Deciphering the Molecular Machinery—Influence of sE-Cadherin on Tumorigenic Traits of Prostate Cancer Cells

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells Lines
2.2. Cell Growth
2.3. Cell Cycling
2.4. Adhesion to Matrix Proteins
2.5. Chemotaxis
2.6. Scratch Wound Assay
2.7. CD44 Expression
2.8. Confocal Laser Scanning Microscopy (CLSM)
2.9. Integrin Surface Expression
2.10. Western Blot Analysis
2.11. Blocking Studies
2.12. Statistics
3. Results
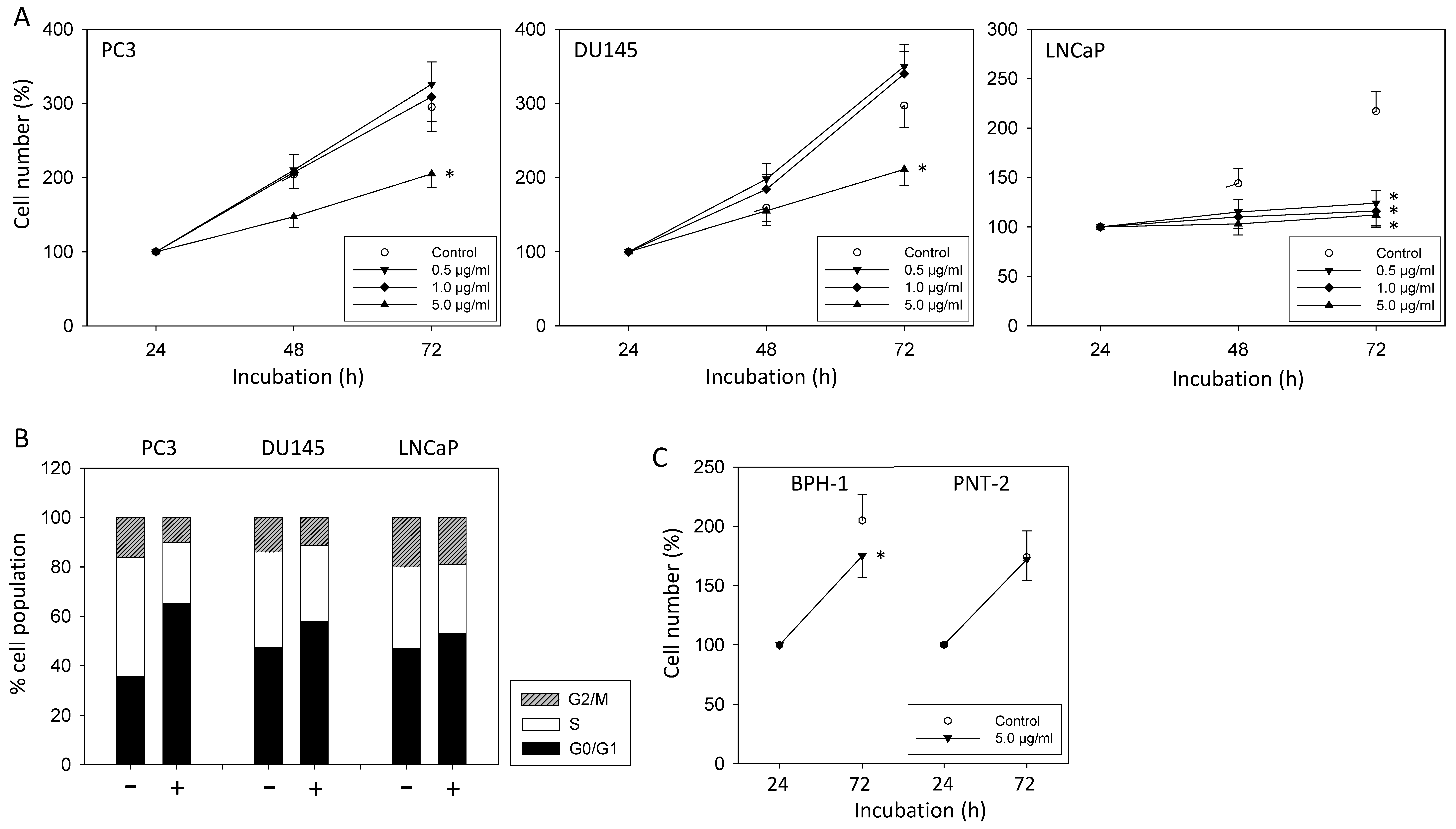
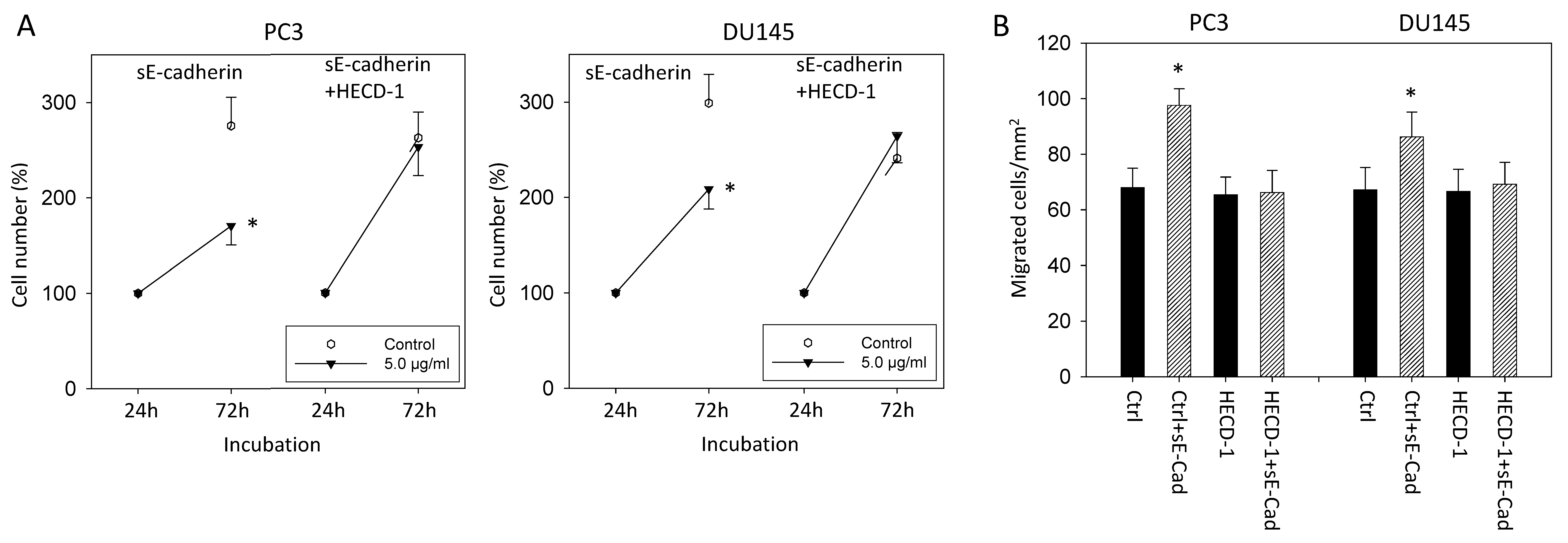
3.1. sE-Cadherin Blocks Tumor Growth
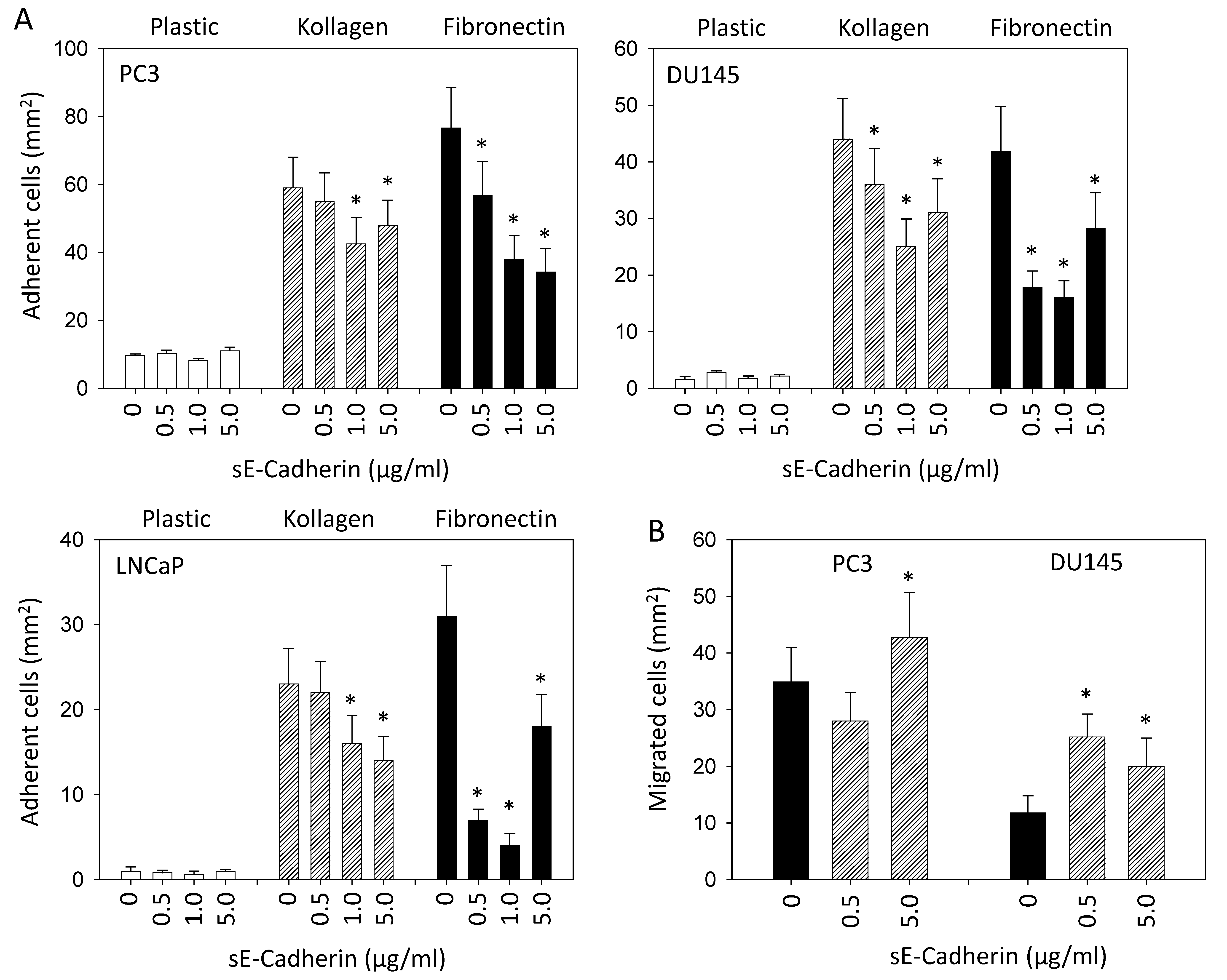
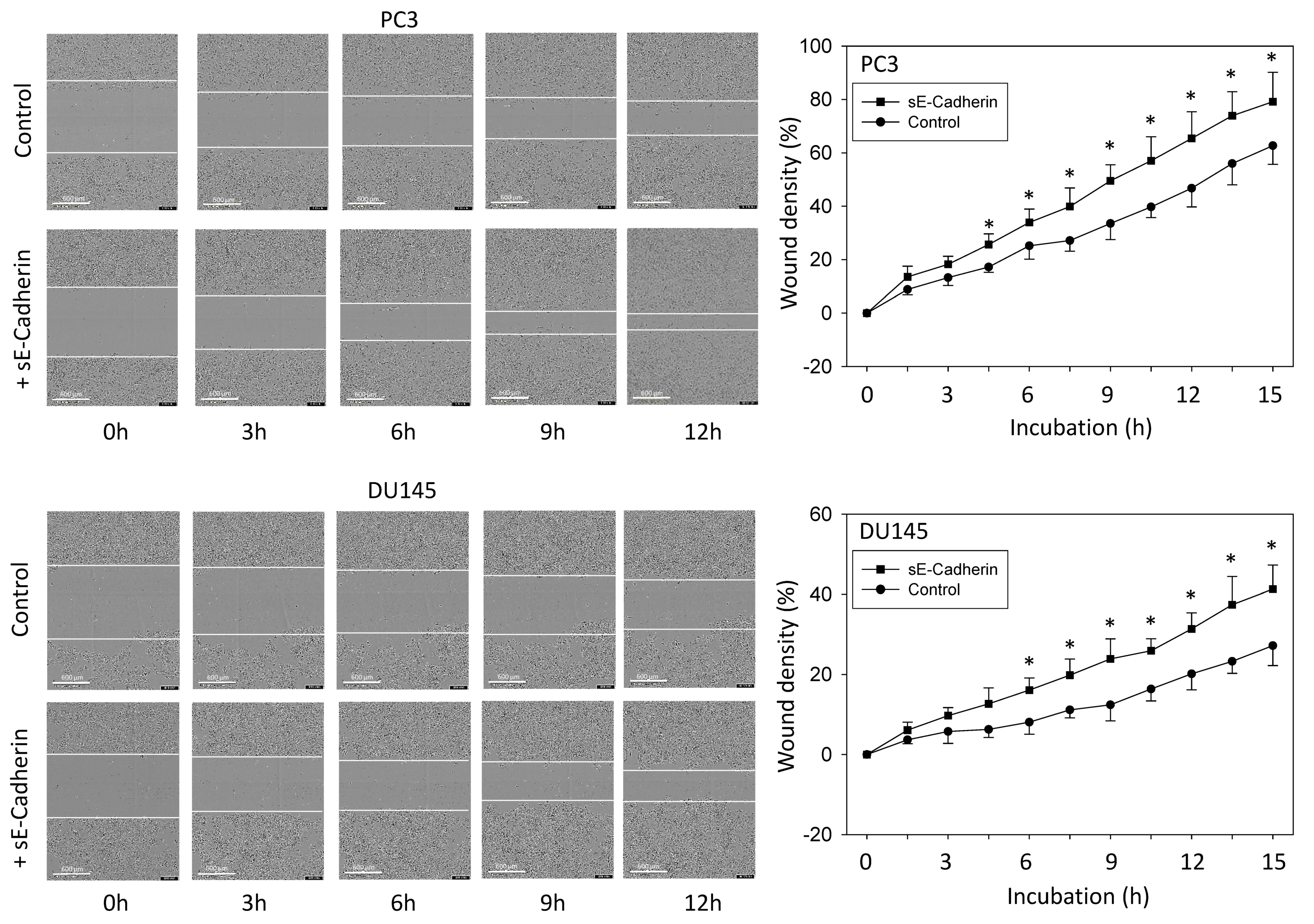
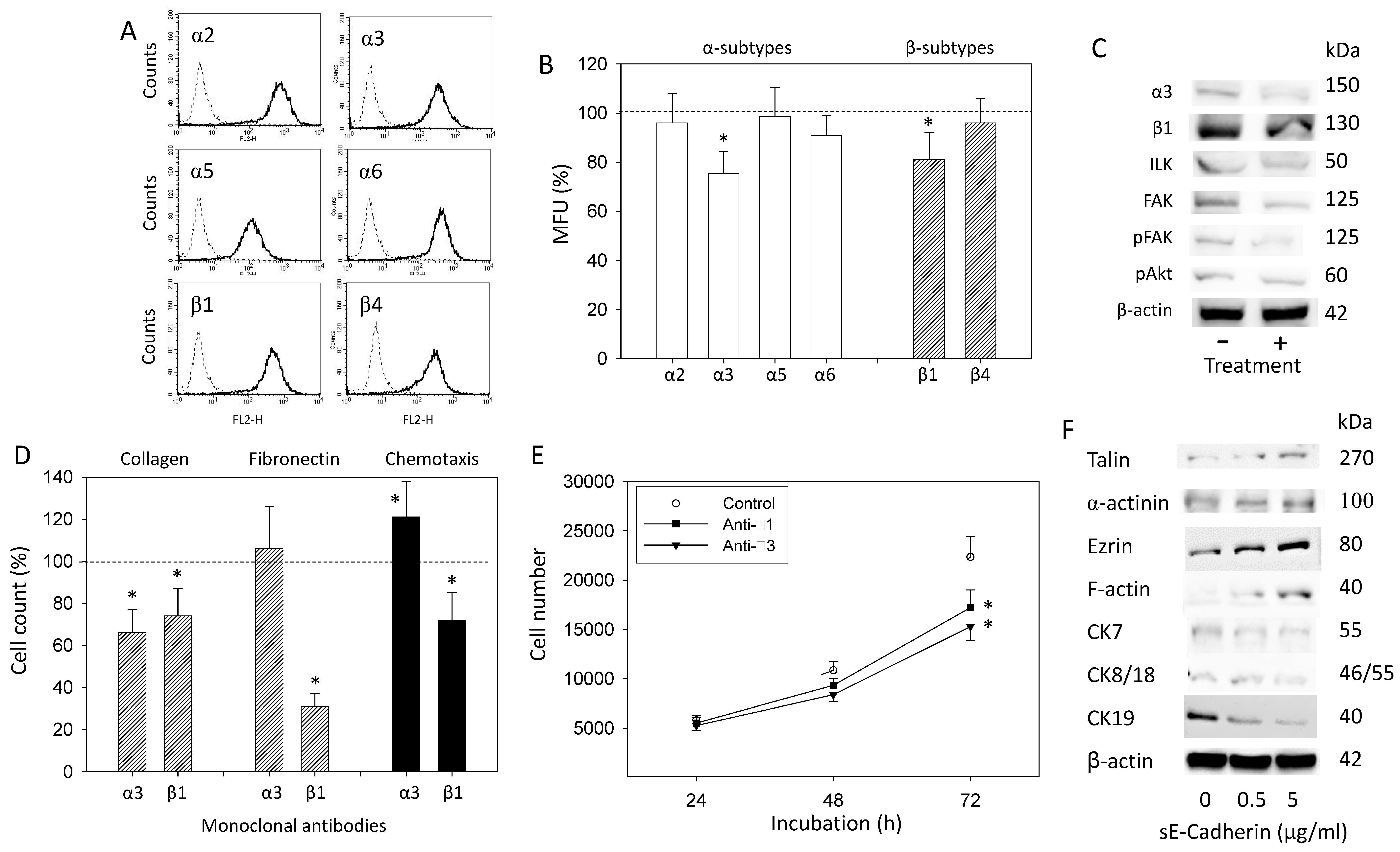
3.2. sE-Cadherin Downregulates Cell Adhesion but Upregulates Cell Migration
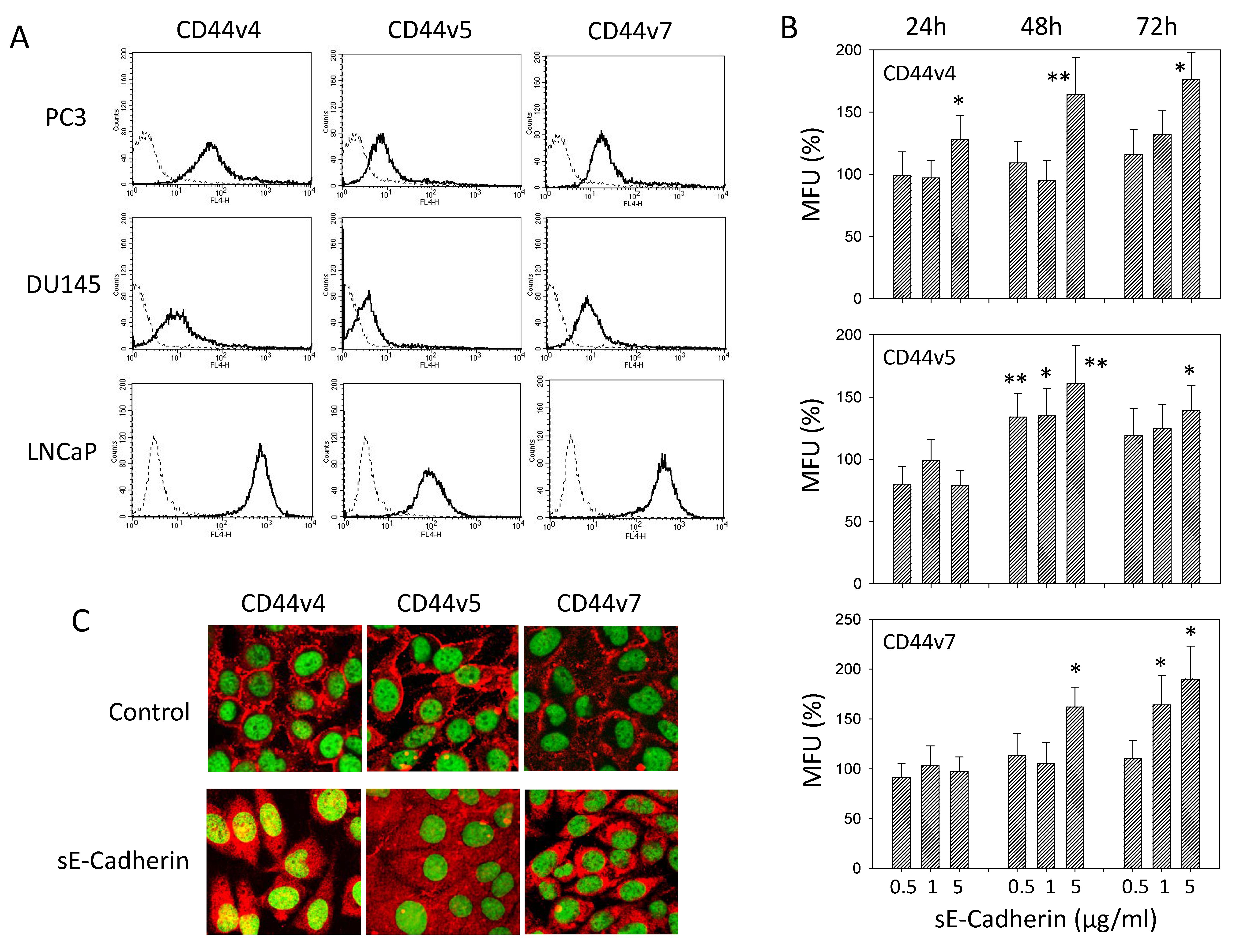
3.3. sE-Cadherin Alters CD44v Expression
3.4. sE-Cadherin Alters Integrin α3 and β1 Expression Level
3.5. sE-Cadherin Influences Cytoskeletal and Focal Adhesion Proteins
3.6. sE-Cadherin Blockade
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: Globocan Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Culp, M.B.; Soerjomataram, I.; Efstathiou, J.A.; Bray, F.; Jemal, A. Recent Global Patterns in Prostate Cancer Incidence and Mortality Rates. Eur. Urol. 2020, 77, 38–52. [Google Scholar] [CrossRef]
- Carioli, G.; Malvezzi, M.; Bertuccio, P.; Boffetta, P.; Levi, F.; La Vecchia, C.; Negri, E. European cancer mortality predictions for the year 2021 with focus on pancreatic and female lung cancer. Ann. Oncol. 2021, 32, 478–487. [Google Scholar] [CrossRef] [PubMed]
