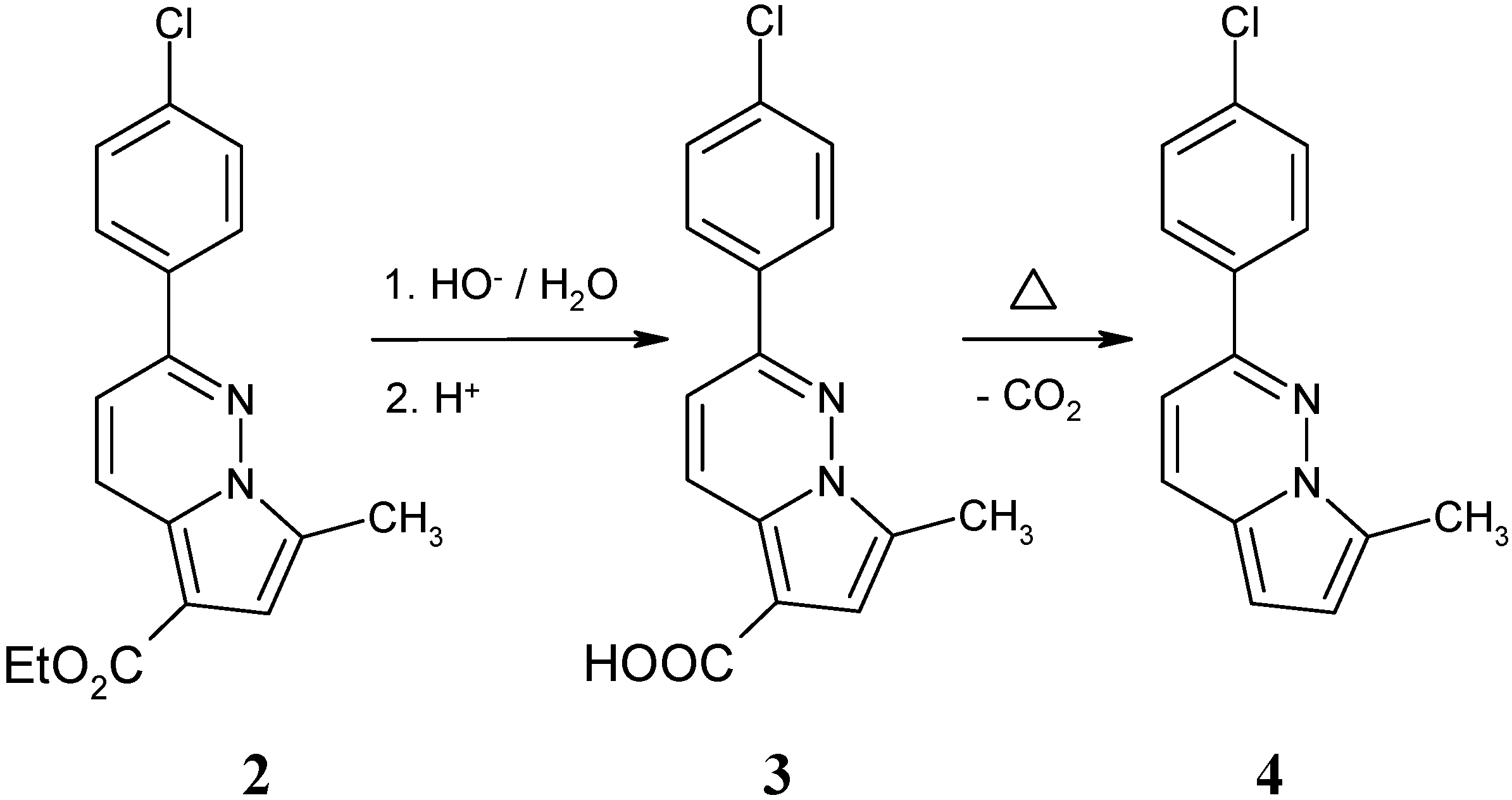
Experimental
General
Syntheses
2-(4-Chlorophenyl)-7-methylpyrrolo[1,2-b]pyridazine-5-carboxylate (3)
2-(4-Chlorophenyl)-7-methylpyrrolo[1,2-b]pyridazine (4).
