
Jmjd3 Mediates Neuropathic Pain by Inducing Macrophage Infiltration and Activation in Lumbar Spinal Stenosis Animal Model

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
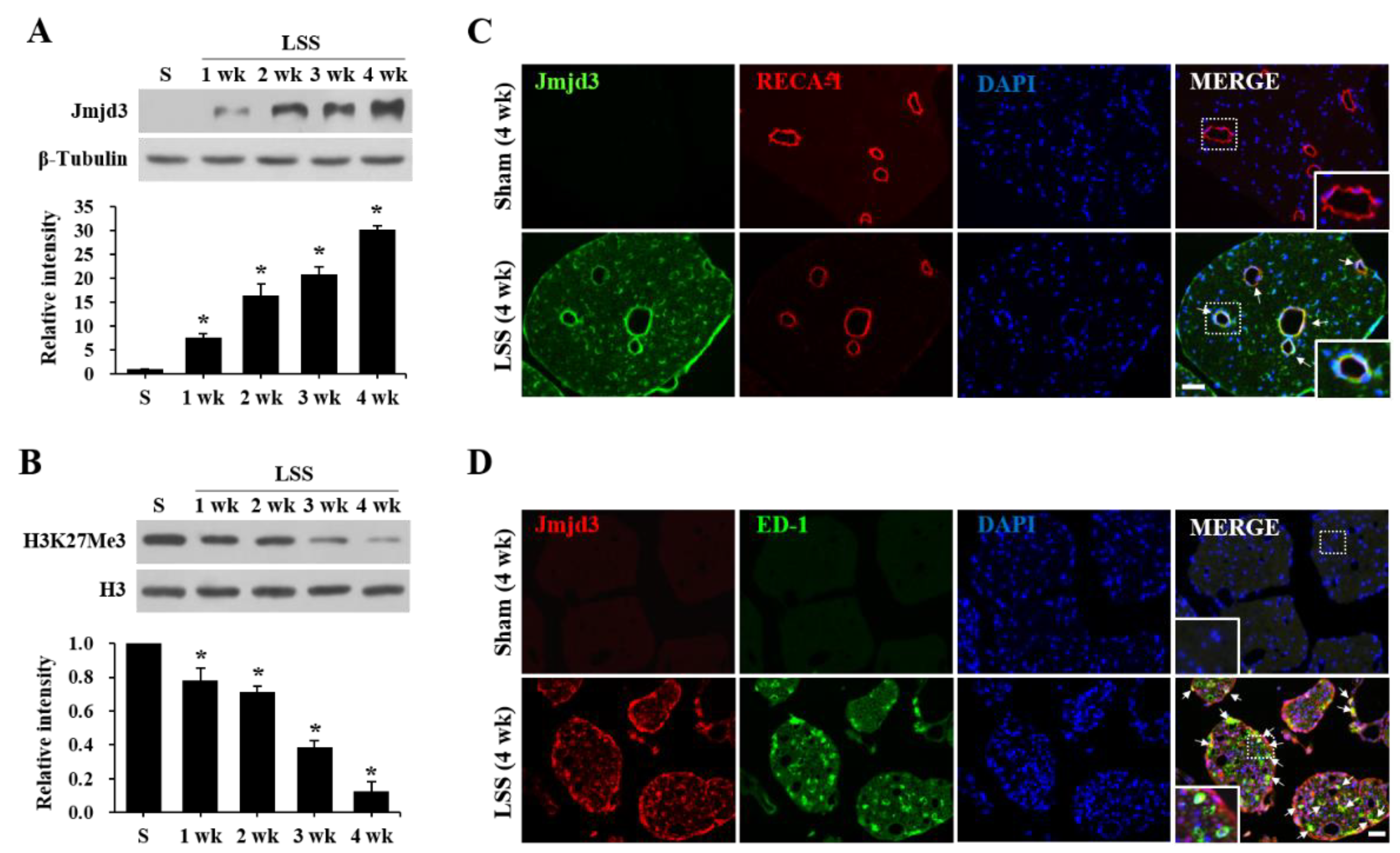
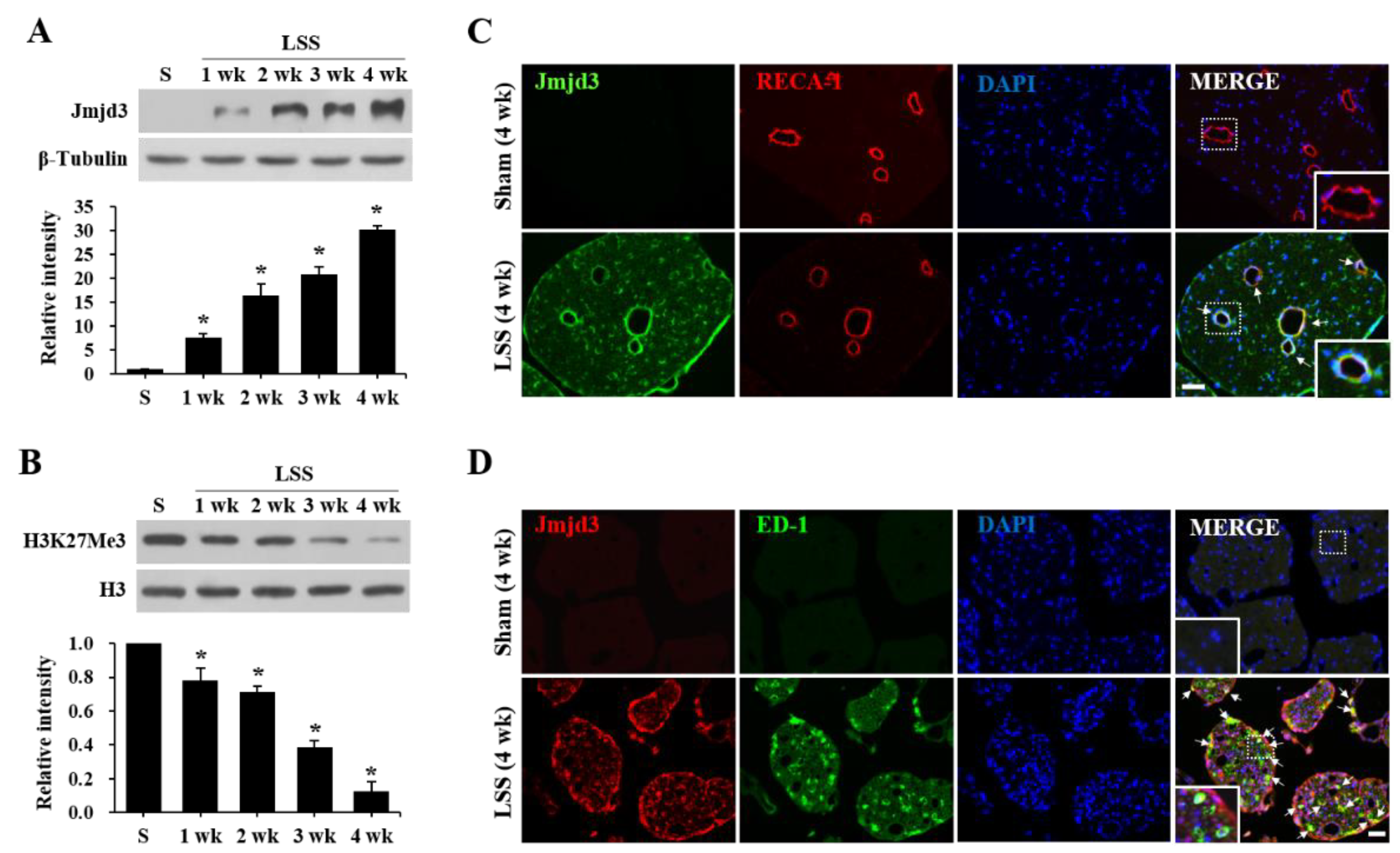
2.1. Jmjd3 Expression and Activity Is Increased after Cauda Equine Compression in an LSS Model
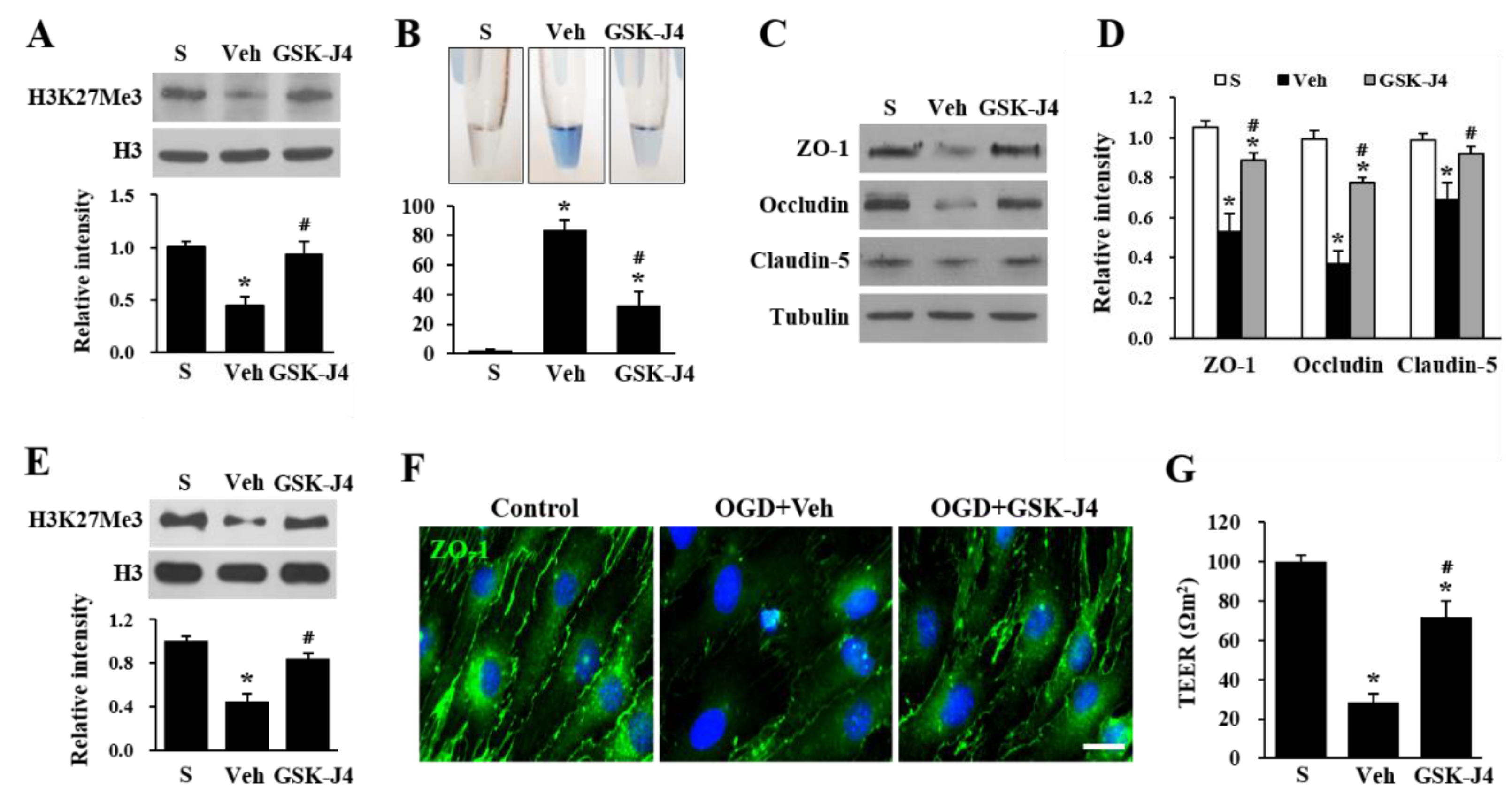
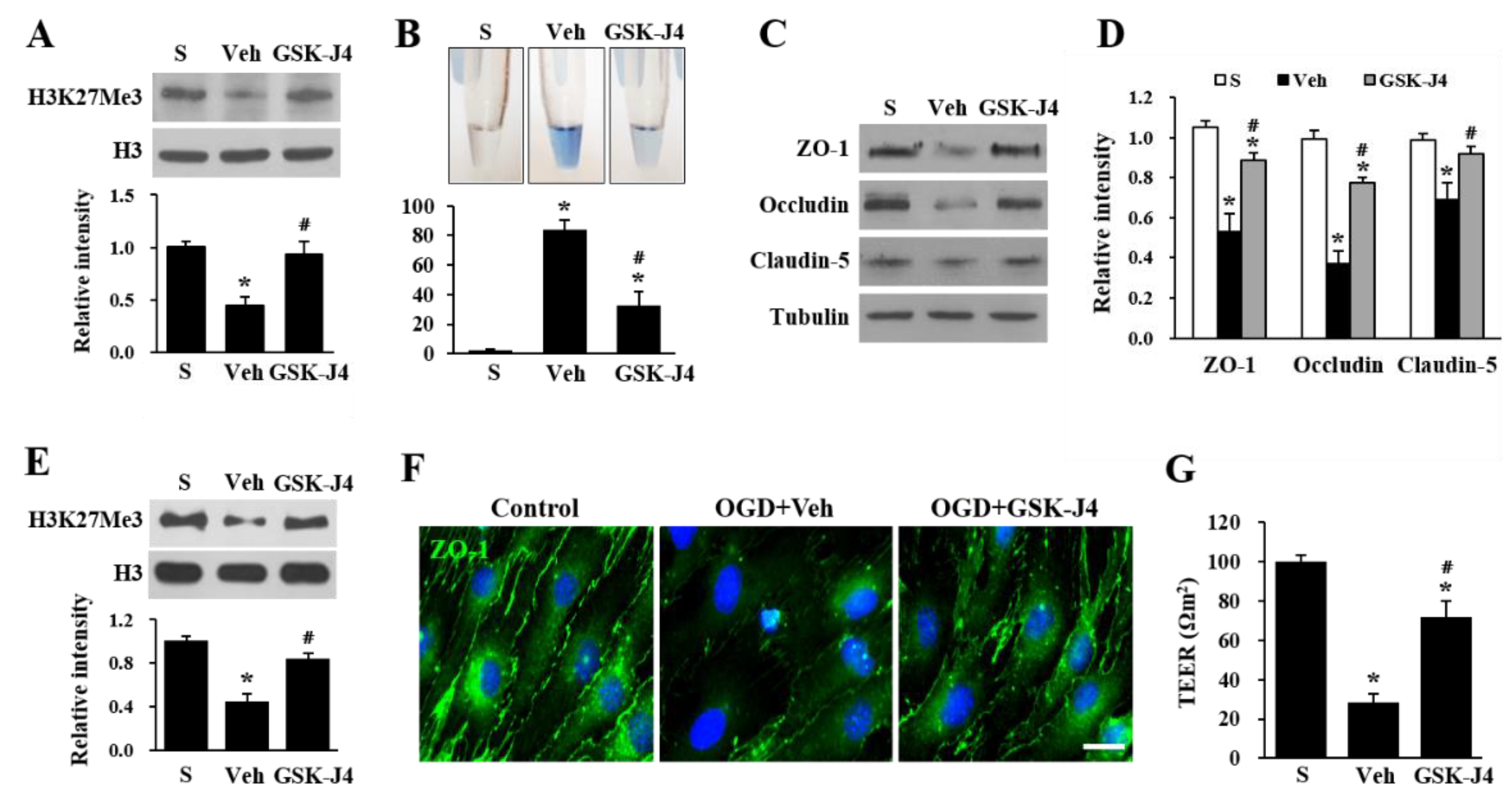
2.2. Jmjd3 Regulates Blood-Nerve Barrier Permeability after Cauda Equine Compression in an LSS Model
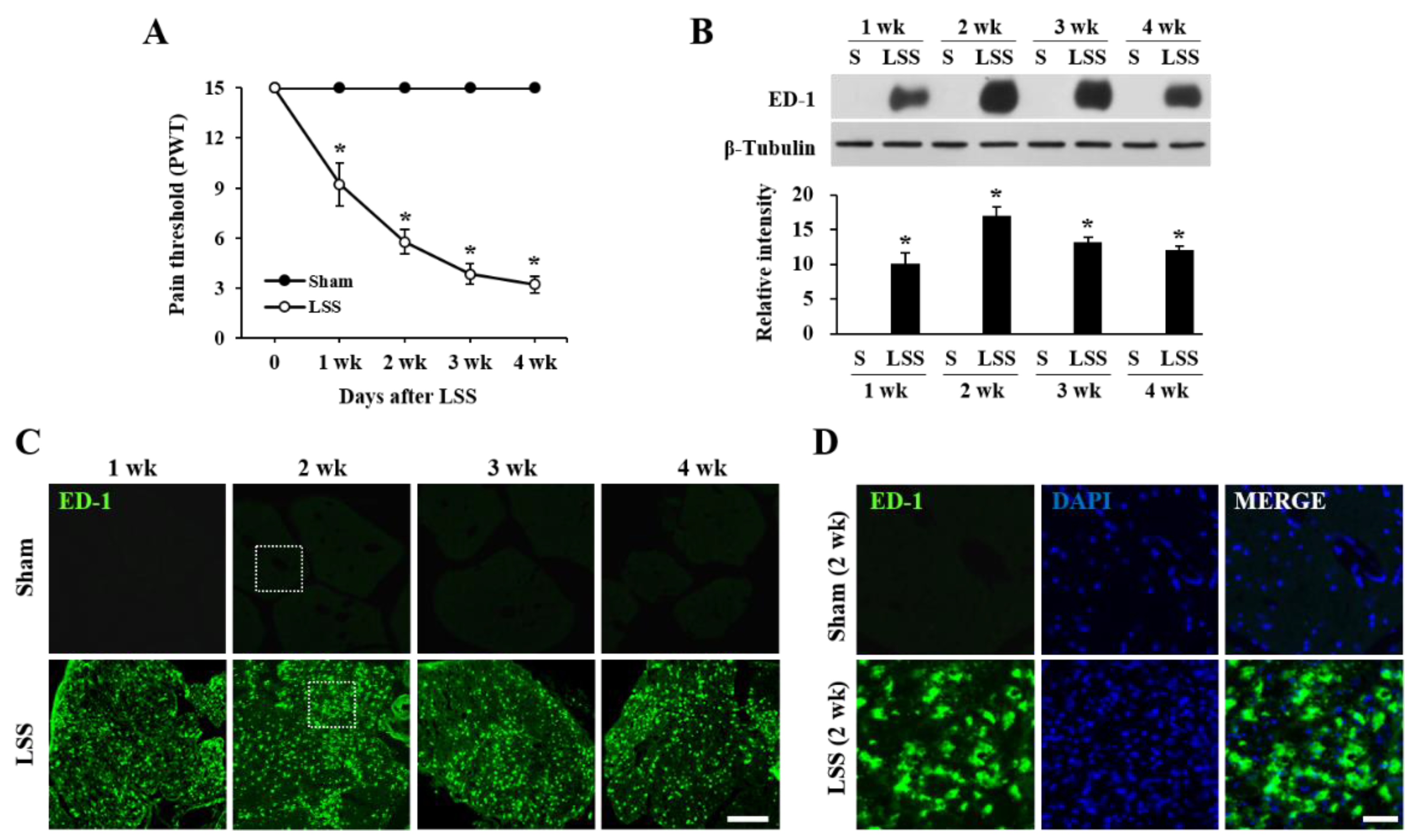
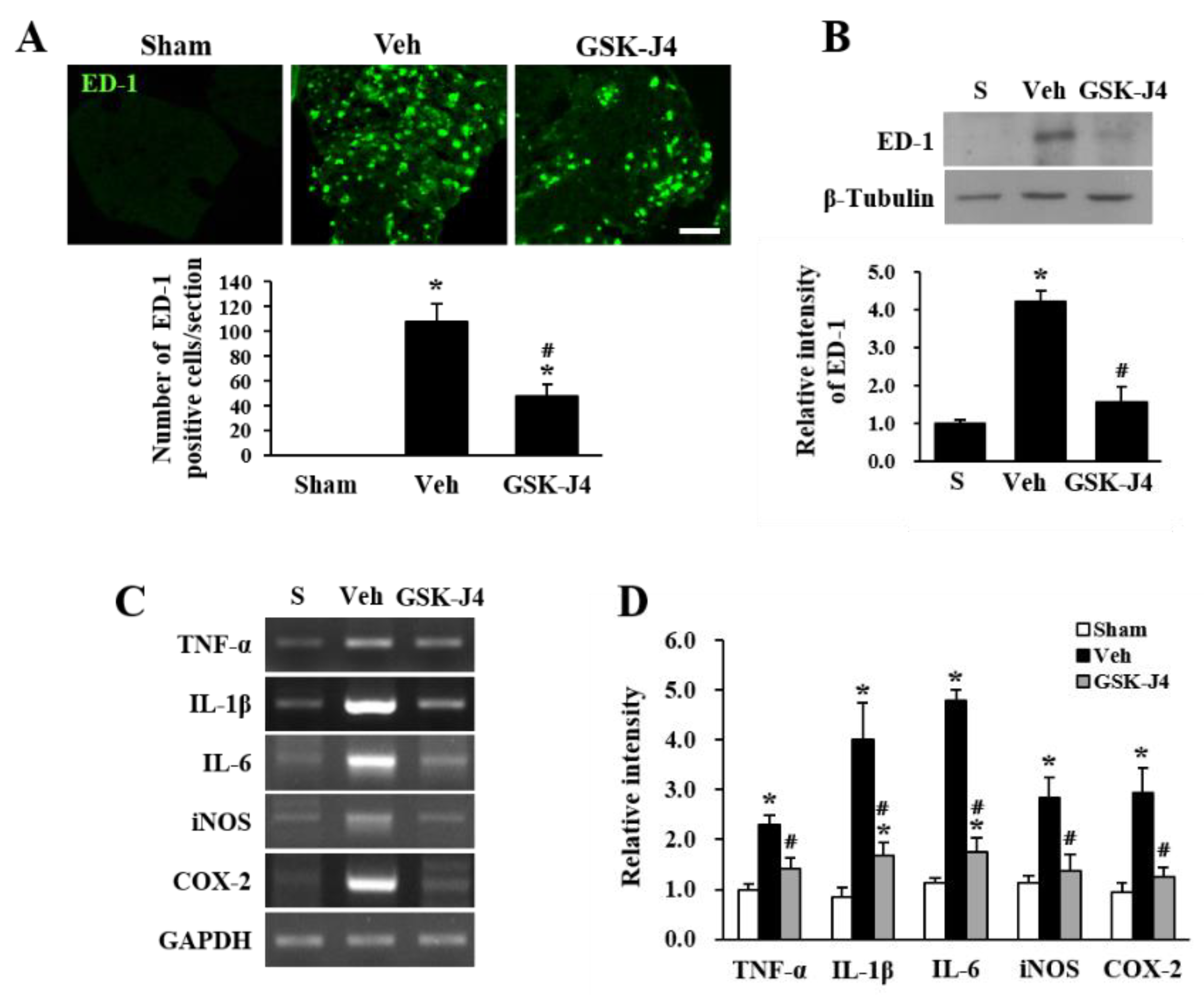
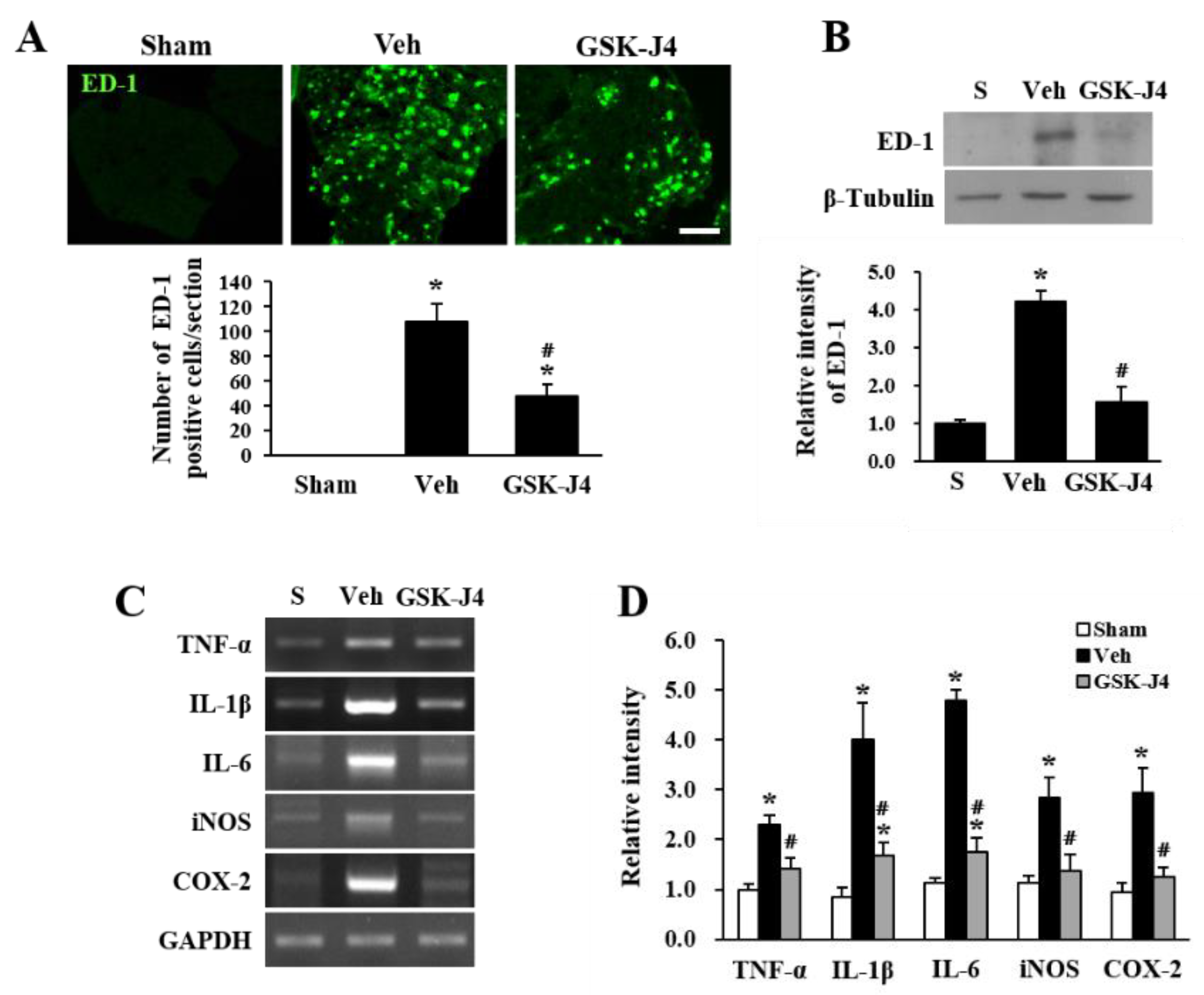
2.3. Jmjd3 Inhibitor Reduces Macrophage Infiltration and Inflammation in Injured Cauda Equina of LSS Rats
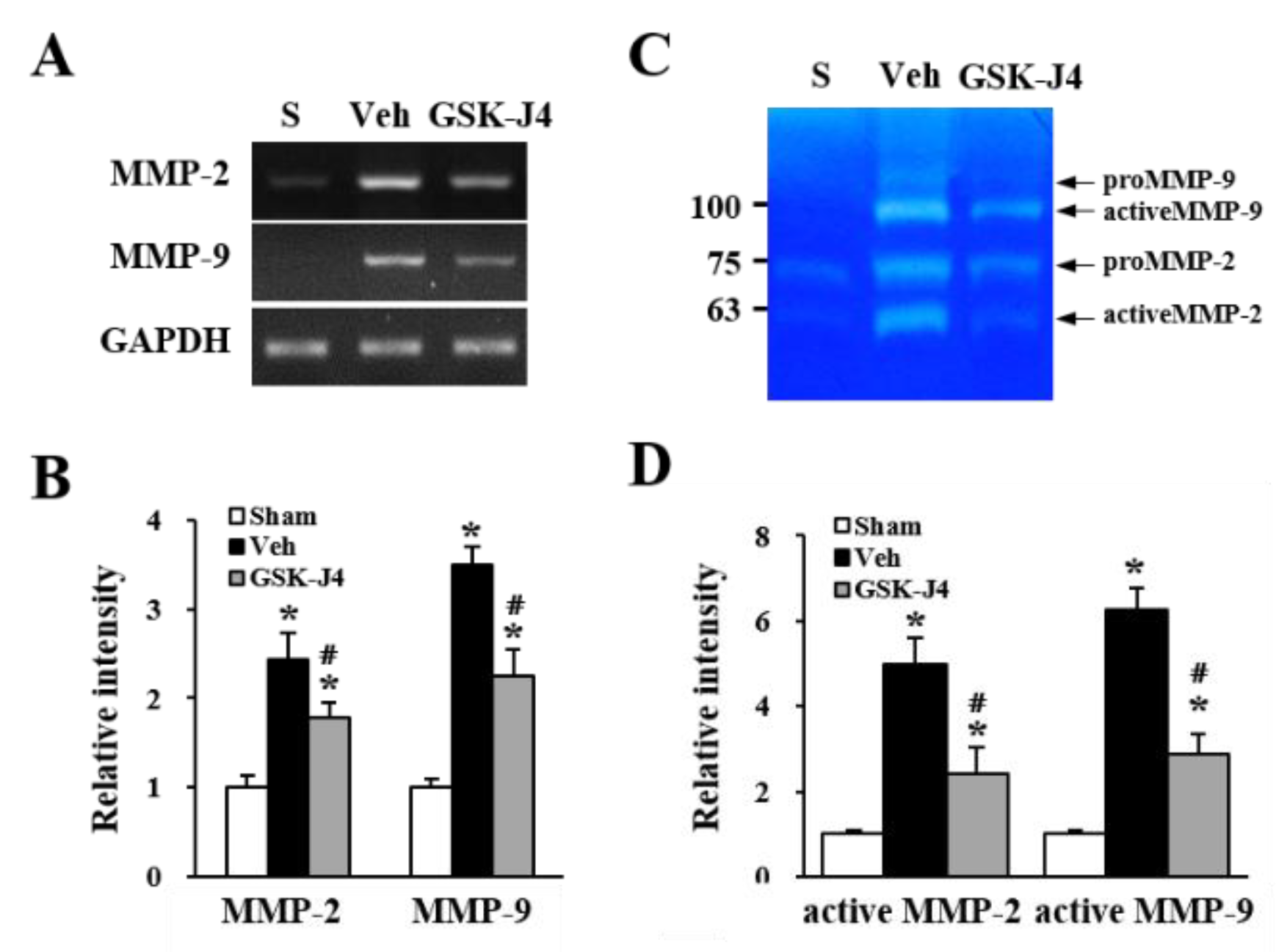
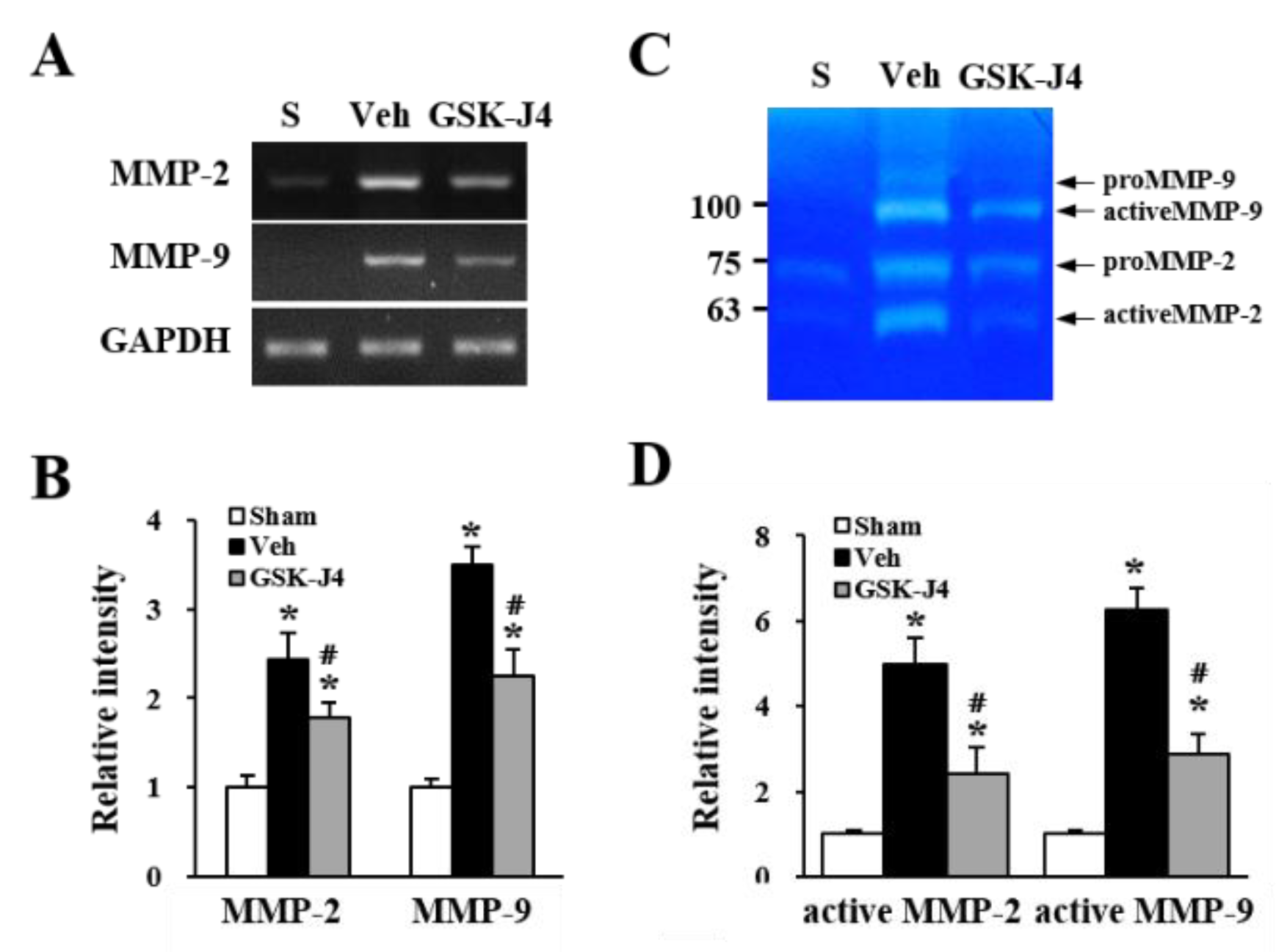
2.4. Jmjd3 Regulates the Expression and Activity of MMP-2/9 in Cauda Equina after Compression Injury
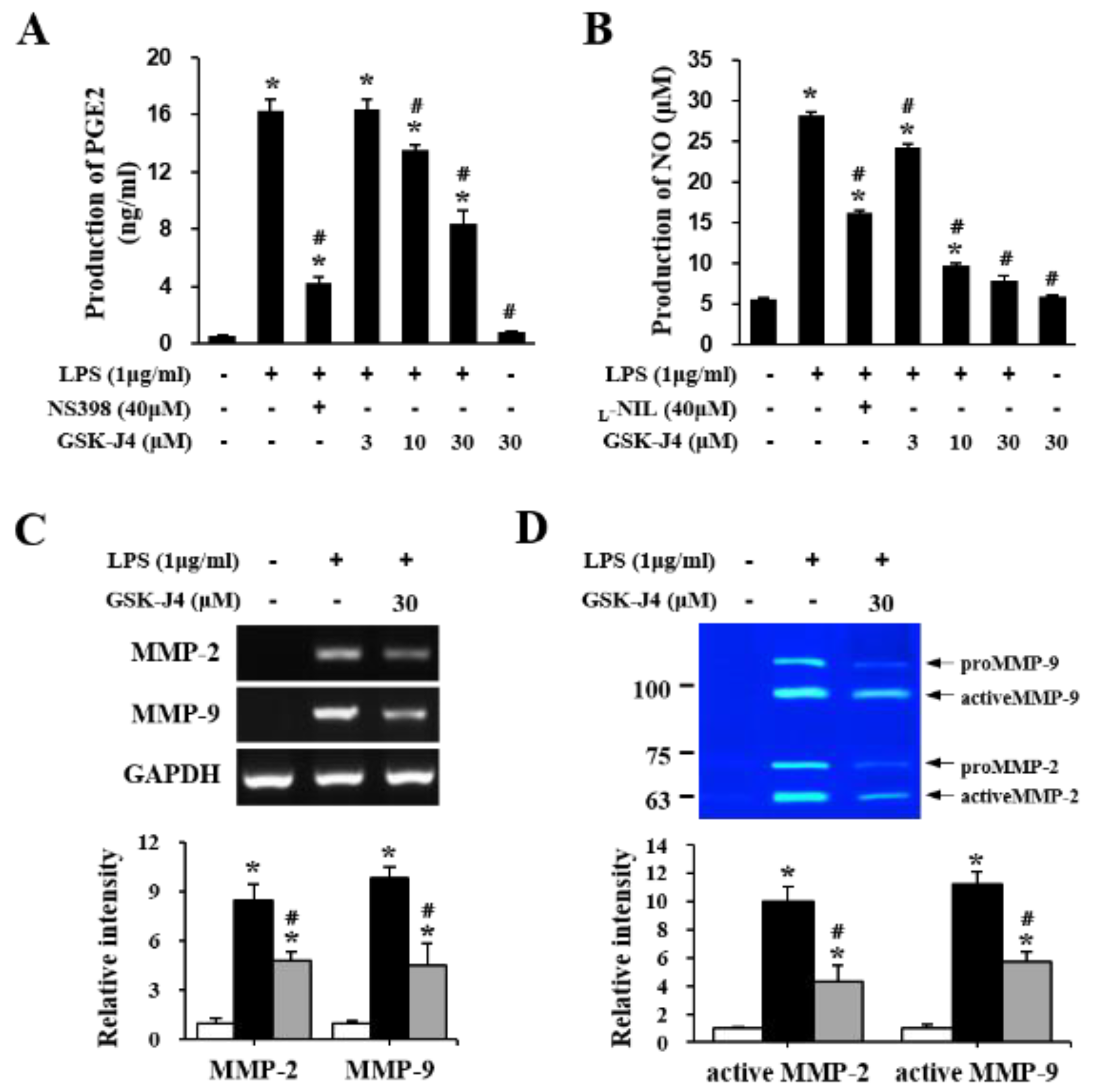
2.5. Jmjd3 Also Regulates Macrophage Activation
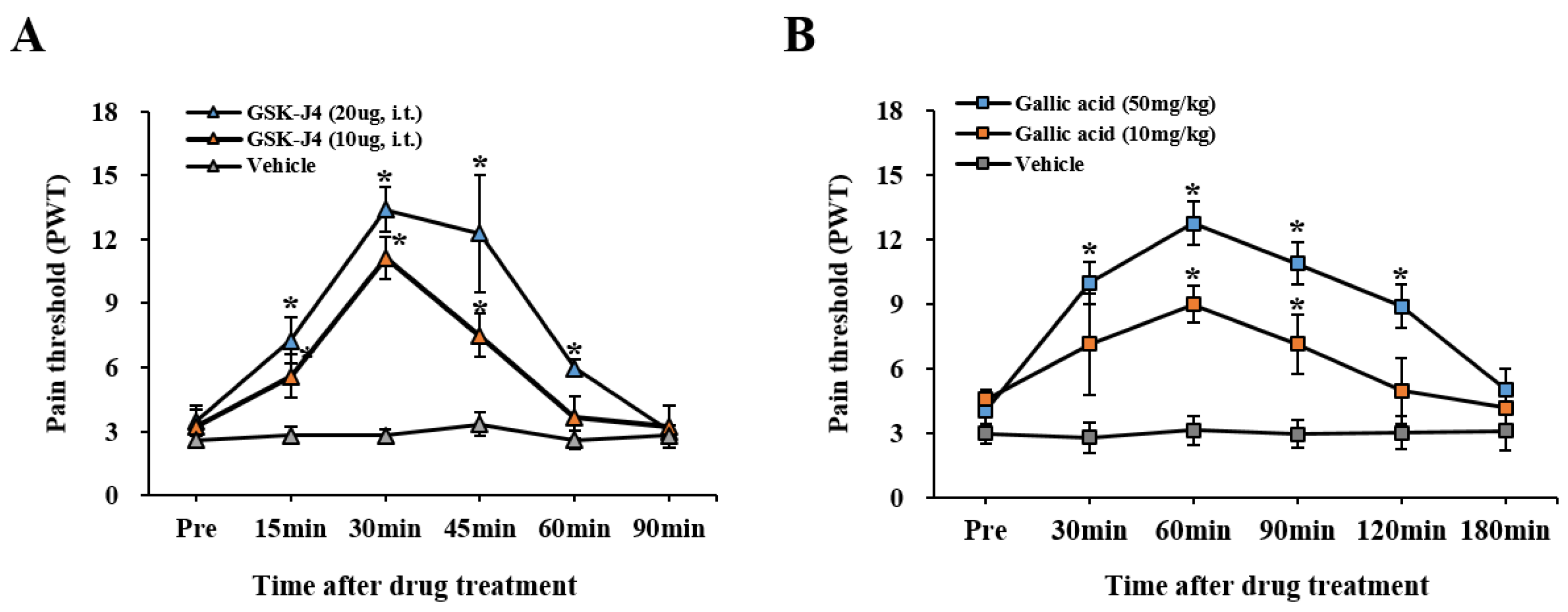
2.6. The Inhibition of Jmjd3 Activity Alleviates LSS-Induced Mechanical Allodynia
3. Discussion
4. Materials and Methods
4.1. Animals and Ethics Statement
4.2. Cauda Equina Compression Injury
4.3. Pain Behavioral Tests
4.4. Drug Administration
4.5. Tissue Preparation
4.6. Immunohistochemistry and Immunocytochemistry
4.7. Western Blot
4.8. Gelatin Zymography
4.9. RNA Isolation and RT-PCR
4.10. Endothelial Cell Culture and OGD/Reperfusion
4.11. Measurement of Transendothelial Electrical Resistance (TEER)
4.12. RAW 264.7 Macrophages Culture
4.13. Determination of NO and PGE2 Production
