
Discovery, Optimization, and Clinical Application of Natural Antimicrobial Peptides

 , , and
, , and 
Abstract
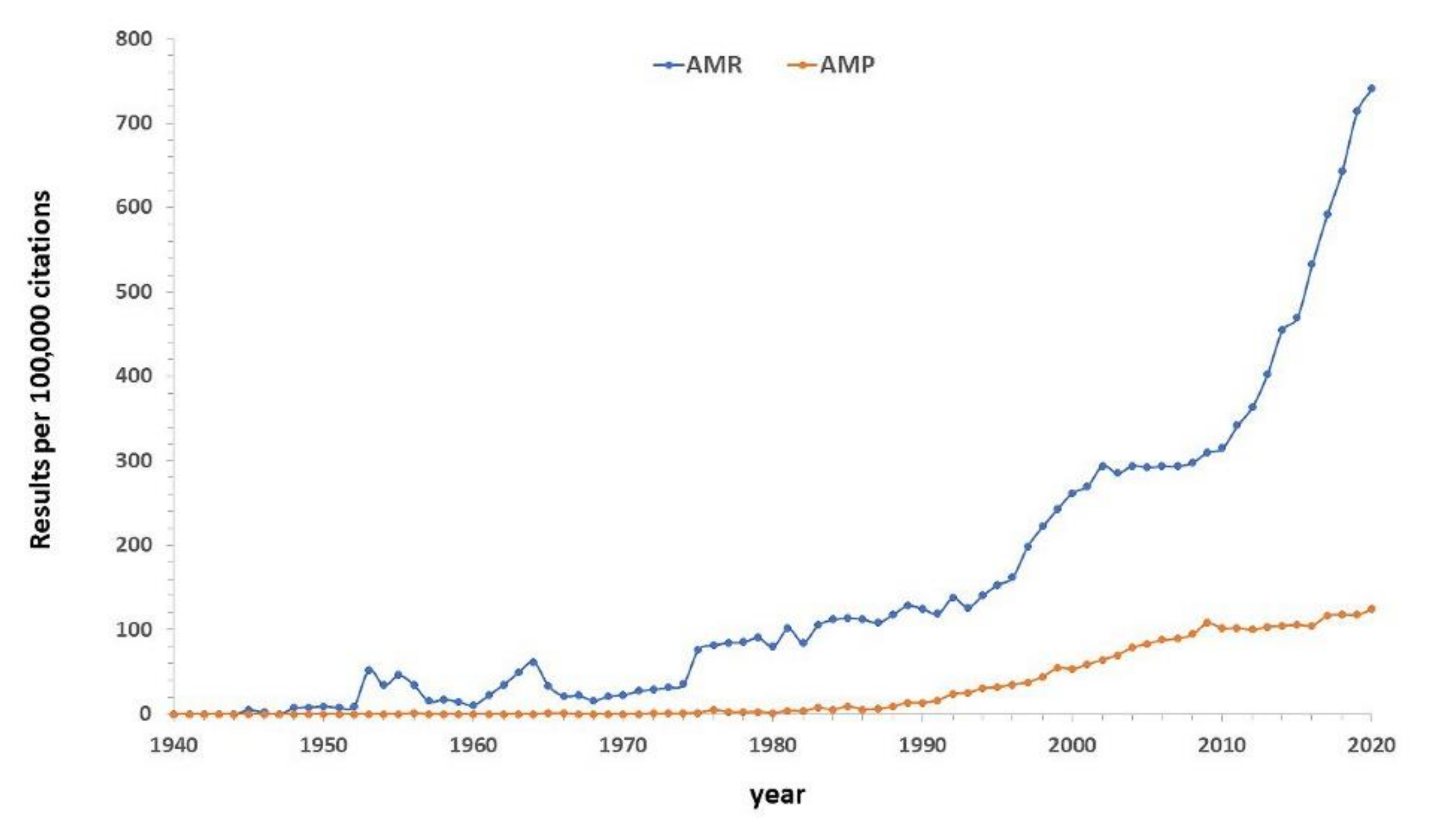
1. Introduction
2. Discovery of Native AMPs: A Historical Overview
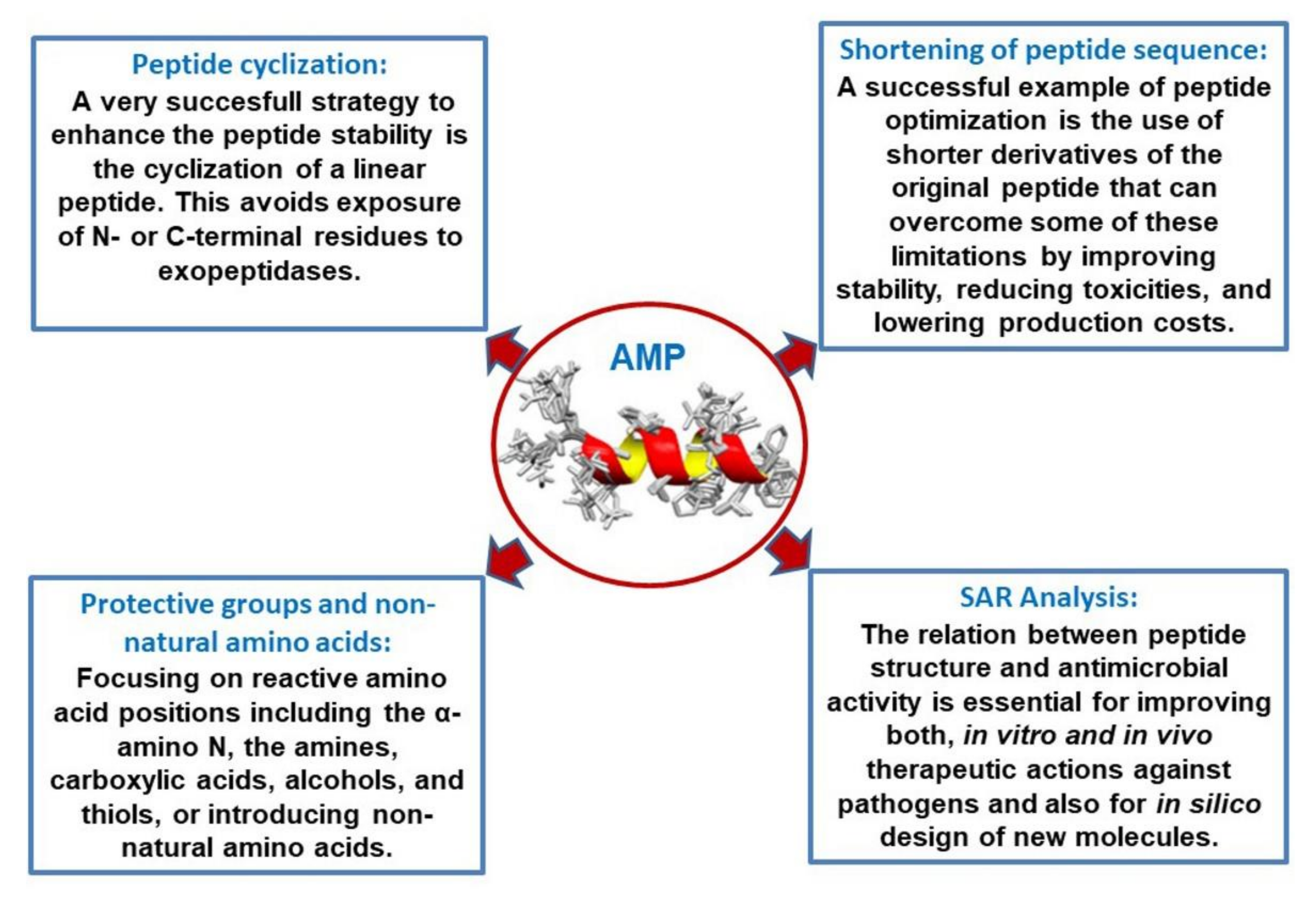
3. Derivatives and Optimization: SAR Analysis
3.1. Shortening of Peptide Sequence
3.2. Introduction of Protective Groups, Non-Natural Amino Acids, and Cyclization: Combination of Derivatizations, and Hybrid Peptides
3.3. Structure–Activity Relationship (SAR) Analysis and In Silico Optimization of AMPs
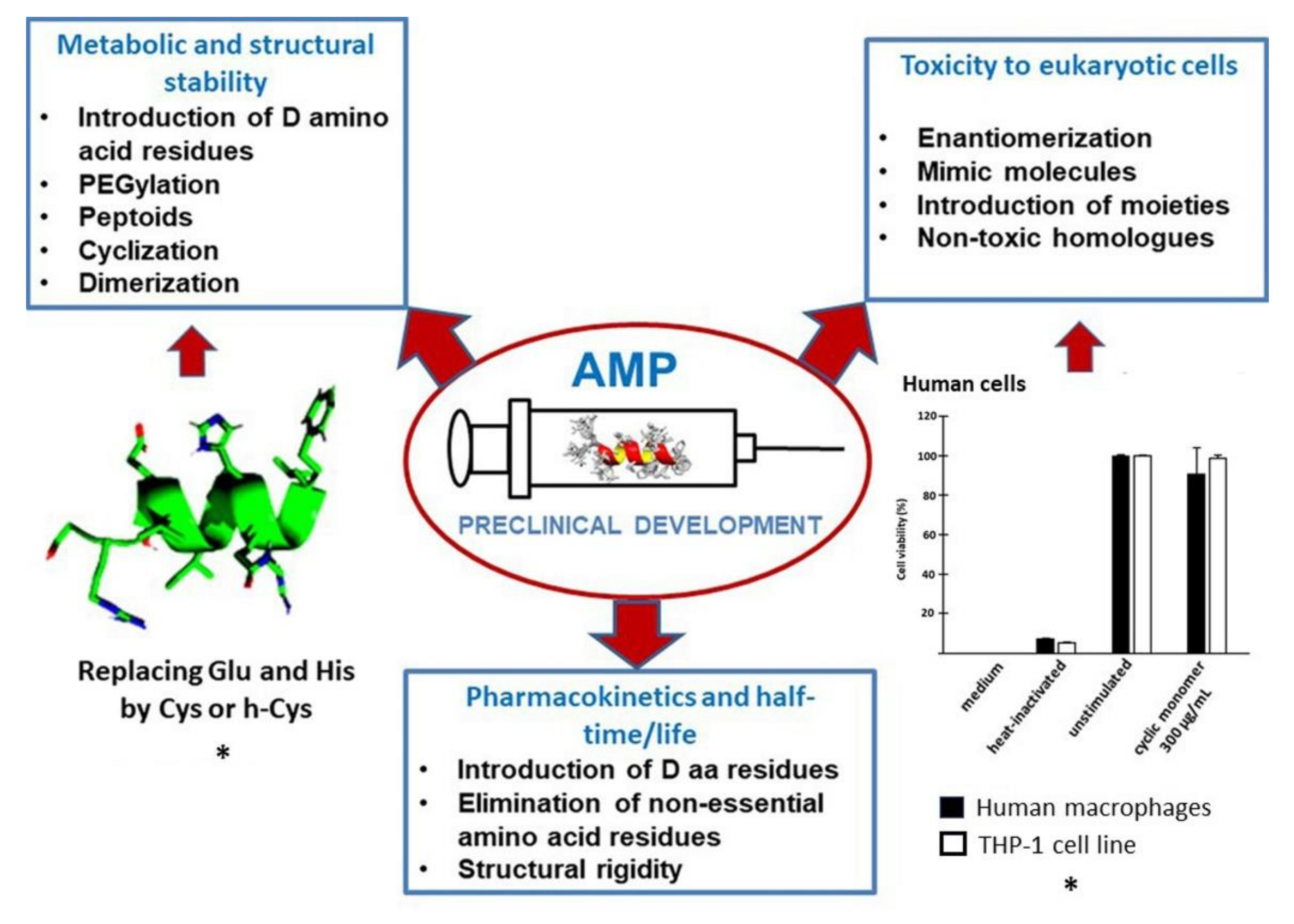
4. Preclinical Development: Stability, Toxicity, and Half-Time
4.1. Metabolic and Structural Stability
4.2. Toxicity to Eukaryotic Cells
4.3. Pharmacokinetics and Half-Time/Life