- Sigurdson, S.S.; Vera-Badillo, F.E.; De Moraes, F.Y. Discussion of Treatment Options for Metastatic Hormone Sensitive Prostate Cancer Patients. Front. Oncol. 2020, 10, 587981. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.K.W.; Chiu, P.K.F.K.F.; Ng, C.F.; Teoh, J.Y.C.Y.C. Novel Strategies for Treating Castration-Resistant Prostate Cancer. Biomedicines 2021, 9, 339. [Google Scholar] [CrossRef]
- Kyriakopoulos, C.E.; Chen, Y.-H.; Carducci, M.A.; Liu, G.; Jarrard, D.F.; Hahn, N.M.; Shevrin, D.H.; Dreicer, R.; Hussain, M.; Eisenberger, M.; et al. Chemohormonal Therapy in Metastatic Hormone-Sensitive Prostate Cancer: Long-Term Survival Analysis of the Randomized Phase III E3805 CHAARTED Trial. J. Clin. Oncol. 2018, 36, 1080–1087. [Google Scholar] [CrossRef] [Green Version]
- Tsaur, I.; Thurn, K.; Juengel, E.; Gust, K.M.; Borgmann, H.; Mager, R.; Bartsch, G.; Oppermann, E.; Ackermann, H.; Nelson, K.; et al. sE-cadherin serves as a diagnostic and predictive parameter in prostate cancer patients. J. Exp. Clin. Cancer Res. 2015, 34, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Tsaur, I.; Hüsch, T.; Jüngel, E.; Schneider, F.; Schneider, M.; Haferkamp, A.; Thomas, C.; Lieb, V.; Wach, S.; Taubert, H.; et al. sE-cadherin is upregulated in serum of patients with renal cell carcinoma and promotes tumor cell dissemination in vitro. Urol. Oncol. Semin. Orig. Investig. 2019, 37, 355.e1–355.e9. [Google Scholar] [CrossRef]