4.14. Measurement of Blood-Nerve Barrier Permeability
4.15. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, Q.; Liu, Y.; Chu, Z.; Chen, J.; Dai, F.; Zhu, X.; Hu, A.; Yun, C. Brain-derived neurotrophic factor expression in dorsal root ganglia of a lumbar spinal stenosis model in rats. Mol. Med. Rep. 2013, 8, 1836–1844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lurie, J.; Tomkins-Lane, C. Management of lumbar spinal stenosis. BMJ 2016, 352, h6234. [Google Scholar] [CrossRef]
- Markman, J.D.; Frazer, M.E.; Rast, S.A.; McDermott, M.P.; Gewandter, J.S.; Chowdhry, A.K.; Czerniecka, K.; Pilcher, W.H.; Simon, L.S.; Dworkin, R.H. Double-blind, randomized, controlled, crossover trial of pregabalin for neurogenic claudication. Neurology 2014, 84, 265–272. [Google Scholar] [CrossRef] [Green Version]
- Megale, R.Z.; Deveza, L.A.; Blyth, F.M.; Naganathan, V.; Ferreira, P.H.; McLachlan, A.J.; Ferreira, M.L. Efficacy and Safety of Oral and Transdermal Opioid Analgesics for Musculoskeletal Pain in Older Adults: A Systematic Review of Randomized, Placebo-Controlled Trials. J. Pain 2018, 19, 475.e1–475.e24. [Google Scholar] [CrossRef]
- Nunley, P.D.; Deer, T.R.; Benyamin, R.M.; Staats, P.S.; E Block, J. Interspinous process decompression is associated with a reduction in opioid analgesia in patients with lumbar spinal stenosis. J. Pain Res. 2018, 11, 2943–2948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hains, B.C.; Waxman, S.G. Activated microglia contribute to the maintenance of chronic pain after spinal cord injury. J. Neurosci. 2006, 26, 4308–4317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, R.-R.; Strichartz, G. Cell Signaling and the Genesis of Neuropathic Pain. Sci. STKE 2004, 2004, re14. [Google Scholar] [CrossRef] [PubMed]