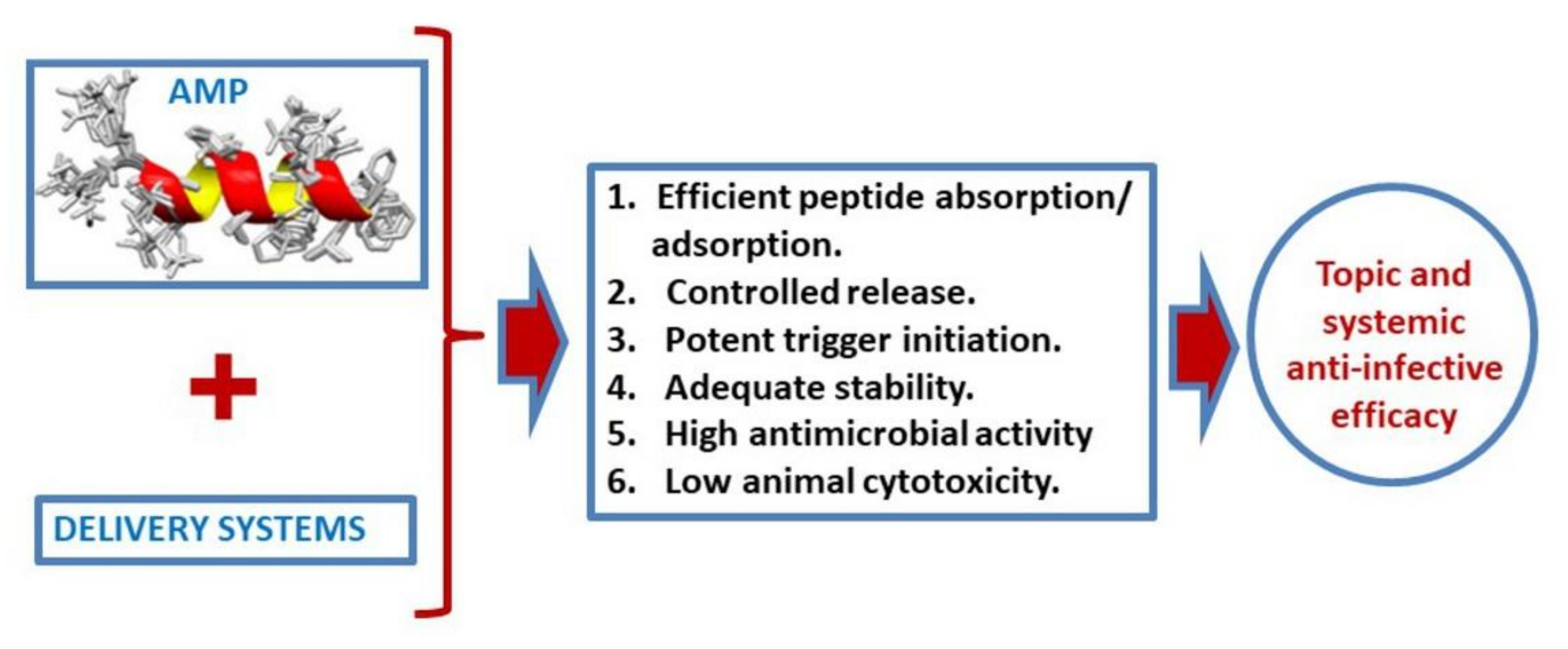
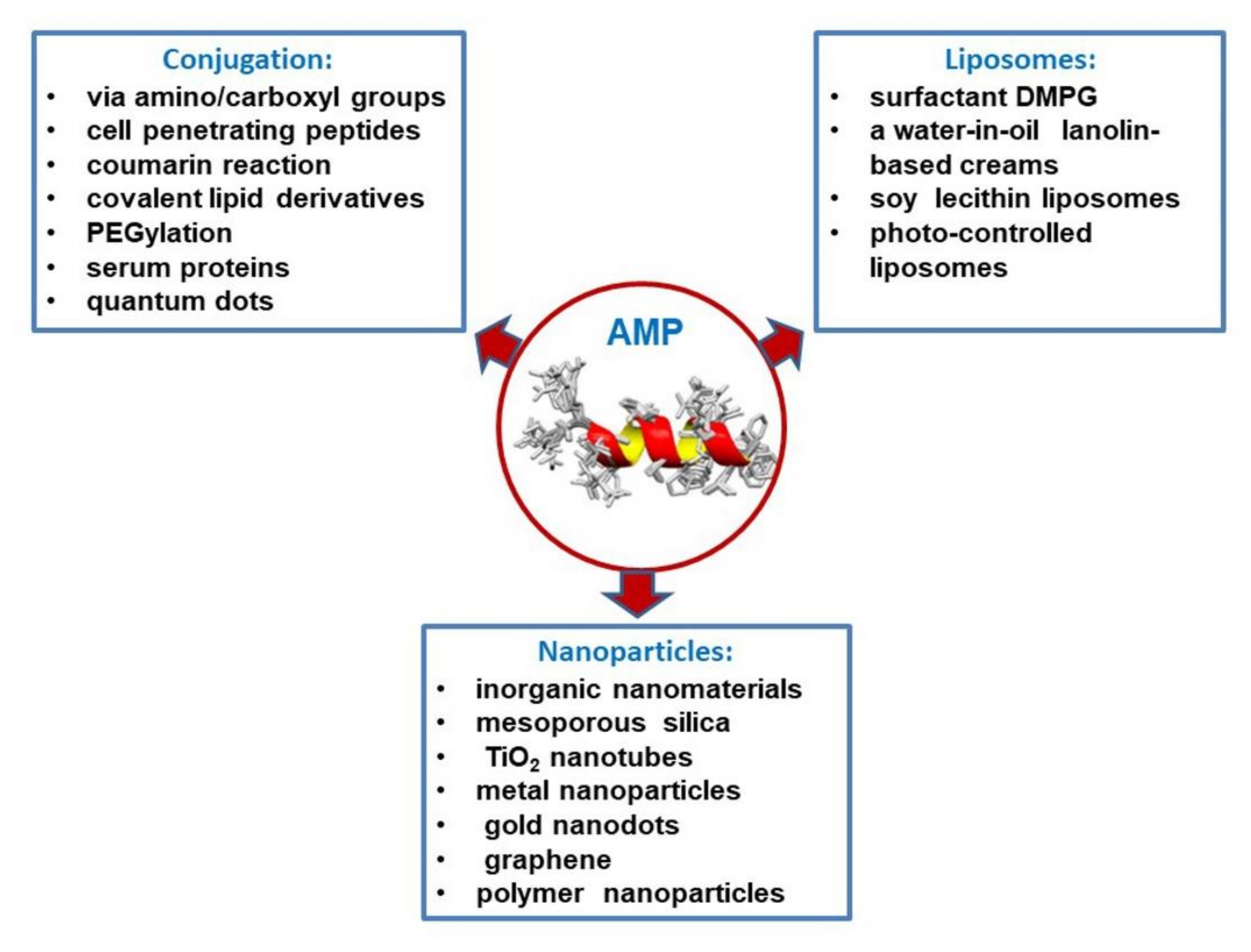
5. Peptide Release Systems (Conjugation, Liposomes, and Nanoparticles)
5.1. Conjugation of Antimicrobial Peptides
5.2. Liposome as Antimicrobial Peptide Delivery Tools
5.3. Nanoparticle-Based Peptide Release Systems
6. Clinical Applications of Antimicrobial Peptides
6.1. AMP with Antibacterial Effects
6.2. AMP with Antiviral Effects
6.3. AMP with Antifungal Effects
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fleming, A. On a remarkable bacteriolytic element found in tissues and secretions. Proc. R. Soc. Lond. Ser. B 1922, 93, 306–317. [Google Scholar]
- Wang, G. APD3: The Antimicrobial Peptide Database. Available online: https://wangapd3.com/main.php (accessed on 21 April 2021).