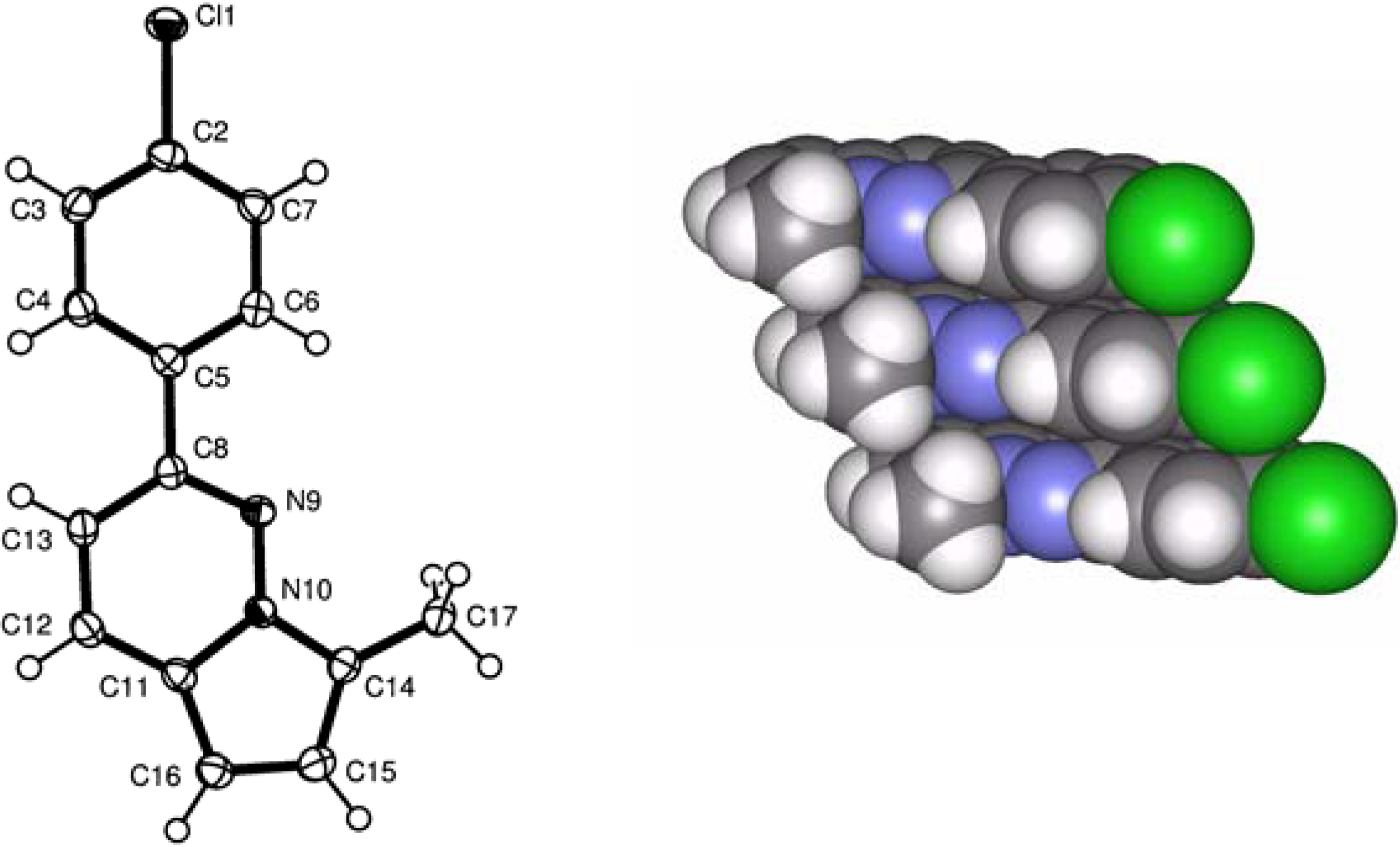
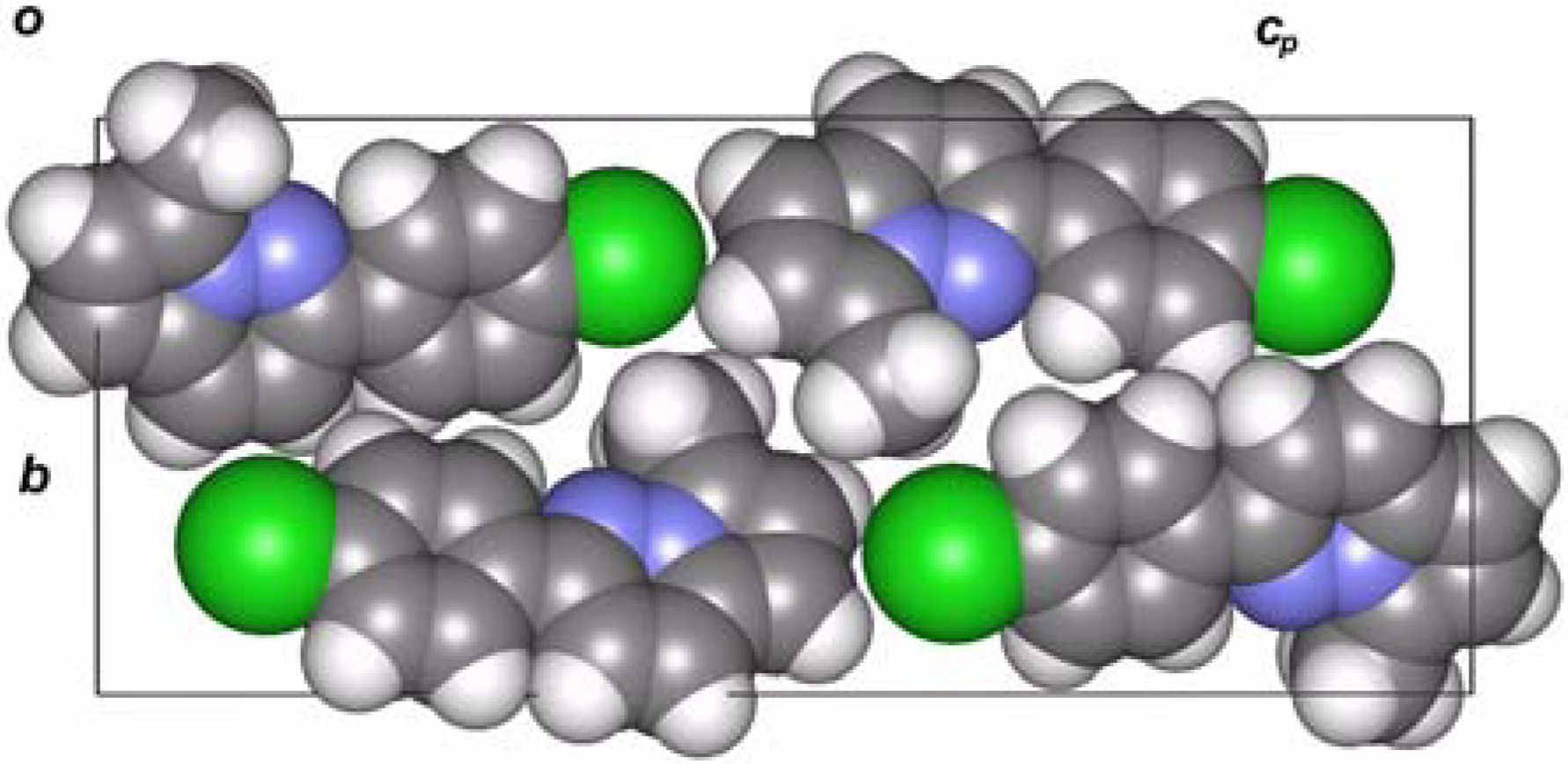
X-ray analysis of 4

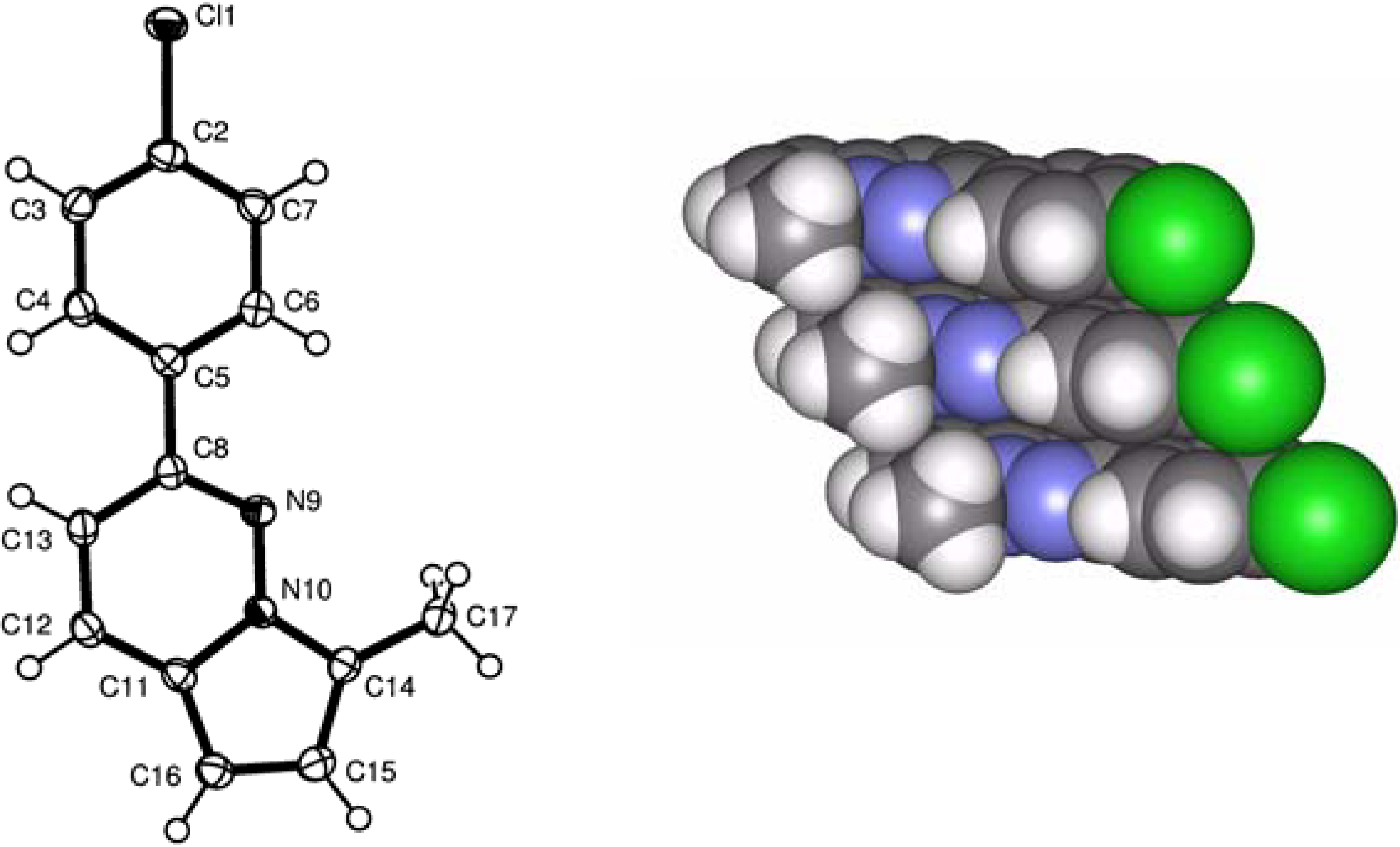{kind=link}
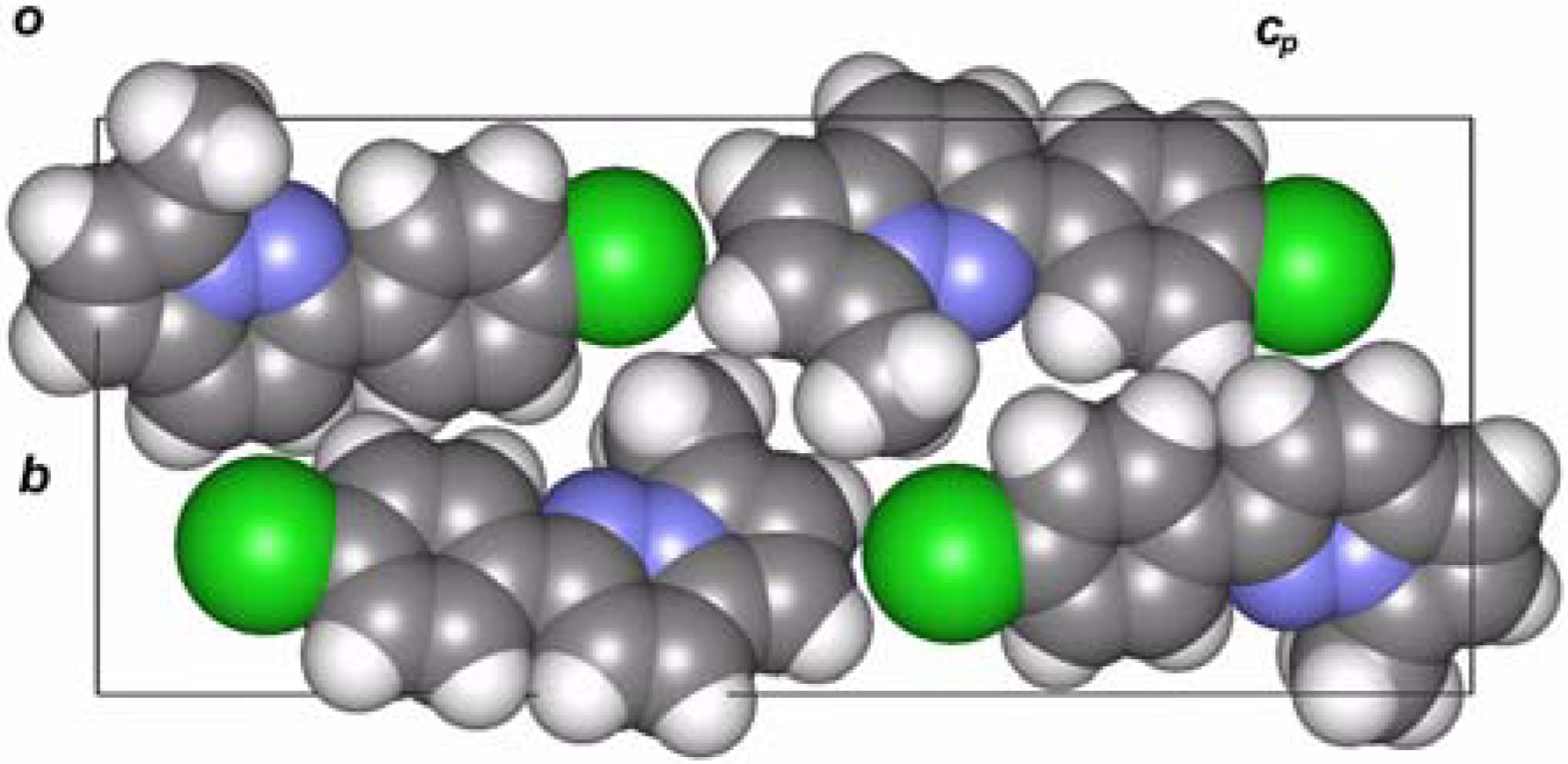{kind=link}
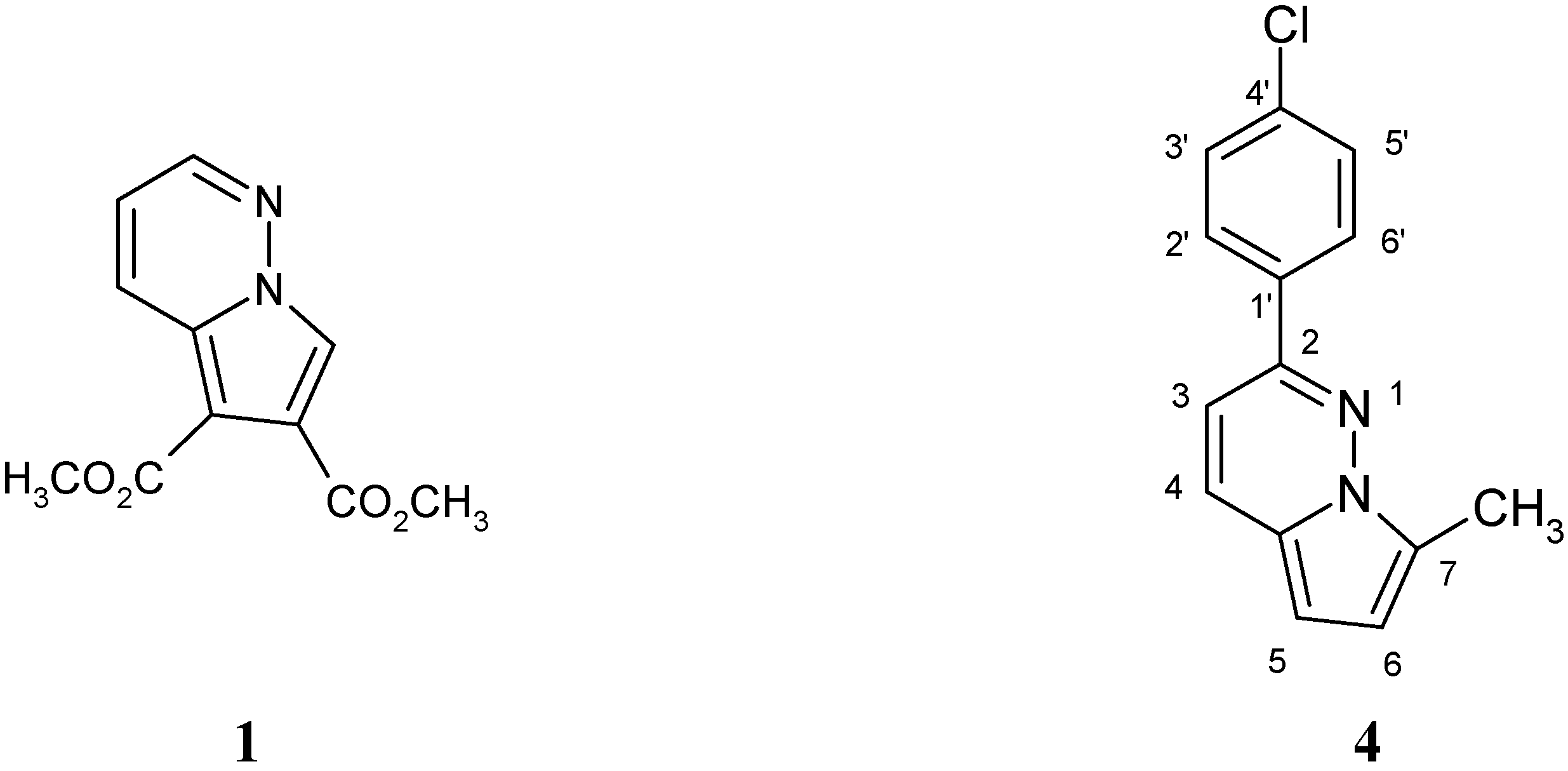{kind=link}