- Waxman, S.G. The molecular pathophysiology of pain: Abnormal expression of sodium channel genes and its contributions to hyperexcitability of primary sensory neurons. Pain 1999, 82 (Suppl. S6), S133–S140. [Google Scholar] [CrossRef]
- Waxman, S.G. Neurobiology: A channel sets the gain on pain. Nat. Cell Biol. 2006, 444, 831–832. [Google Scholar] [CrossRef]
- Shunmugavel, A.; Khan, M.; Martin, M.M.; Copay, A.G.; Subach, B.R.; Schuler, T.C.; Singh, I. S-Nitrosoglutathione Administration Ameliorates Cauda Equina Compression Injury in Rats. Neurosci. Med. 2012, 3, 294–305. [Google Scholar] [CrossRef] [Green Version]
- Shunmugavel, A.; Martin, M.M.; Khan, M.; Copay, A.G.; Subach, B.R.; Schuler, T.C.; Singh, I. Simvastatin Ameliorates Cauda Equina Compression Injury in a Rat Model of Lumbar Spinal Stenosis. J. Neuroimmune Pharmacol. 2013, 8, 274–286. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Choi, H.Y.; Park, C.S.; Jang, C.; Lee, K.T.; Lee, J.Y.; Youn, I.; Yune, T.Y. Inhibition of COX-2 alleviates lumbar spinal stenosis-induced chronic mechanical allodynia in rats. Int. Immunopharmacol. 2019, 75, 105738. [Google Scholar] [CrossRef]
- Kanda, T. Blood-nerve barrier: Structure and function. Brain Nerve 2011, 63, 557–569. [Google Scholar] [PubMed]