- Wang, G. Antimicrobial Peptides: Discovery, Design and Novel Therapeutic Strategies, 2nd ed.; CABI: Wallingford, UK, 2017. [Google Scholar]
- Jenssen, H.; Hamill, P.; Hancock, R. Peptide Antimicrobial Agents. Clin. Microbiol. Rev. 2006, 19, 491–511. [Google Scholar] [CrossRef] [PubMed]
- Gallo, R.L.; Hooper, L.V. Epithelial antimicrobial defence of the skin and intestine. Nat. Rev. Immunol. 2012, 12, 503–516. [Google Scholar] [CrossRef]
- Phoenix, D.A.; Dennison, S.R.; Harris, F. Antimicrobial Peptides: Their History, Evolution, and Functional Promiscuity. In Antimicrobial Peptides, 1st ed.; Wiley-VCH: Weinheim, Germany, 2013; pp. 1–37. [Google Scholar]
- Wang, G.S.; Li, X.; Wang, Z. APD3: The antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016, 44, D1087–D1093. [Google Scholar] [CrossRef]
- Hancock, R.E.W.; Sahl, H.-G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [Google Scholar] [CrossRef] [PubMed]
- Wang, G. APD3: Sequence Statistics. Available online: https://wangapd3.com/statistic/statistic.php (accessed on 21 April 2021).
- Kumar, P.; Kizhakkedathu, J.N.; Straus, S.K. Antimicrobial Peptides: Diversity, Mechanism of Action and Strategies to Improve the Activity and Biocompatibility In Vivo. Biomolecules 2018, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Kang, X.; Dong, F.; Shi, C.; Liu, S.; Sun, J.; Chen, J.; Li, H.; Xu, H.; Lao, X.; Zheng, H. DRAMP 2.0, an updated data repository of antimicrobial peptides. Sci. Data 2019, 6, 148. [Google Scholar] [CrossRef]
- Pirtskhalava, M.; Gabrielian, A.; Cruz, P.; Griggs, H.L.; Squires, R.B.; Hurt, D.E.; Grigolava, M.; Chubinidze, M.; Gogoladze, G.; Vishnepolsky, B.; et al. DBAASP v.2: An enhanced database of structure and antimicrobial/cytotoxic activity of natural and synthetic peptides. Nucleic Acids Res. 2016, 44, D1104–D1112. [Google Scholar] [CrossRef]
- Singh, S.; Chaudhary, K.; Dhanda, S.; Bhalla, S.; Usmani, S.S.; Gautam, A.; Tuknait, A.; Agrawal, P.; Mathur, D.; Raghava, G.P. SATPdb: A database of structurally annotated therapeutic peptides. Nucleic Acids Res. 2016, 44, D1119–D1126. [Google Scholar] [CrossRef]
- Waghu, F.H.; Barai, R.S.; Gurung, P.; Idicula-Thomas, S. CAMPR3: A database on sequences, structures and signatures of antimicrobial peptides. Nucleic Acids Res. 2016, 44, D1094–D1097. [Google Scholar] [CrossRef]
- Qureshi, A.; Thakur, N.; Tandon, H.; Kumar, M. AVPdb: A database of experimentally validated antiviral peptides targeting medically important viruses. Nucleic Acids Res. 2014, 42, D1147–D1153. [Google Scholar] [CrossRef]
- Piotto, S.P.; Sessa, L.; Concilio, S.; Iannelli, P. YADAMP: Yet another database of antimicrobial peptides. Int. J. Antimicrob. Agents 2012, 39, 346–351. [Google Scholar] [CrossRef]
- Tyagi, A.; Pankaj, V.; Singh, S.; Roy, S.; Semwal, M.; Shasany, A.K.; Sharma, A. PlantAFP: A curated database of plant-origin antifungal peptides. Amino Acids 2019, 51, 1561–1568. [Google Scholar] [CrossRef]
- Hammami, R.; Zouhir, A.; Ben Hamida, J.; Fliss, I. BACTIBASE: A new web-accessible database for bacteriocin characterization. BMC Microbiol. 2007, 7, 89. [Google Scholar] [CrossRef]
- Ramos-Martin, F.; Annaval, T.; Buchoux, S.; Sarazin, C.; D’Amelio, N. ADAPTABLE: A comprehensive web platform of antimicrobial peptides tailored to the user’s research. Life Sci. Alliance 2019, 2, e201900512. [Google Scholar] [CrossRef]
- Ye, G.; Wu, H.; Huang, J.; Wang, W.; Ge, K.; Li, G.; Zhong, J.; Huang, Q. LAMP2: A major update of the database linking antimicrobial peptides. Database 2020, 2020, baaa061. [Google Scholar] [CrossRef]
- Aronica, P.G.; Reid, L.M.; Desai, N.; Li, J.; Fox, S.J.; Yadahalli, S.; Essex, J.W.; Verma, C.S. Computational Methods and Tools in Antimicrobial Peptide Research. J. Chem. Inf. Model. 2021, 61, 3172–3196. [Google Scholar] [CrossRef] [PubMed]
- Vilas Boas, L.C.P.; Campos, M.L.; Berlanda, R.L.A.; de Carvalho Neves, N.; Franco, O.L. Antiviral peptides as promising therapeutic drugs. Cell. Mol. Life Sci. 2019, 76, 3525–3542. [Google Scholar] [CrossRef]
- Lou, Z.; Sun, Y.; Rao, Z. Current progress in antiviral strategies. Trends Pharmacol. Sci. 2014, 35, 86–102. [Google Scholar] [CrossRef] [PubMed]
- De Ullivarri, M.F.; Arbulu, S.; Garcia-Gutierrez, E.; Cotter, P.D. Antifungal Peptides as Therapeutic Agents. Front. Cell. Infect. Microbiol. 2020, 10, 105. [Google Scholar] [CrossRef]
- Giovati, L.; Ciociola, T.; Magliani, W.; Conti, S. Antimicrobial peptides with antiprotozoal activity: Current state and future perspectives. Futur. Med. Chem. 2018, 10, 2569–2572. [Google Scholar] [CrossRef] [PubMed]
- Abraham, E.P.; Chain, E. An Enzyme from Bacteria able to Destroy Penicillin. Nature 1940, 146, 837. [Google Scholar] [CrossRef]
- Podolsky, S.H. The evolving response to antibiotic resistance (1945–2018). Palgrave Commun. 2018, 4, 124. [Google Scholar] [CrossRef]
- World Health Organization. Antimicrobial Resistance. Available online: https://www.who.int/news-room/fact-sheets/detail/antimicrobial-resistance (accessed on 21 April 2021).
