
Docosahexaenoic Acid Inhibits Cell Proliferation through a Suppression of c-Myc Protein in Pancreatic Ductal Adenocarcinoma Cells

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents, Chemicals and Antibodies
2.2. Cell Culture and Treatment of DHA
2.3. Assessment of Cell Proliferation
2.4. Analysis of Cell Cycle Distribution
2.5. Annexin-V-Propidium Iodide-Binding Assay
2.6. Protein Extraction and Western Blotting Analysis
2.7. Measurement of Intracellular Reactive Oxygen Species (ROS)
2.8. Analysis of Thiol Compounds and Nucleotides
2.9. Biostatistical Analysis
3. Results
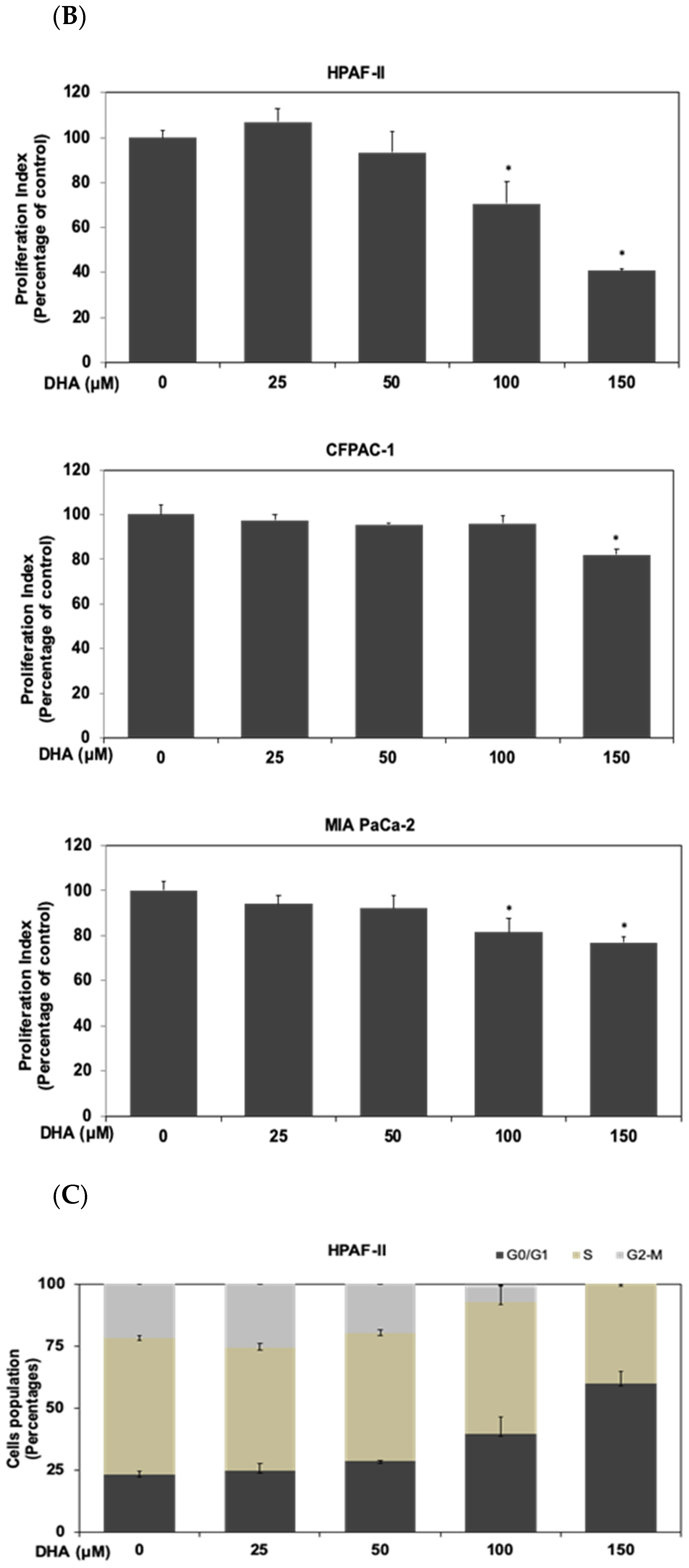
3.1. DHA Inhibited the Proliferation of Human PDAC HPAF-II Cells in Association with Downregulation of Cyclin D1 and c-Myc Proteins
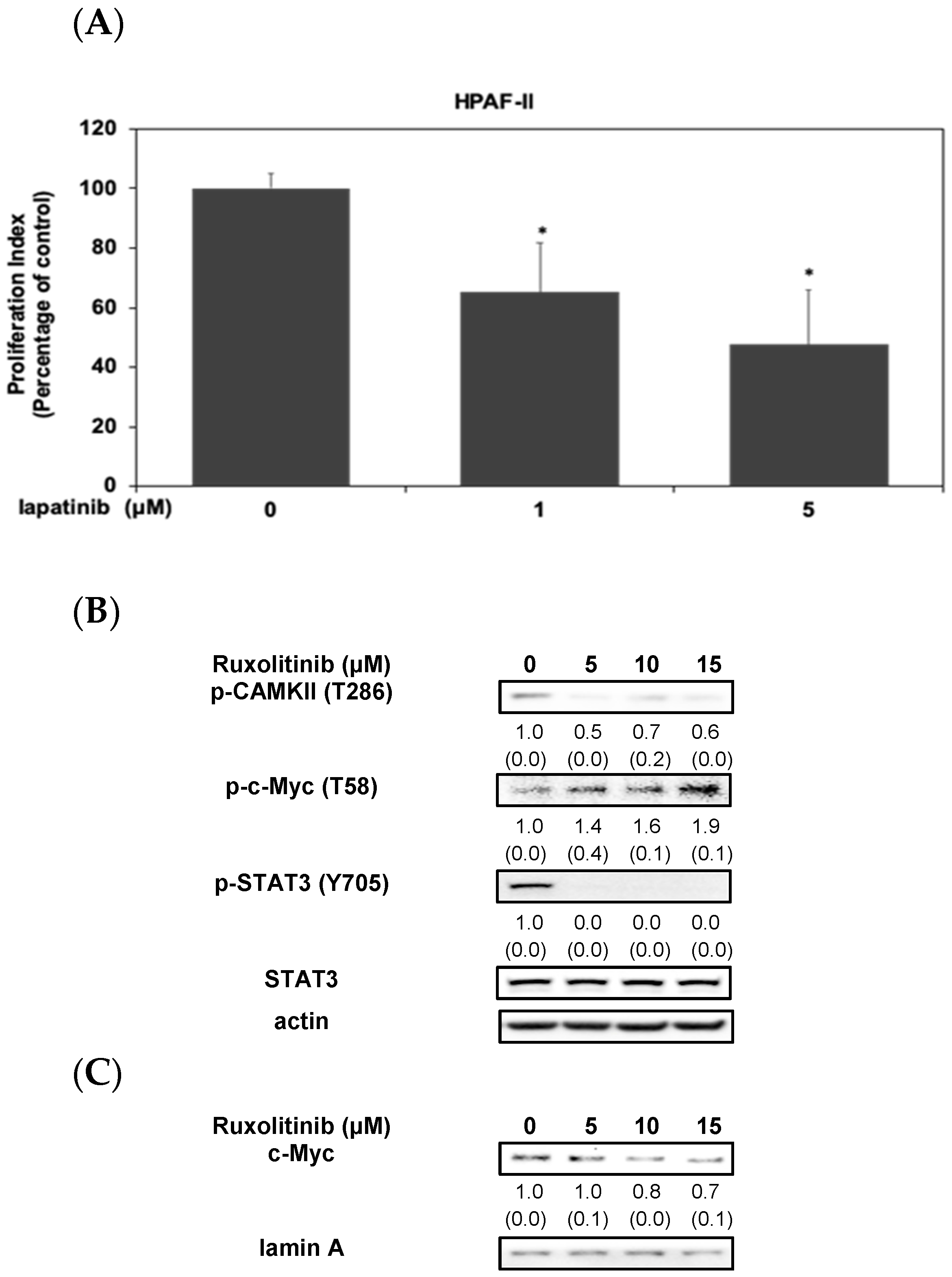
3.2. Activation of STAT3/CAMKII Signaling Pathway Is Involved in the Phosphorylation and Expression of c-Myc Protein in HPAF-II Cells
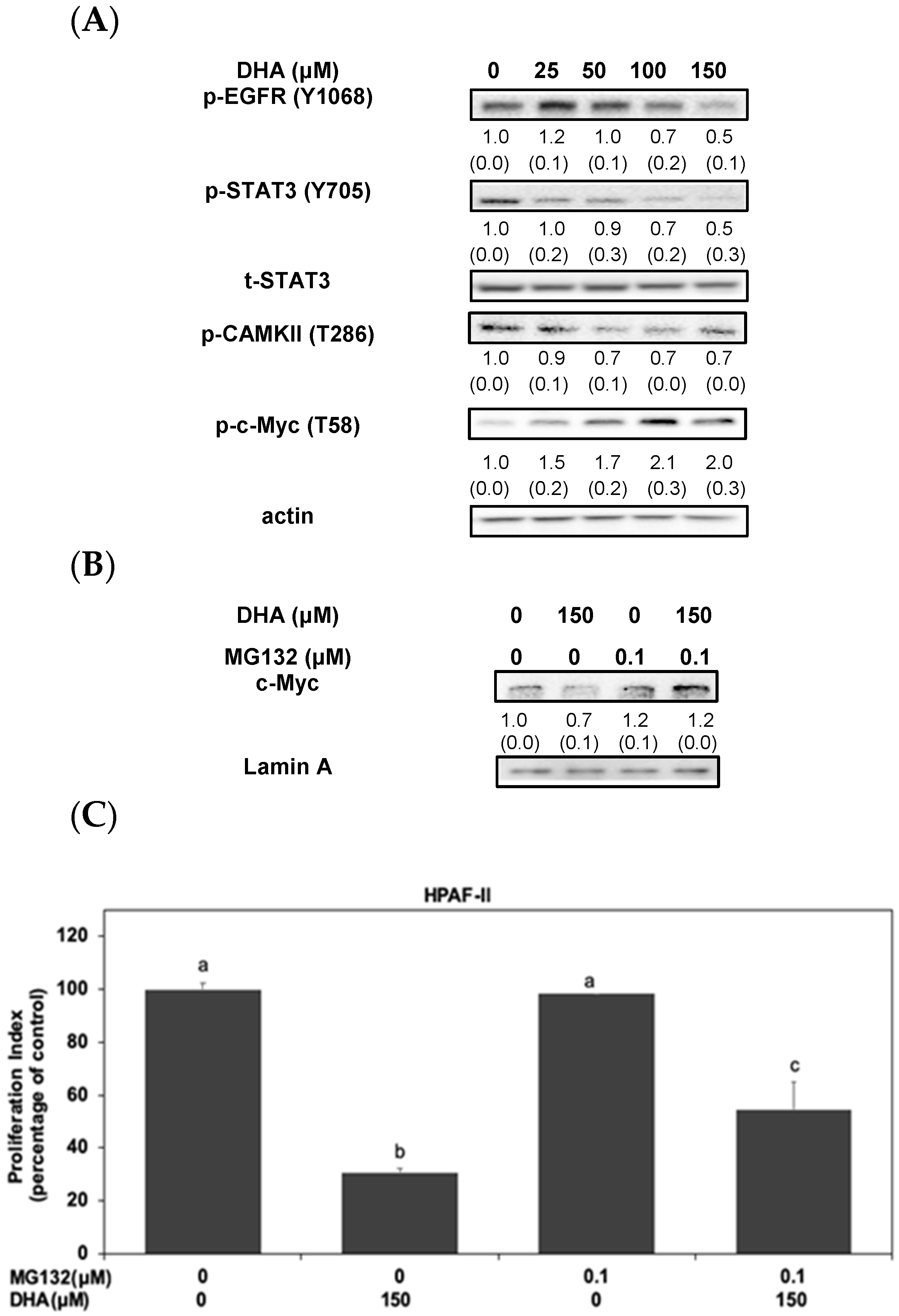
3.3. DHA Induced the Proteasomal Degradation and Instability of c-Myc Protein in HPAF-II Cells
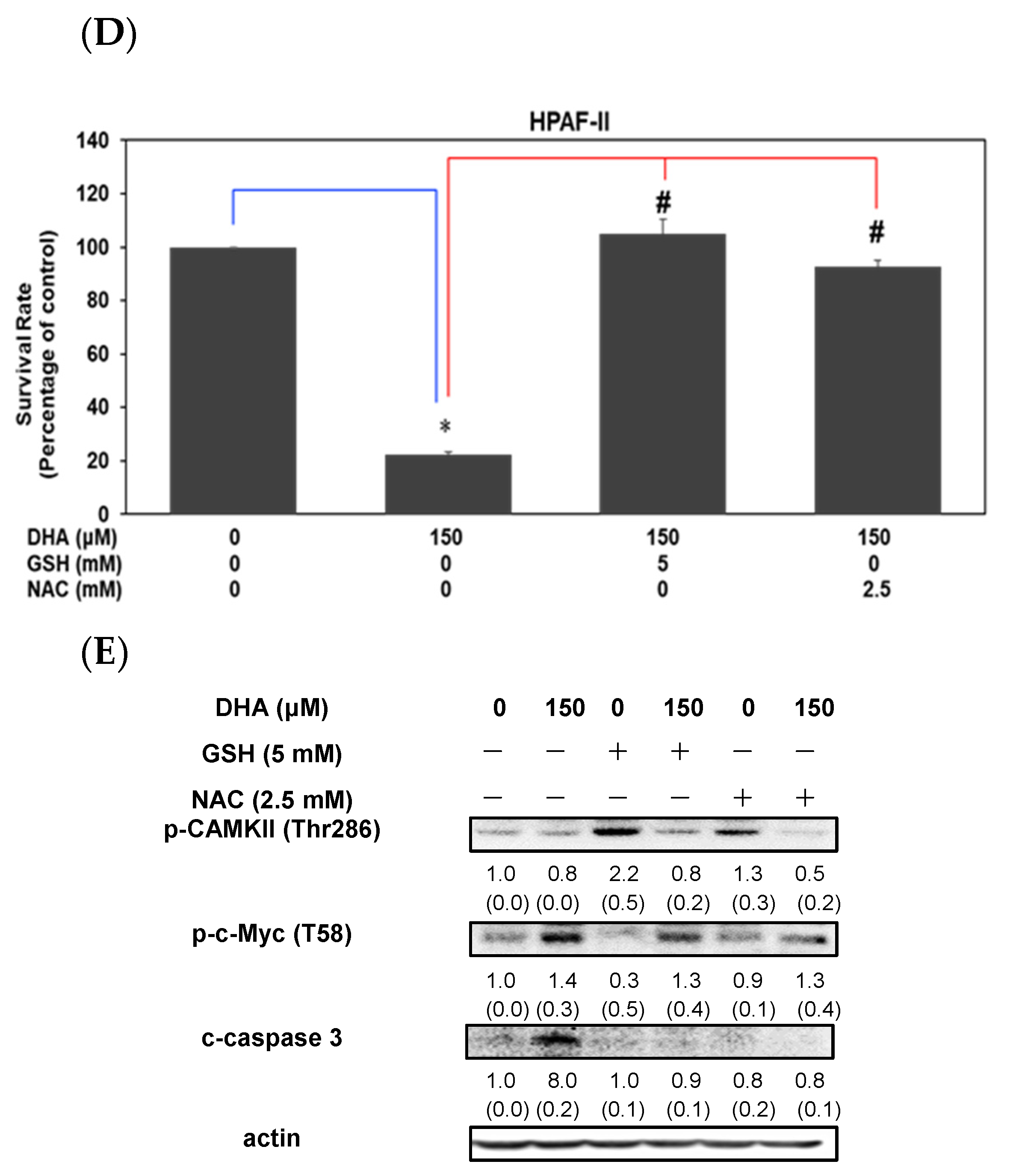
3.4. DHA Induced Induced Cell Death through an Increment of GSSG/GSH Ratio and Reactive Oxygen Species Level in HPAF-II Cells
3.5. DHA Inhibited Cell Survival through an Induction of Apoptosis in HPAF-II Cells
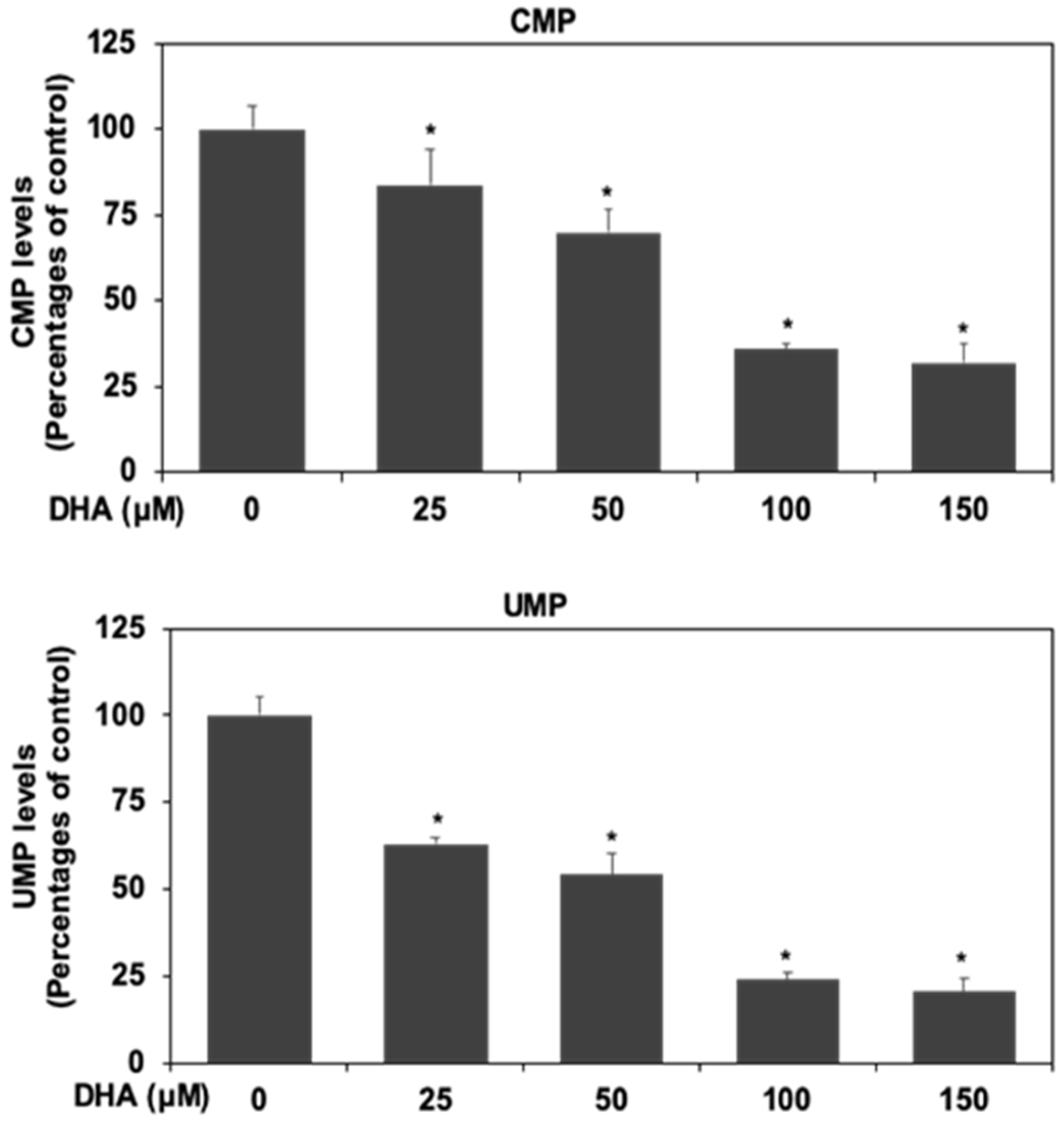