- Hu, Q.-P.; Kuang, J.-Y.; Yang, Q.-K.; Bian, X.-W.; Yu, S.-C. Beyond a tumor suppressor: Soluble E-cadherin promotes the progression of cancer. Int. J. Cancer 2016, 138, 2804–2812. [Google Scholar] [CrossRef] [Green Version]
- Grabowska, M.M.; Day, M.L. Soluble E-cadherin: More than a symptom of disease. Front. Biosci. 2012, 17, 1948–1964. [Google Scholar] [CrossRef] [Green Version]
- De Wever, O.; DeRycke, L.; Hendrix, A.; De Meerleer, G.; Godeau, F.; Depypere, H.; Bracke, M. Soluble cadherins as cancer biomarkers. Clin. Exp. Metastasis 2007, 24, 685–697. [Google Scholar] [CrossRef]
- Zhu, S.; Zhao, G.; Zhao, X.; Zhan, X.; Cai, M.; Geng, C.; Pu, Q.; Zhao, Q.; Fu, Q.; Huang, C.; et al. Elevated soluble E-cadherin during the epithelial-mesenchymal transition process and as a diagnostic marker in colorectal cancer. Gene 2020, 754, 144899. [Google Scholar] [CrossRef]
- He, Y.; Zhang, J.; Wang, S.K.; Huang, W.B.; Chen, X.G.; He, B.S.; Wang, J.S. Serum sCD44v6 and sE-cadherin levels in patients with esophageal squamous cell carcinoma. Zhejiang Da Xue Xue Bao Yi Xue Ban 2012, 41, 305–309. [Google Scholar]
- Johnson, S.K.; Ms, V.C.R.; Hennings, L.; Haun, R.S. Kallikrein 7 enhances pancreatic cancer cell invasion by shedding E-cadherin. Cancer 2007, 109, 1811–1820. [Google Scholar] [CrossRef]
- Lee, Y.; Ko, D.; Yoon, J.; Lee, Y.; Kim, S. TMEM52B suppression promotes cancer cell survival and invasion through modulating E-cadherin stability and EGFR activity. J. Exp. Clin. Cancer Res. 2021, 40, 1–20. [Google Scholar] [CrossRef]