- Peschel, A.; Sahl, H.-G. The co-evolution of host cationic antimicrobial peptides and microbial resistance. Nat. Rev. Microbiol. 2006, 4, 529–536. [Google Scholar] [CrossRef]
- Skarnes, R.C.; Watson, D.W. Antimicrobial factors of normal tissues and fluids. Bacteriol. Rev. 1957, 21, 273–294. [Google Scholar] [CrossRef]
- Abraham, E.P.; Robinson, R. Crystallization of Lysozyme. Nature 1937, 140, 24. [Google Scholar] [CrossRef]
- Roberts, E.A.H. The preparation and properties of purified egg-white lysozyme. Q. J. Exp. Physiol. 1937, 27, 89–98. [Google Scholar] [CrossRef]
- Canfield, R.E. The Amino Acid Sequence of Egg White Lysozyme. J. Biol. Chem. 1963, 238, 2698–2707. [Google Scholar] [CrossRef]
- Rogers, L.A. The inhibiting effect of streptococcus lactis on lactobacillus bulgaricus. J. Bacteriol. 1928, 16, 321–325. [Google Scholar] [CrossRef]
- Berridge, N.J.; Newton, G.G.F.; Abraham, E.P. Purification and nature of the antibiotic nisin. Biochem. J. 1952, 52, 529–535. [Google Scholar] [CrossRef]
- Sahl, H.-G.; Jack, R.W.; Bierbaum, G. Biosynthesis and Biological Activities of Lantibiotics with Unique Post-Translational Modifications. Eur. J. Biol. Chem. 1995, 230, 827–853. [Google Scholar] [CrossRef] [PubMed]
- Dubos, R.J.; Cattaneo, C. Studies on a bactericidal agent extracted from a soil bacillus: III. Preparation and activity of a protein-free fractionj. J. Exp. Med. 1939, 70, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Dubos, R.J.; Hotchkiss, R.D. The production of bactericidal substances by aerobic sporulating bacilli. J. Exp. Med. 1941, 73, 629–640. [Google Scholar] [CrossRef]
- Herrell, W.E.; Heilman, D. Experimental and clinical studies on gramicidin. J. Clin. Investig. 1941, 20, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Craig, L.C.; Gregory, J.D.; Barry, G.T. Studies on polypeptides and amino acids by countercurrent distribution. Cold Spring Harb. Symp. Quant. Biol. 1950, 14, 24–31. [Google Scholar] [CrossRef]
- Sarges, R.; Gramicidin, W.B., VIII. The structure of valine-and isoleucine-gramicidin C. Biochemistry 1965, 4, 2491–2494. [Google Scholar] [CrossRef]
- Sarges, R.; Witkop, B. Gramicidin A. V. The Structure of Valine- and Isoleucine-gramicidin A. J. Am. Chem. Soc. 1965, 87, 2011–2020. [Google Scholar] [CrossRef]
- Sarges, R.; Witkop, B. Gramicidin. VII. The Structure of Valine- and Isoleucine-gramicidin B. J. Am. Chem. Soc. 1965, 87, 2027–2030. [Google Scholar] [CrossRef]
- Van Epps, H.L. René Dubos: Unearthing antibiotics. J. Exp. Med. 2006, 203, 259. [Google Scholar] [CrossRef]
- Nakatsuji, T.; Gallo, R.L. Antimicrobial Peptides: Old Molecules with New Ideas. J. Investig. Dermatol. 2012, 132, 887–895. [Google Scholar] [CrossRef]
- Martin, A.J.P.; Synge, R.L.M. A new form of chromatogram employing two liquid phases: A theory of chromatography. 2. Application to the micro-determination of the higher monoamino-acids in proteins. Biochem. J. 1941, 35, 1358–1368. [Google Scholar] [CrossRef]
- Consden, R.; Gordon, A.H.; Martin, A.J.P. Qualitative analysis of proteins: A partition chromatographic method using paper. Biochem. J. 1944, 38, 224–232. [Google Scholar] [CrossRef]
- Edman, P. A method for the determination of amino acid sequence in peptides. Arch. Biochem. 1949, 22, 475. [Google Scholar] [CrossRef]
- Edman, P.; Begg, G. A Protein Sequenator. Eur. J. Biol. Chem. 1967, 1, 80–91. [Google Scholar] [CrossRef]
- Moore, S.; Stein, W.H. Chromatography of amino acids on sulfonated polystyrene resins. J. Biol. Chem. 1951, 192, 663–681. [Google Scholar] [CrossRef]
- Merrifield, R.B. Solid Phase Peptide Synthesis. I. The Synthesis of a Tetrapeptide. J. Am. Chem. Soc. 1963, 85, 2149–2154. [Google Scholar] [CrossRef]
- Balls, A.K.; Hale, W.S.; Harris, T.H. A Crystalline Protein Obtained from a Lipoprotein of Wheat Flour. Cereal Chem. 1942, 19, 279–288. [Google Scholar]
- Mak, A.S.; Jones, B.L. The amino acid sequence of wheat β-purothionin. Can. J. Biochem. 1976, 54, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, K.; Okada, T.; Yoshizumi, H.; Kagamiyama, H. Complete Primary Structures of Two Subunits of Purothionin A, a Lethal Protein for Brewer’s Yeast from Wheat Flour. J. Biochem. 1977, 82, 753–767. [Google Scholar] [CrossRef]
- Nawrot, R.; Barylski, J.; Nowicki, G.; Broniarczyk, J.; Buchwald, W.; Goździcka-Józefiak, A. Plant antimicrobial peptides. Folia Microbiol. 2014, 59, 181–196. [Google Scholar] [CrossRef]
- Stec, B. Plant thionins—The structural perspective. Cell. Mol. Life Sci. 2006, 63, 1370–1385. [Google Scholar] [CrossRef]
- Sergiev, P. Clinical use of gramicidin S. Lancet 1944, 244, 717–718. [Google Scholar] [CrossRef]
- Gause, G.F.; Brazhnikova, M.G. Gramicidin S and its use in the Treatment of Infected Wounds. Nature 1944, 154, 703. [Google Scholar] [CrossRef]
- Gause, G.F.; Brazhnikova, M.G. Soviet Gramicidin and Wound Healing. Moscow Medgiz (in Russian). 1943, 107. [Google Scholar]
- Gause, G.F. Colistatin: A New Antibiotic Substance With Chemotherapeutic Activity. Science 1946, 104, 289–290. [Google Scholar] [CrossRef]
- Stansly, P.G.; Shepherd, R.G.; White, H.J. Polymyxin: A new chemotherapeutic agent. Bull. Johns Hopkins Hosp. 1947, 81, 43–54. [Google Scholar] [PubMed]
- Poirel, L.; Jayol, A.; Nordmann, P. Polymyxins: Antibacterial Activity, Susceptibility Testing, and Resistance Mechanisms Encoded by Plasmids or Chromosomes. Clin. Microbiol. Rev. 2017, 30, 557–596. [Google Scholar] [CrossRef]
- World Health Organization; WHO Advisory Group on Integrated Surveillance of Antimicrobial Resistance. Critically Important Antimicrobials for Human Medicine: Ranking of Antimicrobial Agents for Risk Management of Antimicrobial Resistance due to Non-Human Use, 6th rev. ed.; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- Chen, J.; Guan, S.-M.; Sun, W.; Fu, H. Melittin, the Major Pain-Producing Substance of Bee Venom. Neurosci. Bull. 2016, 32, 265–272. [Google Scholar] [CrossRef]
- Habermann, E. Bee and Wasp Venoms. Science 1972, 177, 314–322. [Google Scholar] [CrossRef]
- Dorman, L.C.; Markley, L.D. Solid phase synthesis and antibacterial activity of N-terminal sequences of melittin. J. Med. Chem. 1971, 14, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Kiss, G.; Michl, H. Uber das Giftsekret der Gelbbauchunke, Bombina variegata L. Toxicon 1962, 1, 33–34. [Google Scholar] [CrossRef]
- Michl, H. Isolation and structure of an hemolytic polypeptide from the defensive secretion of european bombina species. Mon. Chem. 1970, 101, 182. [Google Scholar]
- Simmaco, M.; Kreil, G.; Barra, D. Bombinins, antimicrobial peptides from Bombina species. Biochim. Biophys. Acta (BBA)—Biomembr. 2009, 1788, 1551–1555. [Google Scholar] [CrossRef] [PubMed]
- Guillarme, D.; Veuthey, J.-L. Chapter 1—Theory and Practice of UHPLC and UHPLC–MS. In Handbook of Advanced Chromatography/Mass Spectrometry Techniques; Holčapek, M., Byrdwell, W.C., Eds.; AOCS Press: Urbana, IL, USA, 2017; pp. 1–38. [Google Scholar]
- Laemmli, U.K. Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Aaij, C.; Borst, P. The gel electrophoresis of DNA. Biochim. Biophys. Acta (BBA)—Nucleic Acids Protein Synth. 1972, 269, 192–200. [Google Scholar] [CrossRef]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef]
- Maxam, A.M.; Gilbert, W. A new method for sequencing DNA. Proc. Natl. Acad. Sci. USA 1977, 74, 560–564. [Google Scholar] [CrossRef]
- Jackson, D.A.; Symons, R.H.; Berg, P. Biochemical Method for Inserting New Genetic Information into DNA of Simian Virus 40: Circular SV40 DNA Molecules Containing Lambda Phage Genes and the Galactose Operon of Escherichia coli. Proc. Natl. Acad. Sci. USA 1972, 69, 2904–2909. [Google Scholar] [CrossRef]
- Gran, L. An oxytocic principle found in Oldenlandia affinis DC, an indigenous Congolese drug “kalata-kalata”, used to accelerate delivery. Medd. Nor. Farm. Selsk. 1970, 12, 8. [Google Scholar]