3.6. DHA Inhibited Nucleotide Synthesis in HPAF-II Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Ryan, D.P.; Hong, T.S.; Bardeesy, N. Pancreatic adenocarcinoma. N. Engl. J. Med. 2014, 371, 1039–1049. [Google Scholar] [CrossRef]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.C.; Mansour, J.; Mollaee, M.; Wagner, K.U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, 6744. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef] [Green Version]
- Santana-Codina, N.; Roeth, A.A.; Zhang, Y.; Yang, A.; Mashadova, O.; Asara, J.M.; Wang, X.; Bronson, R.T.; Lyssiotis, C.A.; Ying, H.; et al. Oncogenic KRAS supports pancreatic cancer through regulation of nucleotide synthesis. Nat. Commun. 2018, 9, 4945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huxley, R.; Ansary-Moghaddam, A.; Berrington de Gonzalez, A.; Barzi, F.; Woodward, M. Type-II diabetes and pancreatic cancer: A meta-analysis of 36 studies. Br. J. Cancer 2005, 92, 2076–2083. [Google Scholar] [CrossRef] [Green Version]
- Austin, M.A.; Kuo, E.; Van Den Eeden, S.K.; Mandelson, M.T.; Brentnall, T.A.; Kamineni, A.; Potter, J.D. Family history of diabetes and pancreatic cancer as risk factors for pancreatic cancer: The PACIFIC study. Cancer Epidemiol. Biomark. Prev. 2013, 22, 1913–1917. [Google Scholar] [CrossRef] [Green Version]
- Elena, J.W.; Steplowski, E.; Yu, K.; Hartge, P.; Tobias, G.S.; Brotzman, M.J.; Chanock, S.J.; Stolzenberg-Solomon, R.Z.; Arslan, A.A.; Bueno-de-Mesquita, H.B.; et al. Diabetes and risk of pancreatic cancer: A pooled analysis from the pancreatic cancer cohort consortium. Cancer Causes Control 2013, 24, 13–25. [Google Scholar] [CrossRef]