{kind=link}
| Chemical formula | C14H11ClN2 |
| Formula weight | 242.71 |
| Crystal Color, Habit | yellow, needle |
| Crystal Dimensions | 0.08 x 0.12 x 0.18 |
| Crystal System | monoclinic |
| Lattice Type | primitive |
| Space Group | P21/c (#14) |
| Lattice Parameters | a = 3.8568(1) Å |
| b = 11.0690(3) Å | |
| c = 26.4243(7) Å | |
| β = 92.777(1)° | |
| Volume | 1126.75(5) Å3 |
| Z value | 4 |
| Dcalc | 1.431 g/cm3 |
| F(000) | 504 |
| μ(MoKα) | 0.314 mm-1 |
| Reflections/restraints/parameters | 2460 / 0 / 156 |
| Residuals: R1, wR2 [I > 2σ(I)] | 0.0578, 0.1305 |
| Goodness of Fit, S | 1.158 |
| Max. shift/error | < 0.001 |
| Max. peak in final Δρ synthesis | 0.49 |
| Min. peak in final Δρ synthesis | -0.32 |
Acknowledgements
References
- Cheng, Y.; Ma, B.; Wudl, F. J. Mater. Chem. 1999, 9, 2183–2188.
- Mitsumori, T.; Bendikov, M.; Sedó, J.; Wudl, F. Chem. Mater. 2003, 15, 3759–3768.
- Vasilescu, M.; Dumitrascu, F.; Lemmetyinen, H.; Tkachenko, N. J. Fluorescence 2004, 14, 443–450.