- Saether, O.; Craik, D.; Campbell, I.D.; Sletten, K.; Juul, J.; Norman, D.G. Elucidation of the Primary and Three-Dimensional Structure of the Uterotonic Polypeptide Kalata B1. Biochemistry 1995, 34, 4147–4158. [Google Scholar] [CrossRef]
- Gran, L. On the Effect of a Polypeptide Isolated from “Kalata-Kalata” (Oldenlandia affinis DC) on the Oestrogen Dominated Uterus. Acta Pharmacol. Toxicol. 2009, 33, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, J.; Vincent, A.T.; Charette, S.J.; Derome, N. A brief history of bioinformatics. Brief. Bioinform. 2019, 20, 1981–1996. [Google Scholar] [CrossRef]
- Williamson, M.P.; Havel, T.F.; Wüthrich, K. Solution conformation of proteinase inhibitor IIA from bull seminal plasma by 1H nuclear magnetic resonance and distance geometry. J. Mol. Biol. 1985, 182, 295–315. [Google Scholar] [CrossRef]
- Karas, M.; Hillenkamp, F. Laser desorption ionization of proteins with molecular masses exceeding 10,000 daltons. Anal. Chem. 1988, 60, 2299–2301. [Google Scholar] [CrossRef]
- Fenn, J.B.; Mann, M.; Meng, C.K.; Wong, S.F.; Whitehouse, C.M. Electrospray ionization for mass spectrometry of large biomolecules. Science 1989, 246, 64–71. [Google Scholar] [CrossRef]
- Hultmark, D.; Steiner, H.; Rasmuson, T.; Boman, H.G. Insect Immunity. Purification and Properties of Three Inducible Bactericidal Proteins from Hemolymph of Immunized Pupae of Hyalophora cecropia. Eur. J. Biol. Chem. 1980, 106, 7–16. [Google Scholar] [CrossRef]
- Steiner, H.; Hultmark, D.; Engström, Å.; Bennich, H.; Boman, H.G. Sequence and specificity of two antibacterial proteins involved in insect immunity. Nature 1981, 292, 246–248. [Google Scholar] [CrossRef]
- Boman, H.G. Antibacterial peptides: Key components needed in immunity. Cell 1991, 65, 205–207. [Google Scholar] [CrossRef]
- Selsted, M.; Harwig, S.S.; Ganz, T.; Schilling, J.W.; Lehrer, R. Primary structures of three human neutrophil defensins. J. Clin. Investig. 1985, 76, 1436–1439. [Google Scholar] [CrossRef]
- Ganz, T.; Selsted, M.; Szklarek, D.; Harwig, S.S.; Daher, K.; Bainton, D.F.; Lehrer, R. Defensins. Natural peptide antibiotics of human neutrophils. J. Clin. Investig. 1985, 76, 1427–1435. [Google Scholar] [CrossRef] [PubMed]
- Zasloff, M. Magainins, a class of antimicrobial peptides from Xenopus skin: Isolation, characterization of two active forms, and partial cDNA sequence of a precursor. Proc. Natl. Acad. Sci. USA 1987, 84, 5449–5453. [Google Scholar] [CrossRef]
- Zanetti, M.; Gennaro, R.; Romeo, D. Cathelicidins: A novel protein family with a common proregion and a variable C-terminal antimicrobial domain. FEBS Lett. 1995, 374, 1–5. [Google Scholar] [CrossRef]
- Romeo, D.; Skerlavaj, B.; Bolognesi, M.; Gennaro, R. Structure and bactericidal activity of an antibiotic dodecapeptide purified from bovine neutrophils. J. Biol. Chem. 1988, 263, 9573–9575. [Google Scholar] [CrossRef]
- Oppenheim, F.G.; Xu, T.; McMillian, F.M.; Levitz, S.M.; Diamond, R.D.; Offner, G.; Troxler, R.F. Histatins, a novel family of histidine-rich proteins in human parotid secretion. Isolation, characterization, primary structure, and fungistatic effects on Candida albicans. J. Biol. Chem. 1988, 263, 7472–7477. [Google Scholar] [CrossRef]
- Pluta, P.; Sokol, R. Changes in the expression of antimicrobial peptide genes in honey bees (Apis mellifera) under the influence of various pathogens. Ann. Parasitol. 2020, 66, 457–465. [Google Scholar] [CrossRef]
- Casteels, P.; Ampe, C.; Jacobs, F.; Vaeck, M.; Tempst, P. Apidaecins: Antibacterial peptides from honeybees. EMBO J. 1989, 8, 2387–2391. [Google Scholar] [CrossRef]
- Matsumoto, K.; Orikasa, Y.; Ichinohe, K.; Hashimoto, S.; Ooi, T.; Taguchi, S. Flow cytometric analysis of the contributing factors for antimicrobial activity enhancement of cell-penetrating type peptides: Case study on engineered apidaecins. Biochem. Biophys. Res. Commun. 2010, 395, 7–10. [Google Scholar] [CrossRef]
- Dutta, R.C.; Nagpal, S.; Salunke, D.M. Functional mapping of apidaecin through secondary structure correlation. Int. J. Biochem. Cell Biol. 2008, 40, 1005–1015. [Google Scholar] [CrossRef]
- Casteels, P.; Ampe, C.; Riviere, L.; Damme, J.; Elicone, C.; Fleming, M.; Jacobs, F.; Tempst, P. Isolation and characterization of abaecin, a major antibacterial response peptide in the honeybee (Apis mellifera). Eur. J. Biol. Chem. 1990, 187, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Casteels, P.; Ampe, C.; Jacobs, F.; Tempst, P. Functional and chemical characterization of Hymenoptaecin, an antibacterial polypeptide that is infection-inducible in the honeybee (Apis mellifera). J. Biol. Chem. 1993, 268, 7044–7054. [Google Scholar] [CrossRef]
- Klaudiny, J.; Albert, S.; Bachanova, K.; Kopernicky, J.; Šimúth, J. Two structurally different defensin genes, one of them encoding a novel defensin isoform, are expressed in honeybee Apis mellifera. Insect Biochem. Mol. Biol. 2005, 35, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Ilyasov, R.; Gaifullina, L.; Saltykova, E.; Poskryakov, A.; Nikolenko, A. Review of the Expression of Antimicrobial Peptide Defensin in Honey Bees Apis mellifera L. J. Apic. Sci. 2012, 56, 115–124. [Google Scholar] [CrossRef]
- Agerberth, B.; Lee, J.-Y.; Bergman, T.; Carlquist, M.; Boman, H.G.; Mutt, V.; Jornvall, H. Amino acid sequence of PR-39. Isolation from pig intestine of a new member of the family of proline-arginine-rich antibacterial peptides. Eur. J. Biol. Chem. 1991, 202, 849–854. [Google Scholar] [CrossRef]
- Veldhuizen, E.J.A.; Schneider, V.A.F.; Agustiandari, H.; Van Dijk, A.; Tjeerdsma-van Bokhoven, J.L.M.; Bikker, F.; Haagsman, H.P. Antimicrobial and Immunomodulatory Activities of PR-39 Derived Peptides. PLoS ONE 2014, 9, e95939. [Google Scholar] [CrossRef]
- Chan, Y.R.; Zanetti, M.; Gennaro, R.; Gallo, R. Anti-Microbial Activity and Cell Binding are Controled by Sequence Determinants in the Anti-Microbial Peptide PR-39. J. Investig. Dermatol. 2001, 116, 230–235. [Google Scholar] [CrossRef]
- Sang, Y.; Blecha, F. Porcine host defense peptides: Expanding repertoire and functions. Dev. Comp. Immunol. 2009, 33, 334–343. [Google Scholar] [CrossRef]
- Diamond, G.; Zasloff, M.; Eck, H.; Brasseur, M.; Maloy, W.L.; Bevins, C.L. Tracheal antimicrobial peptide, a cysteine-rich peptide from mammalian tracheal mucosa: Peptide isolation and cloning of a cDNA. Proc. Natl. Acad. Sci. USA 1991, 88, 3952–3956. [Google Scholar] [CrossRef] [PubMed]
- Lawyer, C.; Watabe, M.; Pai, S.; Bakir, H.; Eagleton, L.; Mashimo, T.; Watabe, K. A synthetic form of tracheal antimicrobial peptide has both bactericidal and antifungal activities. Drug Des. Discov. 1996, 14, 171–178. [Google Scholar]
- Agerberth, B.; Gunne, H.; Odeberg, J.; Kogner, P.; Boman, H.G.; Gudmundsson, G.H. FALL-39, a putative human peptide antibiotic, is cysteine-free and expressed in bone marrow and testis. Proc. Natl. Acad. Sci. USA 1995, 92, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Morizane, S.; Gallo, R. Antimicrobial peptides in the pathogenesis of psoriasis. J. Dermatol. 2012, 39, 225–230. [Google Scholar] [CrossRef]
- Pahar, B.; Madonna, S.; Das, A.; Albanesi, C.; Girolomoni, G. Immunomodulatory Role of the Antimicrobial LL-37 Peptide in Autoimmune Diseases and Viral Infections. Vaccines 2020, 8, 517. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.-Q.; Yuan, J.; Ösapay, G.; Ösapay, K.; Tran, D.; Miller, C.J.; Ouellette, A.J.; Selsted, M.E. A Cyclic Antimicrobial Peptide Produced in Primate Leukocytes by the Ligation of Two Truncated α-Defensins. Science 1999, 286, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Tongaonkar, P.; Tran, P.; Roberts, K.; Schaal, J.; Ösapay, G.; Tran, D.; Ouellette, A.J.; Selsted, M.E. Rhesus macaque θ-defensin isoforms: Expression, antimicrobial activities, and demonstration of a prominent role in neutrophil granule microbicidal activities. J. Leukoc. Biol. 2011, 89, 283–290. [Google Scholar] [CrossRef]