- Sato, K.; Hikita, H.; Myojin, Y.; Fukumoto, K.; Murai, K.; Sakane, S.; Tamura, T.; Yamai, T.; Nozaki, Y.; Yoshioka, T.; et al. Hyperglycemia enhances pancreatic cancer progression accompanied by elevations in phosphorylated STAT3 and MYC levels. PLoS ONE 2020, 15, e0235573. [Google Scholar] [CrossRef]
- Ciardiello, F.; Tortora, G. EGFR antagonists in cancer treatment. N. Engl. J. Med. 2008, 358, 1160–1174. [Google Scholar] [CrossRef] [Green Version]
- Padma, V.V. An overview of targeted cancer therapy. BioMedicine 2015, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Lee, H.; Herrmann, A.; Buettner, R.; Jove, R. Revisiting STAT3 signalling in cancer: New and unexpected biological functions. Nat. Rev. Cancer 2014, 14, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Nagathihalli, N.S.; Castellanos, J.A.; Shi, C.; Beesetty, Y.; Reyzer, M.L.; Caprioli, R.; Chen, X.; Walsh, A.J.; Skala, M.C.; Moses, H.L.; et al. Signal transducer and activator of transcription 3, mediated remodeling of the tumor microenvironment results in enhanced tumor drug delivery in a mouse model of pancreatic cancer. Gastroenterology 2015, 149, 1932–1943.e9. [Google Scholar] [CrossRef] [Green Version]
- Nagathihalli, N.S.; Castellanos, J.A.; Lamichhane, P.; Messaggio, F.; Shi, C.; Dai, X.; Rai, P.; Chen, X.; VanSaun, M.N.; Merchant, N.B. Inverse correlation of STAT3 and MEK signaling mediates resistance to RAS pathway inhibition in pancreatic cancer. Cancer Res. 2018, 78, 6235–6246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Yue, P.; Fletcher, S.; Zhao, W.; Gunning, P.T.; Turkson, J. A novel small-molecule disrupts Stat3 SH2 domain-phosphotyrosine interactions and Stat3-dependent tumor processes. Biochem. Pharmacol. 