- Takeshita, Y.; Sato, R.; Kanda, T. Blood–Nerve Barrier (BNB) Pathology in Diabetic Peripheral Neuropathy and In Vitro Human BNB Model. Int. J. Mol. Sci. 2020, 22, 62. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S. Pathophysiology, diagnosis and treatment of intermittent claudication in patients with lumbar canal stenosis. World J. Orthop. 2014, 5, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, S.; Myers, R.R.; Janes, J.; Shubayev, V. Cytokine regulation of MMP-9 in peripheral glia: Implications for pathological processes and pain in injured nerve. Brain Behav. Immun. 2007, 21, 561–568. [Google Scholar] [CrossRef] [Green Version]
- Remacle, A.G.; Hullugundi, S.K.; Dolkas, J.; Angert, M.; Chernov, A.V.; Strongin, A.Y.; Shubayev, V.I. Acute- and late-phase matrix metalloproteinase (MMP)-9 activity is comparable in female and male rats after peripheral nerve injury. J. Neuroinflamm. 2018, 15, 89. [Google Scholar] [CrossRef] [Green Version]
- Shubayev, V.I.; Angert, M.; Dolkas, J.; Campana, W.M.; Palenscar, K.; Myers, R.R. TNFalpha-induced MMP-9 promotes macrophage recruitment into injured peripheral nerve. Mol. Cell. Neurosci. 2006, 31, 407–415. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Na, W.H.; Choi, H.Y.; Lee, K.H.; Ju, B.G.; Yune, T.Y. Jmjd3 mediates blood–spinal cord barrier disruption after spinal cord injury by regulating MMP-3 and MMP-9 expressions. Neurobiol. Dis. 2016, 95, 66–81. [Google Scholar] [CrossRef] [PubMed]
- Park, W.Y.; Hong, B.J.; Lee, J.; Choi, C.; Kim, M.Y. H3K27 Demethylase JMJD3 Employs the NF-kappaB and BMP Signaling Pathways to Modulate the Tumor Microenvironment and Promote Melanoma Progression and Metastasis. Cancer Res. 2016, 76, 161–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.Y.; Kim, H.S.; Choi, H.Y.; Oh, T.H.; Yune, T.Y. Fluoxetine inhibits matrix metalloprotease activation and prevents disruption of blood–spinal cord barrier after spinal cord injury. Brain 2012, 135, 2375–2389. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhou, Y.; Qiu, L.B.; Ding, G.R.; Pang, X.F. Altered expression of matrix metalloproteinases and tight junction proteins in rats following PEMF-induced BBB permeability change. Biomed. Environ. Sci. 2012, 25, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Kruidenier, L.; Chung, C.-W.; Cheng, Z.; Liddle, J.; Che, K.; Joberty, G.; Bantscheff, M.; Bountra, C.; Bridges, A.; Diallo, H.; et al. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nat. Cell Biol. 2012, 488, 404–408. [Google Scholar] [CrossRef] [Green Version]
- Salminen, A.; Kaarniranta, K.; Hiltunen, M.; Kauppinen, A. Histone demethylase Jumonji D3 (JMJD3/KDM6B) at the nexus of epigenetic regulation of inflammation and the aging process. J. Mol. Med. 2014, 92, 1035–1043. [Google Scholar] [CrossRef] [PubMed]
- Park, C.S.; Lee, J.Y.; Choi, H.Y.; Lee, K.; Heo, Y.; Ju, B.G.; Choo, H.-Y.P.; Yune, T.Y. Gallic acid attenuates blood-spinal cord barrier disruption by inhibiting Jmjd3 expression and activation after spinal cord injury. Neurobiol. Dis. 2020, 145, 105077. [Google Scholar] [CrossRef] [PubMed]
- Gibson, W.; Wand, B.M.; E O’Connell, N. Transcutaneous Electrical Nerve Stimulation (TENS) for neuropathic pain in adults. Cochrane Database Syst. Rev. 2015, 9, CD011976. [Google Scholar] [CrossRef] [Green Version]
- Chavan, S.S.; Ma, P.; Chiu, I.M. Neuro-immune interactions in inflammation and host defense: Implications for transplantation. Am. J. Transplant. 2017, 18, 556–563. [Google Scholar] [CrossRef]