- Weiß, J.V.; Klein-Scory, S.; Kübler, S.; Reinacher-Schick, A.; Stricker, I.; Schmiegel, W.; Schwarte-Waldhoff, I. Soluble E-cadherin as a serum biomarker candidate: Elevated levels in patients with late-stage colorectal carcinoma and FAP. Int. J. Cancer 2011, 128, 1384–1392. [Google Scholar] [CrossRef]
- Okugawa, Y.; Toiyama, Y.; Inoue, Y.; Iwata, T.; Fujikawa, H.; Saigusa, S.; Konishi, N.; Tanaka, K.; Uchida, K.; Kusunoki, M. Clinical Significance of Serum Soluble E-cadherin in Colorectal Carcinoma. J. Surg. Res. 2012, 175, e67–e73. [Google Scholar] [CrossRef] [PubMed]
- Shariat, S.F.; Matsumoto, K.; Casella, R.; Jian, W.; Lerner, S.P. Urinary Levels of Soluble E-Cadherin in the Detection of Transitional Cell Carcinoma of the Urinary Bladder. Eur. Urol. 2005, 48, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Shariat, S.F.; Casella, R.; Wheeler, T.M.; Slawin, K.M.; Lerner, S.P. Preoperative Plasma Soluble E-Cadherin Predicts Metastases to Lymph Nodes and Prognosis in Patients Undergoing Radical Cystectomy. J. Urol. 2003, 170, 2248–2252. [Google Scholar] [CrossRef] [PubMed]
- Rutz, J.; Thaler, S.; Maxeiner, S.; Chun, F.K.; Blaheta, R.A. Sulforaphane Reduces Prostate Cancer Cell Growth and Proliferation In Vitro by Modulating the Cdk-Cyclin Axis and Expression of the CD44 Variants 4, 5, and 7. Int. J. Mol. Sci. 2020, 21, 8724. [Google Scholar] [CrossRef]