2010, 79, 1398–1409. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Seth, A.; Davis, R.J. Transactivation of gene expression by Myc is inhibited by mutation at the phosphorylation sites Thr-58 and Ser-62. Proc. Natl. Acad. Sci. USA 1993, 90, 3216–3220. [Google Scholar] [CrossRef] [Green Version]
- Kazi, A.; Xiang, S.; Yang, H.; Delitto, D.; Trevino, J.; Jiang, R.H.Y.; Ayaz, M.; Lawrence, H.R.; Kennedy, P.; Sebti, S.M. GSK3 suppression upregulates β-catenin and c-Myc to abrogate KRas-dependent tumors. Nat. Commun. 2018, 9, 5154. [Google Scholar] [CrossRef]
- Welcker, M.; Orian, A.; Jin, J.; Grim, J.E.; Harper, J.W.; Eisenman, R.N.; Clurman, B.E. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc. Natl. Acad. Sci. USA 2004, 101, 9085–9090. [Google Scholar] [CrossRef] [Green Version]
- Qin, Y.; Hu, Q.; Xu, J.; Ji, S.; Dai, W.; Liu, W.; Xu, W.; Sun, Q.; Zhang, Z.; Ni, Q.; et al. PRMT5 enhances tumorigenicity and glycolysis in pancreatic cancer via the FBW7/cMyc axis. Cell Commun. Signal. 2019, 17, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, C.V.; Le, A.; Gao, P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin. Cancer Res. 2009, 15, 6479–6483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, Y.; Zhang, J.; Ma, X.; Kim, B.W.; Wang, H.; Li, J.; Pan, Y.; Xu, Y.; Ding, L.; Yang, L.; et al. Stabilization of the c-Myc protein by CAMKIIγ promotes T cell lymphoma. Cancer Cell 2017, 32, 115–128.e7. [Google Scholar] [CrossRef] [Green Version]