- Moalem, G.; Tracey, D.J. Immune and inflammatory mechanisms in neuropathic pain. Brain Res. Rev. 2006, 51, 240–264. [Google Scholar] [CrossRef] [PubMed]
- Sweitzer, S.M.; Hickey, W.F.; Rutkowski, M.D.; Pahl, J.L.; A DeLeo, J. Focal peripheral nerve injury induces leukocyte trafficking into the central nervous system: Potential relationship to neuropathic pain. Pain 2002, 100, 163–170. [Google Scholar] [CrossRef]
- Scholz, J.; Woolf, C.J. The neuropathic pain triad: Neurons, immune cells and glia. Nat. Neurosci. 2007, 10, 1361–1368. [Google Scholar] [CrossRef]
- Sunderkotter, C.; Steinbrink, K.; Goebeler, M.; Bhardwaj, R.; Sorg, C. Macrophages and angiogenesis. J. Leukoc. Biol. 1994, 55, 410–422. [Google Scholar] [CrossRef]
- Mizisin, A.P.; Kalichman, M.W.; Myers, R.R.; Powell, H.C. Role of the Blood-Nerve Barrier in Experimental Nerve Edema. Toxicol. Pathol. 1990, 18, 170–185. [Google Scholar] [CrossRef] [Green Version]
- Omura, K.; Ohbayashi, M.; Sano, M.; Omura, T.; Hasegawa, T.; Nagano, A. The recovery of blood–nerve barrier in crush nerve injury—a quantitative analysis utilizing immunohistochemistry. Brain Res. 2004, 1001, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Seitz, R.J.; Lipfert, P.; Willrich, A.; Himmelmann, F. Toxic effects of triethyldodecylammoniumbromide (TEA-C12) on myelinated nerve fibers and blood-nerve barrier in the mouse. Exp. Brain Res. 1989, 74, 293–302. [Google Scholar] [CrossRef]
- Lim, T.K.; Shi, X.Q.; Martin, H.C.; Huang, H.; Luheshi, G.; Rivest, S.; Zhang, J. Blood-Nerve barrier dysfunction contributes to the generation of neuropathic pain and allows targeting of injured nerves for pain relief. Pain 2014, 155, 954–967. [Google Scholar] [CrossRef]