- Luxton, H.J.; Simpson, B.S.; Mills, I.G.; Brindle, N.R.; Ahmed, Z.; Stavrinides, V.; Heavey, S.; Stamm, S.; Whitaker, H.C. The Oncogene Metadherin Interacts with the Known Splicing Proteins YTHDC1, Sam68 and T-STAR and Plays a Novel Role in Alternative mRNA Splicing. Cancers 2019, 11, 1233. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.; Handorean, A.M.; Iczkowski, K.A. MicroRNAs 373 and 520c Are Downregulated in Prostate Cancer, Suppress CD44 Translation and Enhance Invasion of Prostate Cancer Cells in vitro. Int. J. Clin. Exp. Pathol. 2009, 2, 361–369. [Google Scholar]
- Das, L.; Anderson, T.A.; Gard, J.M.; Sroka, I.C.; Strautman, S.R.; Nagle, R.B.; Morrissey, C.; Knudsen, B.S.; Cress, A.E. Characterization of Laminin Binding Integrin Internalization in Prostate Cancer Cells. J. Cell. Biochem. 2017, 118, 1038–1049. [Google Scholar] [CrossRef] [Green Version]
- Kurozumi, A.; Goto, Y.; Matsushita, R.; Fukumoto, I.; Kato, M.; Nishikawa, R.; Sakamoto, S.; Enokida, H.; Nakagawa, M.; Ichikawa, T.; et al. Tumor-suppressive micro RNA -223 inhibits cancer cell migration and invasion by targeting ITGA 3/ ITGB 1 signaling in prostate cancer. Cancer Sci. 2016, 107, 84–94. [Google Scholar] [CrossRef] [Green Version]
- Kwon, O.Y.; Ryu, S.; Choi, J.K.; Lee, S.H. Smilax glabra Roxb. Inhibits Collagen Induced Adhesion and Migration of PC3 and LNCaP Prostate Cancer Cells through the Inhibition of Beta 1 Integrin Expression. Molecules 2020, 25, 3006. [Google Scholar] [CrossRef]