- Werner, K.; Kullenberg de Gaudry, D.; Taylor, L.A.; Keck, T.; Unger, C.; Hopt, U.T.; Massing, U. Dietary supplementation with n-3-fatty acids in patients with pancreatic cancer and cachexia: Marine phospholipids versus fish oil-a randomized controlled double-blind trial. Lipids Health Dis. 2017, 16, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, W.C.; Lee, D.Y.; Chiang, E.I.; Syu, J.N.; Chao, C.Y.; Yang, M.D.; Tsai, S.Y.; Tang, F.Y. Docosahexaenoic acid inhibits the proliferation of Kras/TP53 double mutant pancreatic ductal adenocarcinoma cells through modulation of glutathione level and suppression of nucleotide synthesis. PLoS ONE 2020, 15, e0241186. [Google Scholar] [CrossRef]
- Chiu, S.C.; Chiang, E.P.; Tsai, S.Y.; Wang, F.Y.; Pai, M.H.; Syu, J.N.; Cheng, C.C.; Rodriguez, R.L.; Tang, F.Y. Eicosapentaenoic acid induces neovasculogenesis in human endothelial progenitor cells by modulating c-kit protein and PI3-K/Akt/eNOS signaling pathways. J. Nutr. Biochem. 2014, 25, 934–945. [Google Scholar] [CrossRef] [Green Version]
- Robles-Escajeda, E.; Das, U.; Ortega, N.M.; Parra, K.; Francia, G.; Dimmock, J.R.; Varela-Ramirez, A.; Aguilera, R.J. A novel curcumin-like dienone induces apoptosis in triple-negative breast cancer cells. Cell. Oncol. 2016, 39, 265–277. [Google Scholar] [CrossRef]
- Robles-Escajeda, E.; Lerma, D.; Nyakeriga, A.M.; Ross, J.A.; Kirken, R.A.; Aguilera, R.J.; Varela-Ramirez, A. Searching in mother nature for anti-cancer activity: Anti-proliferative and pro-apoptotic effect elicited by green barley on leukemia/lymphoma cells. PLoS ONE 2013, 8, e73508. [Google Scholar] [CrossRef] [Green Version]