- Moreau, N.; Mauborgne, A.; Bourgoin, S.; Couraud, P.-O.; Romero, I.A.; Weksler, B.B.; Villanueva, L.; Pohl, M.; Boucher, Y. Early alterations of Hedgehog signaling pathway in vascular endothelial cells after peripheral nerve injury elicit blood-nerve barrier disruption, nerve inflammation, and neuropathic pain development. Pain 2016, 157, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Uchida, K.; Takeno, K.; Baba, H.; Suzuki, Y.; Hayakawa, K.; Yoshizawa, H. Imaging of Cauda Equina Edema in Lumbar Canal Stenosis By Using Gadolinium-Enhanced MR Imaging: Experimental Constriction Injury. Am. J. Neuroradiol. 2006, 27, 346–353. [Google Scholar]
- Nakano, M.; Matsui, H.; Miaki, K.; Yamagami, T.; Tsuji, H. Postlaminectomy adhesion of the cauda equina. Changes of postoperative vascular permeability of the equina in rats. Spine 1997, 22, 1105–1114. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Li, C.; Cao, Y.; Qin, T.; Jiang, L.; Xu, Y.; Li, M.; Luo, Z.; Hu, J.; Lu, H. UTX/KDM6A deletion promotes the recovery of spinal cord injury by epigenetically triggering intrinsic neural regeneration. Mol. Ther. Methods Clin. Dev. 2021, 20, 337–349. [Google Scholar] [CrossRef]
- Ni, S.; Luo, Z.; Jiang, L.; Guo, Z.; Li, P.; Xu, X.; Cao, Y.; Duan, C.; Wu, T.; Li, C.; et al. UTX/KDM6A Deletion Promotes Recovery of Spinal Cord Injury by Epigenetically Regulating Vascular Regeneration. Mol. Ther. J. Am. Soc. Gene Ther. 2019, 27, 2134–2146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmermann, M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain 1983, 16, 109–110. [Google Scholar] [CrossRef]