- Hyder, C.L.; Lazaro, G.; Pylvänäinen, J.W.; Roberts, M.; Rosenberg, S.M.; Eriksson, J.E. Nestin regulates prostate cancer cell invasion by influencing the localisation and functions of FAK and integrins. J. Cell Sci. 2014, 127, 2161–2173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goto, Y.; Ando, T.; Izumi, H.; Feng, X.; Arang, N.; Gilardi, M.; Wang, Z.; Ando, K.; Gutkind, J.S. Muscarinic receptors promote castration-resistant growth of prostate cancer through a FAK–YAP signaling axis. Oncogene 2020, 39, 4014–4027. [Google Scholar] [CrossRef] [PubMed]
- Uwada, J.; Mukai, S.; Terada, N.; Nakazawa, H.; Islam, M.S.; Nagai, T.; Fujii, M.; Yamasaki, K.; Taniguchi, T.; Kamoto, T.; et al. Pleiotropic effects of probenecid on three-dimensional cultures of prostate cancer cells. Life Sci. 2021, 278, 119554. [Google Scholar] [CrossRef] [PubMed]
- Pal, M. Tumor metastasis suppressor functions of Ets transcription factor through integrin β3-mediated signaling pathway. J. Cell. Physiol. 2019, 234, 20266–20274. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Chen, H.-J.; Chen, S.-H.; Xue, X.-Y.; Chen, H.; Zheng, Q.-S.; Wei, Y.; Li, X.-D.; Huang, J.-B.; Cai, H.; et al. Upregulation of Talin-1 expression associates with advanced pathological features and predicts lymph node metastases and biochemical recurrence of prostate cancer. Medicine 2016, 95, e4326. [Google Scholar] [CrossRef] [PubMed]
- McFarlane, S.; McFarlane, C.; Montgomery, N.; Hill, A.; Waugh, D.J. CD44-mediated activation of α5β1-integrin, cortactin and paxillin signaling underpins adhesion of basal-like breast cancer cells to endothelium and Fibronectin-enriched matrices. Oncotarget 2015, 6, 36762–36773. [Google Scholar] [CrossRef] [Green Version]