- Eruslanov, E.; Kusmartsev, S. Identification of ROS using oxidized DCFDA and flow-cytometry. Methods Mol. Biol. 2010, 594, 57–72. [Google Scholar]
- Kapur, A.; Felder, M.; Fass, L.; Kaur, J.; Czarnecki, A.; Rathi, K.; Zeng, S.; Osowski, K.K.; Howell, C.; Xiong, M.P.; et al. Modulation of oxidative stress and subsequent induction of apoptosis and endoplasmic reticulum stress allows citral to decrease cancer cell proliferation. Sci. Rep. 2016, 6, 27530. [Google Scholar] [CrossRef]
- Barfeld, S.J.; Fazli, L.; Persson, M.; Marjavaara, L.; Urbanucci, A.; Kaukoniemi, K.M.; Rennie, P.S.; Ceder, Y.; Chabes, A.; Visakorpi, T.; et al. Myc-dependent purine biosynthesis affects nucleolar stress and therapy response in prostate cancer. Oncotarget 2015, 6, 12587–12602. [Google Scholar] [CrossRef] [Green Version]
- Geryk, J.; Krsička, D.; Vlčková, M.; Havlovicová, M.; Macek, M., Jr.; Kremlíková Pourová, R. The key role of purine metabolism in the folate-dependent phenotype of autism spectrum disorders: An in silico analysis. Metabolites 2020, 10, 184. [Google Scholar] [CrossRef]
- Zhou, Q.; Lin, M.; Feng, X.; Ma, F.; Zhu, Y.; Liu, X.; Qu, C.; Sui, H.; Sun, B.; Zhu, A.; et al. Targeting CLK3 inhibits the progression of cholangiocarcinoma by reprogramming nucleotide metabolism. J. Exp. Med. 2020, 217, e20191779. [Google Scholar] [CrossRef]
- Liu, Y.C.; Li, F.; Handler, J.; Huang, C.R.; Xiang, Y.; Neretti, N.; Sedivy, J.M.; Zeller, K.I.; Dang, C.V. Global regulation of nucleotide biosynthetic genes by c-Myc. PLoS ONE 2008, 3, e2722. [Google Scholar] [CrossRef]