- Schittek, B.; Hipfel, R.; Sauer, B.; Bauer, J.; Kalbacher, H.; Stevanovic, S.; Schirle, M.; Schroeder, K.; Blin, N.; Meier, F.; et al. Dermcidin: A novel human antibiotic peptide secreted by sweat glands. Nat. Immunol. 2001, 2, 1133–1137. [Google Scholar] [CrossRef] [PubMed]
- Zeth, K.; Sancho-Vaello, E. The Human Antimicrobial Peptides Dermcidin and LL-37 Show Novel Distinct Pathways in Membrane Interactions. Front. Chem. 2017, 5, 86. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Weichbrodt, C.; Salnikov, E.; Dynowski, M.; Forsberg, B.; Bechinger, B.; Steinem, C.; de Groot, B.L.; Zachariae, U.; Zeth, K. Crystal structure and functional mechanism of a human antimicrobial membrane channel. Proc. Natl. Acad. Sci. USA 2013, 110, 4586–4591. [Google Scholar] [CrossRef]
- Mygind, P.H.; Fischer, R.L.; Schnorr, K.M.; Hansen, M.T.; Sönksen, C.P.; Ludvigsen, S.; Raventós, D.; Buskov, S.; Christensen, B.; De Maria, L.; et al. Plectasin is a peptide antibiotic with therapeutic potential from a saprophytic fungus. Nature 2005, 437, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Xiang, F.; Xie, Z.; Feng, J.; Yang, W.; Cao, Z.; Li, W.; Chen, Z.; Wu, Y. Plectasin, First Animal Toxin-Like Fungal Defensin Blocking Potassium Channels through Recognizing Channel Pore Region. Toxins 2015, 7, 34–42. [Google Scholar] [CrossRef]
- Tian, C.; Zhu, R.; Zhu, L.; Qiu, T.; Cao, Z.; Kang, T. Potassium Channels: Structures, Diseases, and Modulators. Chem. Biol. Drug Des. 2014, 83, 1–26. [Google Scholar] [CrossRef]
- Tam, C.; Mun, J.J.; Evans, D.; Fleiszig, S.M. Cytokeratins mediate epithelial innate defense through their antimicrobial properties. J. Clin. Investig. 2012, 122, 3665–3677. [Google Scholar] [CrossRef]
- Chan, J.K.; Yuen, D.; Too, P.H.-M.; Sun, Y.; Willard, B.; Man, D.; Tam, C. Keratin 6a reorganization for ubiquitin–proteasomal processing is a direct antimicrobial response. J. Cell Biol. 2018, 217, 731–744. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.K.; Too, P.; Tam, K.P.C. The Ubiquitin-Proteasome Pathway Generates Keratin 6A-Derived Antimicrobial Peptides to Mediate Antimicrobial Activities in Human Corneal Epithelial Cells. Investig. Ophthalmol. Vis. Sci. 2016, 57. [Google Scholar]
- Münch, J.; Ständker, L.; Adermann, K.; Schulz, A.; Schindler, M.; Chinnadurai, R.; Pöhlmann, S.; Chaipan, C.; Biet, T.; Peters, T.; et al. Discovery and Optimization of a Natural HIV-1 Entry Inhibitor Targeting the gp41 Fusion Peptide. Cell 2007, 129, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Zirafi, O.; Kim, K.-A.; Ständker, L.; Mohr, K.B.; Sauter, D.; Heigele, A.; Kluge, S.F.; Wiercinska, E.; Chudziak, D.; Richter, R.; et al. Discovery and Characterization of an Endogenous CXCR4 Antagonist. Cell Rep. 2015, 11, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Hayn, M.; Blötz, A.; Rodríguez, A.; Vidal, S.; Preising, N.; Ständker, L.; Wiese, S.; Stürzel, C.M.; Harms, M.; Gross, R.; et al. Natural cystatin C fragments inhibit GPR15-mediated HIV and SIV infection without interfering with GPR15L signaling. Proc. Natl. Acad. Sci. USA 2021, 118, e2023776118. [Google Scholar] [CrossRef] [PubMed]
- Liepke, C.; Baxmann, S.; Heine, C.; Breithaupt, N.; Ständker, L.; Forssmann, W.-G. Human hemoglobin-derived peptides exhibit antimicrobial activity: A class of host defense peptides. J. Chromatogr. B 2003, 791, 345–356. [Google Scholar] [CrossRef]
- Groß, R.; Bauer, R.; Krüger, F.; Rücker-Braun, E.; Olari, L.-R.; Ständker, L.; Preising, N.; Rodríguez, A.A.; Conzelmann, C.; Gerbl, F.; et al. A Placenta Derived C-Terminal Fragment of β-Hemoglobin With Combined Antibacterial and Antiviral Activity. Front. Microbiol. 2020, 11, 508. [Google Scholar] [CrossRef]
- Nichols, D.; Cahoon, N.; Trakhtenberg, E.M.; Pham, L.; Mehta, A.; Belanger, A.; Kanigan, T.; Lewis, K.; Epstein, S.S. Use of Ichip for High-Throughput In Situ Cultivation of “Uncultivable” Microbial Species. Appl. Environ. Microbiol. 2010, 76, 2445–2450. [Google Scholar] [CrossRef]
- Ling, L.L.; Schneider, T.; Peoples, A.J.; Spoering, A.L.; Engels, I.; Conlon, B.P.; Mueller, A.; Schäberle, T.F.; Hughes, D.E.; Epstein, S.; et al. A new antibiotic kills pathogens without detectable resistance. Nature 2015, 517, 455–459. [Google Scholar] [CrossRef]
- Gunjal, V.B.; Thakare, R.P.; Chopra, S.; Reddy, D.S. Teixobactin: A Paving Stone toward a New Class of Antibiotics? J. Med. Chem. 2020, 63, 12171–12195. [Google Scholar] [CrossRef] [PubMed]
- Öster, C.; Walkowiak, G.P.; Hughes, D.E.; Spoering, A.L.; Peoples, A.J.; Catherwood, A.C.; Tod, J.A.; Lloyd, A.J.; Herrmann, T.; Lewis, K.; et al. Structural studies suggest aggregation as one of the modes of action for teixobactin. Chem. Sci. 2018, 9, 8850–8859. [Google Scholar] [CrossRef]
- Imai, Y.; Meyer, K.J.; Iinishi, A.; Favre-Godal, Q.; Green, R.; Manuse, S.; Caboni, M.; Mori, M.; Niles, S.; Ghiglieri, M.; et al. A new antibiotic selectively kills Gram-negative pathogens. Nature 2019, 576, 459–464. [Google Scholar] [CrossRef]
- Kaur, H.; Jakob, R.P.; Marzinek, J.K.; Green, R.; Imai, Y.; Bolla, J.R.; Agustoni, E.; Robinson, C.V.; Bond, P.J.; Lewis, K.; et al. The antibiotic darobactin mimics a β-strand to inhibit outer membrane insertase. Nature 2021, 593, 125–129. [Google Scholar] [CrossRef]
- Paterson, D.J.; Tassieri, M.; Reboud, J.; Wilson, R.; Cooper, J.M. Lipid topology and electrostatic interactions underpin lytic activity of linear cationic antimicrobial peptides in membranes. Proc. Natl. Acad. Sci. USA 2017, 114, E8324–E8332. [Google Scholar] [CrossRef]
- Alba, A.; López-Abarrategui, C.; Otero-González, A.J. Host defense peptides: An alternative as antiinfective and immunomodulatory therapeutics. Biopolymers 2012, 98, 251–267. [Google Scholar] [CrossRef]
- Bowdish, D.; Davidson, D.J.; Lau, Y.E.; Lee, K.; Scott, M.G.; Hancock, R. Impact of LL-37 on anti-infective immunity. J. Leukoc. Biol. 2005, 77, 451–459. [Google Scholar] [CrossRef]
- Hollands, A.; Gonzalez, D.; Leire, E.; Donald, C.; Gallo, R.; Sanderson-Smith, M.; Dorrestein, P.C.; Nizet, V. A Bacterial Pathogen Co-opts Host Plasmin to Resist Killing by Cathelicidin Antimicrobial Peptides. J. Biol. Chem. 2012, 287, 40891–40897. [Google Scholar] [CrossRef]
- Moncla, B.J.; Pryke, K.; Rohan, L.C.; Graebing, P.W. Degradation of naturally occurring and engineered antimicrobial peptides by proteases. Adv. Biosci. Biotechnol. 2011, 2, 404–408. [Google Scholar] [CrossRef] [PubMed]
- Shurko, J.F.; Galega, R.S.; Li, C.; Lee, G.C. Evaluation of LL-37 antimicrobial peptide derivatives alone and in combination with vancomycin against S. aureus. J. Antibiot. 2018, 71, 971–974. [Google Scholar] [CrossRef] [PubMed]
- Nell, M.J.; Tjabringa, G.S.; Wafelman, A.R.; Verrijk, R.; Hiemstra, P.; Drijfhout, J.W.; Grote, J.J. Development of novel LL-37 derived antimicrobial peptides with LPS and LTA neutralizing and antimicrobial activities for therapeutic application. Peptides 2006, 27, 649–660. [Google Scholar] [CrossRef] [PubMed]
- Peek, F.; Nell, M.J.; Brand, R.; Jansen-Werkhoven, T.; Van Hoogdalem, E.; Frijns, J. Double-blind placebo-controlled study of the novel peptide drug P60.4Ac in cronic middle ear infection. In Proceedings of the 49th Interscience Conference on Antimicrobial Agents, San Francisco, CA, USA, 12–15 September 2009; pp. L1–L337. [Google Scholar]
- Fuscaldi, L.L.; de Avelar, J.T., Jr.; dos Santos, D.M.; Boff, D.; de Oliveira, V.L.S.; Gomes, K.A.G.G.; Cruz, R.D.C.; de Oliveira, P.L.; Magalhães, P.P.; Cisalpino, P.S.; et al. Shortened derivatives from native antimicrobial peptide LyeTx I: In vitro and in vivo biological activity assessment. Exp. Biol. Med. 2021, 246, 414–425. [Google Scholar] [CrossRef] [PubMed]