- Pang, J.; Liu, W.-P.; Liu, X.-P.; Li, L.-Y.; Fang, Y.-Q.; Sun, Q.-P.; Liu, S.-J.; Li, M.-T.; Su, Z.-L.; Gao, X. Profiling Protein Markers Associated with Lymph Node Metastasis in Prostate Cancer by DIGE-based Proteomics Analysis. J. Proteome Res. 2010, 9, 216–226. [Google Scholar] [CrossRef]
- Singh, R.; Kapur, N.; Mir, H.; Singh, N.; Lillard, J.W., Jr.; Singh, S. CXCR6-CXCL16 axis promotes prostate cancer by mediating cytoskeleton rearrangement via Ezrin activation and αvβ3 integrin clustering. Oncotarget 2016, 7, 7343–7353. [Google Scholar] [CrossRef]
- Shipitsin, M.; Small, C.E.; Choudhury, S.; Giladi, E.; Friedlander, S.F.; Nardone, J.; Hussain, S.; Hurley, A.D.; Ernst, C.; Huang, Y.E.; et al. Identification of proteomic biomarkers predicting prostate cancer aggressiveness and lethality despite biopsy-sampling error. Br. J. Cancer 2014, 111, 1201–1212. [Google Scholar] [CrossRef]
- Virtanen, S.S.; Ishizu, T.; Sandholm, J.; Löyttyniemi, E.; Väänänen, H.K.; Tuomela, J.M.; Härkönen, P.L. Alendronate-induced disruption of actin cytoskeleton and inhibition of migration/invasion are associated with cofilin downregulation in PC-3 prostate cancer cells. Oncotarget 2018, 9, 32593–32608. [Google Scholar] [CrossRef]
- Bizzarro, V.; Belvedere, R.; Migliaro, V.; Romano, E.; Parente, L.; Petrella, A. Hypoxia regulates ANXA1 expression to support prostate cancer cell invasion and aggressiveness. Cell Adhes. Migr. 2017, 11, 