- Duvall, M.G.; Levy, B.D. DHA- and EPA-derived resolvins, protectins, and maresins in airway inflammation. Eur. J. Pharmacol. 2016, 785, 144–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkins, R.A.; Sangster, K.; Arends, M.J. Apoptotic death of pancreatic cancer cells induced by polyunsaturated fatty acids varies with double bond number and involves an oxidative mechanism. J. Pathol. 1998, 185, 61–70. [Google Scholar] [CrossRef]
- Park, M.; Kim, H. Anti-cancer mechanism of docosahexaenoic acid in pancreatic carcinogenesis: A mini-review. J. Cancer Prev. 2017, 22, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, C.M.; Tien, S.C.; Hsieh, P.K.; Jeng, Y.M.; Chang, M.C.; Chang, Y.T.; Chen, Y.J.; Chen, Y.J.; Lee, E.Y.P.; Lee, W.H. High glucose triggers nucleotide imbalance through O-GlcNAcylation of key enzymes and induces KRAS mutation in pancreatic cells. Cell Metab. 2019, 29, 1334–1349.e10. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; Zhang, H.; Mei, G. Synergistic antitumor activity of DHA and JQ1 in colorectal carcinoma. Eur. J. Pharmacol. 2020, 885, 173500. [Google Scholar] [CrossRef] [PubMed]
- Dosch, A.R.; Dai, X.; Reyzer, M.L.; Mehra, S.; Srinivasan, S.; Willobee, B.A.; Kwon, D.; Kashikar, N.; Caprioli, R.; Merchant, N.B.; et al. Combined Src/EGFR inhibition targets STAT3 signaling and induces stromal remodeling to improve survival in pancreatic cancer. Mol. Cancer Res. 2020, 18, 623–631. [Google Scholar] [CrossRef] [Green Version]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative stress in cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef]