- Solstad, R.G.; Johansen, C.; Stensvåg, K.; Strøm, M.B.; Haug, T. Structure-activity relationship studies of shortened analogues of the antimicrobial peptide EeCentrocin 1 from the sea urchin Echinus esculentus. J. Pept. Sci. 2020, 26, e3233. [Google Scholar] [CrossRef] [PubMed]
- Conda-Sheridan, M.; Krishnaiah, M. Protecting Groups in Peptide Synthesis. Methods Mol. Biol. 2020, 2103, 111–128. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, H.; Matsumoto, H.; Hashimoto, K.; Teraguchi, S.; Takase, M.; Hayasawa, H. N-Acylated and d Enantiomer Derivatives of a Nonamer Core Peptide of Lactoferricin B Showing Improved Antimicrobial Activity. Antimicrob. Agents Chemother. 1999, 43, 1267–1269. [Google Scholar] [CrossRef]
- Postma, T.M.; Liskamp, R.M.J. Highly potent antimicrobial peptide derivatives of bovine cateslytin. RSC Adv. 2016, 6, 94840–94844. [Google Scholar] [CrossRef]
- Wenzel, M.; Rautenbach, M.; Vosloo, J.A.; Siersma, T.; Aisenbrey, C.H.M.; Zaitseva, E.; Laubscher, W.E.; van Rensburg, W.; Behrends, J.C.; Bechinger, B.; et al. The Multifaceted Antibacterial Mechanisms of the Pioneering Peptide Antibiotics Tyrocidine and Gramicidin S. mBio 2018, 9, e00802-18. [Google Scholar] [CrossRef]
- Grein, F.; Müller, A.; Scherer, K.M.; Liu, X.; Ludwig, K.C.; Klöckner, A.; Strach, M.; Sahl, H.-G.; Kubitscheck, U.; Schneider, T. Ca(2+)-Daptomycin targets cell wall biosynthesis by forming a tripartite complex with undecaprenyl-coupled intermediates and membrane lipids. Nat. Commun. 2020, 11, 1455. [Google Scholar] [CrossRef] [PubMed]
- Wilmes, M.; Stockem, M.; Bierbaum, G.; Schlag, M.; Götz, F.; Tran, D.Q.; Schaal, J.B.; Ouellette, A.J.; Selsted, M.E.; Sahl, H.-G. Killing of Staphylococci by θ-Defensins Involves Membrane Impairment and Activation of Autolytic Enzymes. Antibiotics 2014, 3, 617–631. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Gu, L.; Hussain, M.A.; Chen, L.; Lin, L.; Wang, H.; Pang, S.; Jiang, C.; Jiang, Z.; Hou, J. Characterization of the Bioactivity and Mechanism of Bactenecin Derivatives Against Food-Pathogens. Front. Microbiol. 2019, 10, 2593. [Google Scholar] [CrossRef] [PubMed]
- Niyonsaba, F.; Madera, L.; Afacan, N.; Okumura, K.; Ogawa, H.; Hancock, R. The innate defense regulator peptides IDR-HH2, IDR-1002, and IDR-1018 modulate human neutrophil functions. J. Leukoc. Biol. 2013, 94, 159–170. [Google Scholar] [CrossRef]
- Dathe, M.; Nikolenko, H.; Klose, J.; Bienert, M. Cyclization Increases the Antimicrobial Activity and Selectivity of Arginine- and Tryptophan-Containing Hexapeptides. Biochemistry 2004, 43, 9140–9150. [Google Scholar] [CrossRef]
- Badosa, E.; Moiset, G.; Montesinos, L.; Talleda, M.; Bardají, E.; Feliu, L.; Planas, M.; Montesinos, E. Derivatives of the Antimicrobial Peptide BP100 for Expression in Plant Systems. PLoS ONE 2013, 8, e85515. [Google Scholar] [CrossRef]
- Klubthawee, N.; Adisakwattana, P.; Hanpithakpong, W.; Somsri, S.; Aunpad, R. A novel, rationally designed, hybrid antimicrobial peptide, inspired by cathelicidin and aurein, exhibits membrane-active mechanisms against Pseudomonas aeruginosa. Sci. Rep. 2020, 10, 9117. [Google Scholar] [CrossRef]
- Schäfer, A.-B.; Wenzel, M. A How-To Guide for Mode of Action Analysis of Antimicrobial Peptides. Front. Cell. Infect. Microbiol. 2020, 10, 540898. [Google Scholar] [CrossRef]
- Liu, Y.; Du, Q.; Ma, C.; Xi, X.; Wang, L.; Zhou, M.; Burrows, J.F.; Chen, T.; Wang, H. Structure–activity relationship of an antimicrobial peptide, Phylloseptin-PHa: Balance of hydrophobicity and charge determines the selectivity of bioactivities. Drug Des. Dev. Ther. 2019, 13, 447–458. [Google Scholar] [CrossRef]
- Waghu, F.; Idicula-Thomas, S. Collection of antimicrobial peptides database and its derivatives: Applications and beyond. Protein Sci. 2020, 29, 36–42. [Google Scholar] [CrossRef]
- Amso, Z.; Hayouka, Z. Antimicrobial random peptide cocktails: A new approach to fight pathogenic bacteria. Chem. Commun. 2019, 55, 2007–2014. [Google Scholar] [CrossRef] [PubMed]
- Bahar, A.A.; Ren, D. Antimicrobial Peptides. Pharmaceuticals 2013, 6, 1543–1575. [Google Scholar] [CrossRef] [PubMed]
- Starr, C.G.; Maderdrut, J.L.; He, J.; Coy, D.H.; Wimley, W.C. Pituitary adenylate cyclase-activating polypeptide is a potent broad-spectrum antimicrobial peptide: Structure-activity relationships. Pepides 2018, 104, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Sun, L.; Huang, S.; Zhu, C.; Li, P.; He, J.; Mackey, V.; Coy, D.H.; He, Q. The antimicrobial peptides and their potential clinical applications. Am. J. Transl. Res. 2019, 11, 3919–3931. [Google Scholar]
- Yount, N.Y.; Bayer, A.S.; Xiong, Y.Q.; Yeaman, M.R. Advances in antimicrobial peptide immunobiology. Biopolymers 2006, 84, 435–458. [Google Scholar] [CrossRef]
- Deslouches, B.; Phadke, S.M.; Lazarevic, V.; Cascio, M.; Islam, K.; Montelaro, R.C.; Mietzner, T.A. De Novo Generation of Cationic Antimicrobial Peptides: Influence of Length and Tryptophan Substitution on Antimicrobial Activity. Antimicrob. Agents Chemother. 2005, 49, 316–322. [Google Scholar] [CrossRef]
- Wang, J.; Yadav, V.; Smart, A.L.; Tajiri, S.; Basit, A.W. Stability of peptide drugs in the colon. Eur. J. Pharm. Sci. 2015, 78, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yadav, V.; Smart, A.L.; Tajiri, S.; Basit, A.W. Toward Oral Delivery of Biopharmaceuticals: An Assessment of the Gastrointestinal Stability of 17 Peptide Drugs. Mol. Pharm. 2015, 12, 966–973. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Chau, J.K.; Perry, N.A.; De Boer, L.; Zaat, S.A.J.; Vogel, H.J. Serum Stabilities of Short Tryptophan- and Arginine-Rich Antimicrobial Peptide Analogs. PLoS ONE 2010, 5, e12684. [Google Scholar] [CrossRef] [PubMed]
- Vicente, F.E.M.; González-Garcia, M.; Pico, E.D.; Castillo, E.M.; Garay, H.E.; Rosi, P.E.; Jimenez, A.M.; Delgado, J.A.C.; Rivera, D.G.; Chinea, G.; et al. Design of a Helical-Stabilized, Cyclic, and Nontoxic Analogue of the Peptide Cm-p5 with Improved Antifungal Activity. ACS Omega 2019, 4, 19081–19095. [Google Scholar] [CrossRef]
- Mahlapuu, M.; Björn, C.; Ekblom, J. Antimicrobial peptides as therapeutic agents: Opportunities and challenges. Crit. Rev. Biotechnol. 2020, 40, 978–992. [Google Scholar] [CrossRef] [PubMed]
- Chongsiriwatana, N.P.; Patch, J.A.; Czyzewski, A.M.; Dohm, M.T.; Ivankin, A.; Gidalevitz, D.; Zuckermann, R.N.; Barron, A.E. Peptoids that mimic the structure, function, and mechanism of helical antimicrobial peptides. Proc. Natl. Acad. Sci. USA 2008, 105, 2794–2799. [Google Scholar] [CrossRef]
- Thaker, H.D.; Som, A.; Ayaz, F.; Lui, D.; Pan, W.; Scott, R.W.; Anguita, J.; Tew, G.N. Synthetic Mimics of Antimicrobial Peptides with Immunomodulatory Responses. J. Am. Chem. Soc. 2012, 134, 11088–11091. [Google Scholar] [CrossRef]
- Pfalzgraff, A.; Brandenburg, K.; Weindl, G. Antimicrobial Peptides and Their Therapeutic Potential for Bacterial Skin Infections and Wounds. Front. Pharmacol. 2018, 9, 281. [Google Scholar] [CrossRef]
- Browne, K.; Chakraborty, S.; Chen, R.; Willcox, M.D.; Black, D.S.; Walsh, W.R.; Kumar, N. A New Era of Antibiotics: The Clinical Potential of Antimicrobial Peptides. Int. J. Mol. Sci. 2020, 21, 7047. [Google Scholar] [CrossRef]
- López-Abarrategui, C.; McBeth, C.; Mandal, S.M.; Sun, Z.J.; Heffron, G.; Alba-Menéndez, A.; Migliolo, L.; Reyes-Acosta, O.; García-Villarino, M.; Nolasco, D.O.; et al. Cm-p5: An antifungal hydrophilic peptide derived from the coastal mollusk Cenchritis muricatus (Gastropoda: Littorinidae). FASEB J. 2015, 29, 3315–3325. [Google Scholar] [CrossRef]
- Di, Y.P.; Lin, Q.; Chen, C.; Montelaro, R.C.; Doi, Y.; Deslouches, B. Enhanced therapeutic index of an antimicrobial peptide in mice by increasing safety and activity against multidrug-resistant bacteria. Sci. Adv. 2020, 6, eaay6817. [Google Scholar] [CrossRef]