247–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberti, I.; Barboro, P.; Barbesino, M.; Sanna, P.; Pisciotta, L.; Parodi, S.; Nicolò, G.; Boccardo, F.; Galli, S.; Balbi, C.; et al. Changes in the expression of cytokeratins and nuclear matrix proteins are correlated with the level of differentiation in human prostate cancer. J. Cell Biochem. 2000, 79, 471–485. [Google Scholar] [CrossRef]
- Heatley, M.; Maxwell, P.; Whiteside, C.; Toner, P. Vimentin and cytokeratin expression in nodular hyperplasia and carcinoma of the prostate. J. Clin. Pathol. 1995, 48, 1031–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsaur, I.; Thomas, A.; Juengel, E.; Maxeiner, S.; Grein, T.; Le, Q.C.; Muschta, V.; Rutz, J.; Chun, F.K.H.; Blaheta, R.A. Deciphering the Molecular Machinery—Influence of sE-Cadherin on Tumorigenic Traits of Prostate Cancer Cells. Biology 2021, 10, 1007. https://doi.org/10.3390/biology10101007
Tsaur I, Thomas A, Juengel E, Maxeiner S, Grein T, Le QC, Muschta V, Rutz J, Chun FKH, Blaheta RA. Deciphering the Molecular Machinery—Influence of sE-Cadherin on Tumorigenic Traits of Prostate Cancer Cells. Biology. 2021; 10(10):1007. https://doi.org/10.3390/biology10101007
Chicago/Turabian StyleTsaur, Igor, Anita Thomas, Eva Juengel, Sebastian Maxeiner, Timothy Grein, Quynh Chi Le, Veronika Muschta, Jochen Rutz, Felix K. H. Chun, and Roman A. Blaheta. 2021. "Deciphering the Molecular Machinery—Influence of sE-Cadherin on Tumorigenic Traits of Prostate Cancer Cells" Biology 10, no. 10: 1007. https://doi.org/10.3390/biology10101007