 : induction;
: induction;  : suppression.
: induction; : suppression.
: suppression.
: induction; : suppression.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Syu, J.-N.; Lee, D.-Y.; Hung, H.-C.; Li, C.-Y.; Lin, H.-Y.; Chiang, E.-P.I.; Chen, Y.-H.; Huang, S.-M.; Tang, F.-Y. Docosahexaenoic Acid Inhibits Cell Proliferation through a Suppression of c-Myc Protein in Pancreatic Ductal Adenocarcinoma Cells. Antioxidants 2021, 10, 1721. https://doi.org/10.3390/antiox10111721
Syu J-N, Lee D-Y, Hung H-C, Li C-Y, Lin H-Y, Chiang E-PI, Chen Y-H, Huang S-M, Tang F-Y. Docosahexaenoic Acid Inhibits Cell Proliferation through a Suppression of c-Myc Protein in Pancreatic Ductal Adenocarcinoma Cells. Antioxidants. 2021; 10(11):1721. https://doi.org/10.3390/antiox10111721
Chicago/Turabian StyleSyu, Jia-Ning, Der-Yen Lee, Hung-Chang Hung, Chia-Ying Li, Hung-Yu Lin, En-Pei Isabel Chiang, Yi-Heng Chen, Shu-Ming Huang, and Feng-Yao Tang. 2021. "Docosahexaenoic Acid Inhibits Cell Proliferation through a Suppression of c-Myc Protein in Pancreatic Ductal Adenocarcinoma Cells" Antioxidants 10, no. 11: 1721. https://doi.org/10.3390/antiox10111721
APA StyleSyu, J.-N., Lee, D.-Y., Hung, H.-C., Li, C.-Y., Lin, H.-Y., Chiang, E.-P. I., Chen, Y.-H., Huang, S.-M., & Tang, F.-Y. (2021). Docosahexaenoic Acid Inhibits Cell Proliferation through a Suppression of c-Myc Protein in Pancreatic Ductal Adenocarcinoma Cells. Antioxidants, 10(11), 1721. https://doi.org/10.3390/antiox10111721