- Lee, J.Y.; Choi, D.C.; Oh, T.H.; Yune, T.Y. Analgesic Effect of Acupuncture Is Mediated via Inhibition of JNK Activation in Astrocytes after Spinal Cord Injury. PLoS ONE 2013, 8, e73948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonin, R.P.; Bories, C.; De Koninck, Y. A Simplified up-down Method (SUDO) for Measuring Mechanical Nociception in Rodents Using von Frey Filaments. Mol. Pain 2014, 10, 26. [Google Scholar] [CrossRef]
- Yun, K.J.; Kim, J.Y.; Kim, J.B.; Lee, K.W.; Jeong, S.Y.; Park, H.J.; Jung, H.J.; Cho, Y.W.; Yun, K.; Lee, K.T. Inhibition of LPS-Induced NO and PGE2 production by asiatic acid via NF-kappa B inactivation in RAW 264.7 macrophages: Possible involvement of the IKK and MAPK pathways. Int. Immunopharmacol. 2008, 8, 431–441. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.; Choi, H.; Park, C.; Jeon, S.; Yune, T. Jmjd3 Mediates Neuropathic Pain by Inducing Macrophage Infiltration and Activation in Lumbar Spinal Stenosis Animal Model. Int. J. Mol. Sci. 2021, 22, 13426. https://doi.org/10.3390/ijms222413426
Lee J, Choi H, Park C, Jeon S, Yune T. Jmjd3 Mediates Neuropathic Pain by Inducing Macrophage Infiltration and Activation in Lumbar Spinal Stenosis Animal Model. International Journal of Molecular Sciences. 2021; 22(24):13426. https://doi.org/10.3390/ijms222413426
Chicago/Turabian StyleLee, Jeeyoun, Haeyoung Choi, Chansol Park, Sangryong Jeon, and Taeyoung Yune. 2021. "Jmjd3 Mediates Neuropathic Pain by Inducing Macrophage Infiltration and Activation in Lumbar Spinal Stenosis Animal Model" International Journal of Molecular Sciences 22, no. 24: 13426. https://doi.org/10.3390/ijms222413426