- Ruxer, J. M.; Lachoux, C.; Ousset, J. B.; Torregrosa, J. L.; Mattioda, G. J. Heterocycl. Chem. 1994, 31, 409–417.
- Ungureanu, M.; Mangalagiu, I.; Grosu, G.; Petrovanu, M. Ann. Pharm. Fr. 1997, 55, 69–72, [Chem. Abstr. 1997, 126, 303587].
- Nassir, A. I.; Gundersen, L.-L.; Rise, F.; Antonsen, Ø; Kristensen, T.; Langhelle, B.; Bast, A.; Custers, I.; Haenen, G. R. M. M.; Wikström, H. Bioorg. Med. Chem. Lett. 1998, 8, 1829–1832. [CrossRef]
- Østby, O. B.; Dalhus, B.; Gundersen, L.-L.; Rise, F.; Bast, A.; Haenen, G. R. M. M. Eur. J. Org. Chem. 2000, 9, 3763–3770.
- Ohtani, M.; Fuji, M.; Fukui, Y.; Adachi, M. World Patent WO 9959999 A1, 1999. [Chem. Abstr. 2000, 132, 12318].
- Østby, O. B.; Gundersen, L.-L.; Rise, F.; Antonsen, Ø.; Fosnes, K.; Larsen, V.; Bast, A.; Custers, I.; Haenen, G. R. M. M. Arch. Pharm. Pharm. Med. Chem. 2001, 334, 21–24.
- Pal, M.; Batchu, V. R.; Khanna, S.; Yeleswarapu, K. R. Tetrahedron 2002, 58, 9933–9940.
- Dumitraşcu, F.; Mitan, C. I.; Dumitrescu, D.; Drăghici, C.; Căproiu, M. T.; Vasilescu, M. Book of Abstracts, 10th Blue Danube Symposium on Heterocyclic Chemistry; PO-126; Vienna, Austria, September 3-6 2003. [Google Scholar]
- Farrugia, L. J. ORTEP-3 for windows - a version of ORTEP-III with a graphical user interface (GUI). J. Appl. Crystallogr. 2000, 30, 565. [Google Scholar]
- COLLECT. In Nonius 2000; Nonius BV: Delft, The Netherlands, 2000.
- Otwinowski, Z.; Minor, W. Methods Enzymol. 1997, 276, 307–326.
- Sheldrick, G. M. SADABS. Program for Empirical Absorption Correction of Area Detector Data; University of Göttingen: Göttingen, Germany, 1996. [Google Scholar]
- Sheldrick, G. M. Acta Crystallogr. 1990, A46, 467–473.
- Sheldrick, G. M. SHELXL97; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Spek, A. L. PLATON, A multipurpose Crystallographic Tool, Version 10500 © 1980-2000;
- WebLab ViewerPro Version 3.5; Molecular Simulations Inc.: San Diego, CA, USA, © 1999.
- Sample availability: Samples of compound 4 are available from the authors.
© 2005 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Caira, M.R.; Dumitrascu, F.; Draghici, C.; Dumitrescu, D.; Cristea, M. Synthesis and X-ray Structure of a New Pyrrolo[1,2-b]-pyridazine Derivative. Molecules 2005, 10, 360-366. https://doi.org/10.3390/10020360
Caira MR, Dumitrascu F, Draghici C, Dumitrescu D, Cristea M. Synthesis and X-ray Structure of a New Pyrrolo[1,2-b]-pyridazine Derivative. Molecules. 2005; 10(2):360-366. https://doi.org/10.3390/10020360
Chicago/Turabian StyleCaira, Mino R., Florea Dumitrascu, Constantin Draghici, Denisa Dumitrescu, and Mihaela Cristea. 2005. "Synthesis and X-ray Structure of a New Pyrrolo[1,2-b]-pyridazine Derivative" Molecules 10, no. 2: 360-366. https://doi.org/10.3390/10020360
APA StyleCaira, M. R., Dumitrascu, F., Draghici, C., Dumitrescu, D., & Cristea, M. (2005). Synthesis and X-ray Structure of a New Pyrrolo[1,2-b]-pyridazine Derivative. Molecules, 10(2), 360-366. https://doi.org/10.3390/10020360