- Qureshi, Z.A.; Hittle, L.E.; O’Hara, J.A.; Rivera, J.I.; Syed, A.; Shields, R.K.; Pasculle, A.W.; Ernst, R.; Doi, Y. Colistin-Resistant Acinetobacter baumannii: Beyond Carbapenem Resistance. Clin. Infect. Dis. 2015, 60, 1295–1303. [Google Scholar] [CrossRef]
- Mahlapuu, M.; Håkansson, J.; Ringstad, L.; Björn, C. Antimicrobial Peptides: An Emerging Category of Therapeutic Agents. Front. Cell. Infect. Microbiol. 2016, 6, 194. [Google Scholar] [CrossRef]
- Tincho, M.; Morris, T.; Meyer, M.; Pretorius, A. Antibacterial Activity of Rationally Designed Antimicrobial Peptides. Int. J. Microbiol. 2020, 2020, 2131535. [Google Scholar] [CrossRef]
- Mourtada, R.; Herce, H.D.; Yin, D.J.; Moroco, J.A.; Wales, T.E.; Engen, J.R.; Walensky, L.D. Design of stapled antimicrobial peptides that are stable, nontoxic and kill antibiotic-resistant bacteria in mice. Nat. Biotechnol. 2019, 37, 1186–1197. [Google Scholar] [CrossRef]
- Brandenburg, K.; Andrä, J.; Garidel, P.; Gutsmann, T. Peptide-based treatment of sepsis. Appl. Microbiol. Biotechnol. 2011, 90, 799–808. [Google Scholar] [CrossRef]
- Brandenburg, K.; Heinbockel, L.; Correa, W.; Lohner, K. Peptides with dual mode of action: Killing bacteria and preventing endotoxin-induced sepsis. Biochim. Biophys. Acta (BBA)—Biomembr. 2016, 1858, 971–979. [Google Scholar] [CrossRef]
- Gai, Z.; Samodelov, S.L.; Kullak-Ublick, G.A.; Visentin, M. Molecular Mechanisms of Colistin-Induced Nephrotoxicity. Molecules 2019, 24, 653. [Google Scholar] [CrossRef]
- Li, F.F.; Brimble, M.A. Using chemical synthesis to optimise antimicrobial peptides in the fight against antimicrobial resistance. Pure Appl. Chem. 2019, 91, 181–198. [Google Scholar] [CrossRef]
- Merrifield, R.B.; Merrifield, E.L.; Juvvadi, P.; Andreu, D.; Boman, H.G. Design and synthesis of antimicrobial peptides. Ciba Found. Symp. 1994, 186, 5–20. [Google Scholar]
- Werle, M.; Bernkop-Schnürch, A. Strategies to improve plasma half life time of peptide and protein drugs. Amino Acids 2006, 30, 351–367. [Google Scholar] [CrossRef]
- Johnson, I. Human insulin from recombinant DNA technology. Science 1983, 219, 632–637. [Google Scholar] [CrossRef]
- Fields, F.R.; Carothers, K.E.; Balsara, R.D.; Ploplis, V.A.; Castellino, F.J.; Lee, S.W. Rational design of syn-safencin, a novel linear antimicrobial peptide derived from the circular bacteriocin safencin AS-48. J. Antibiot. 2018, 71, 592–600. [Google Scholar] [CrossRef]
- Duckworth, W.C.; Bennett, R.G.; Hamel, F.G. Insulin Degradation: Progress and Potential. Endocr. Rev. 1998, 19, 608–624. [Google Scholar] [CrossRef] [PubMed]
- Hamamoto, K.; Kida, Y.; Zhang, Y.; Shimizu, T.; Kuwano, K. Antimicrobial Activity and Stability to Proteolysis of Small Linear Cationic Peptides with D-Amino Acid Substitutions. Microbiol. Immunol. 2002, 46, 741–749. [Google Scholar] [CrossRef]
- Lee, A.C.-L.; Harris, J.L.; Khanna, K.K.; Hong, J.-H. A Comprehensive Review on Current Advances in Peptide Drug Development and Design. Int. J. Mol. Sci. 2019, 20, 2383. [Google Scholar] [CrossRef]
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorg. Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef] [PubMed]
- Zorzi, A.; Deyle, K.; Heinis, C. Cyclic peptide therapeutics: Past, present and future. Curr. Opin. Chem. Biol. 2017, 38, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Bogdanowich-Knipp, S.J.; Chakrabarti, S.; Siahaan, T.J.; Williams, T.D.; Dillman, R.K. Solution stability of linear vs. cyclic RGD peptides. J. Pept. Res. 1999, 53, 530–541. [Google Scholar] [CrossRef]
- Rink, R.; Arkema-Meter, A.; Baudoin, I.; Post, E.; Kuipers, A.; Nelemans, S.; Akanbi, M.H.J.; Moll, G. To protect peptide pharmaceuticals against peptidases. J. Pharmacol. Toxicol. Methods 2010, 61, 210–218. [Google Scholar] [CrossRef]
- Knappe, D.; Henklein, P.; Hoffmann, R.; Hilpert, K. Easy Strategy To Protect Antimicrobial Peptides from Fast Degradation in Serum. Antimicrob. Agents Chemother. 2010, 54, 4003–4005. [Google Scholar] [CrossRef]
- Mathur, D.; Prakash, S.; Anand, P.; Kaur, H.; Agrawal, P.; Mehta, A.; Kumar, R.; Singh, S.; Raghava, G.P.S. PEPlife: A Repository of the Half-life of Peptides. Sci. Rep. 2016, 6, 36617. [Google Scholar] [CrossRef]
- Schütz, D.; Ruiz-Blanco, Y.B.; Münch, J.; Kirchhoff, F.; Sanchez-Garcia, E.; Müller, J.A. Peptide and peptide-based inhibitors of SARS-CoV-2 entry. Adv. Drug Deliv. Rev. 2020, 167, 47–65. [Google Scholar] [CrossRef]
- Splith, K.; Neundorf, I. Antimicrobial peptides with cell-penetrating peptide properties and vice versa. Eur. Biophys. J. 2011, 40, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Brogden, N.K.; Brogden, K.A. Will new generations of modified antimicrobial peptides improve their potential as pharmaceuticals? Int. J. Antimicrob. Agents 2011, 38, 217–225. [Google Scholar] [CrossRef] [PubMed]
- De La Torre, B.G.; Albericio, F. Peptide Therapeutics 2.0. Molecules 2020, 25, 2293. [Google Scholar] [CrossRef] [PubMed]
- Vagner, J.; Qu, H.; Hruby, V.J. Peptidomimetics, a synthetic tool of drug discovery. Curr. Opin. Chem. Biol. 2008, 12, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Nordström, R.; Malmsten, M. Delivery systems for antimicrobial peptides. Adv. Colloid Interface Sci. 2017, 242, 17–34. [Google Scholar] [CrossRef]
- Recio, C.; Maione, F.; Iqbal, A.; Mascolo, N.; De Feo, V. The Potential Therapeutic Application of Peptides and Peptidomimetics in Cardiovascular Disease. Front. Pharmacol. 2016, 7, 526. [Google Scholar] [CrossRef]
- Nordström, R.; Nyström, L.; Ilyas, H.; Atreya, H.S.; Borro, B.C.; Bhunia, A.; Malmsten, M. Microgels as carriers of antimicrobial peptides—Effects of peptide PEGylation. Colloids Surf. A Physicochem. Eng. Asp. 2019, 565, 8–15. [Google Scholar] [CrossRef]
- Drayton, M.; Kizhakkedathu, J.N.; Straus, S.K. Towards Robust Delivery of Antimicrobial Peptides to Combat Bacterial Resistance. Molecules 2020, 25, 3048. [Google Scholar] [CrossRef] [PubMed]
- David, A.A.; Park, S.E.; Parang, K.; Tiwari, R.K. Antibiotics-Peptide Conjugates Against Multidrug-resistant Bacterial Pathogens. Curr. Top. Med. Chem. 2018, 18, 1926–1936. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, A.; Neundorf, I. Design and Application of Antimicrobial Peptide Conjugates. Int. J. Mol. Sci. 2016, 17, 701. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Lim, S.I.; Shin, S.-H.; Lim, Y.; Koh, J.W.; Yang, S. Conjugation of Cell-Penetrating Peptides to Antimicrobial Peptides Enhances Antibacterial Activity. ACS Omega 2019, 4, 15694–15701. [Google Scholar] [CrossRef]
- Al-Rifai, A.A.; Ayoub, M.T.; Shakya, A.K.; Abu Safieh, K.A.; Mubarak, M.S. Synthesis, characterization, and antimicrobial activity of some new coumarin derivatives. Med. Chem. Res. 2012, 21, 468–476. [Google Scholar] [CrossRef]
- Ferreira, S.Z.; Carneiro, H.C.; Lara, H.A.; Alves, R.; Resende, J.; Oliveira, H.M.; Silva, L.M.; Santos, D.; Freitas, R. Synthesis of a New Peptide–Coumarin Conjugate: A Potential Agent against Cryptococcosis. ACS Med. Chem. Lett. 2015, 6, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Salomone, F.; Cardarelli, F.; Di Luca, M.; Boccardi, C.; Nifosì, R.; Bardi, G.; Di Bari, L.; Serresi, M.; Beltram, F. A novel chimeric cell-penetrating peptide with membrane-disruptive properties for efficient endosomal escape. J. Control. Release 2012, 163, 293–303. [Google Scholar] [CrossRef]
- Luan, L.; Meng, Q.; Xu, L.; Meng, Z.; Yan, H.; Liu, K. Peptide amphiphiles with multifunctional fragments promoting cellular uptake and endosomal escape as efficient gene vectors. J. Mater. Chem. B 2015, 3, 1068–1078. [Google Scholar] [CrossRef]
- Hu, Y.; Amin, M.N.; Padhee, S.; Wang, R.; Qiao, Q.; Bai, G.; Li, Y.; Mathew, A.; Cao, C.; Cai, J. Lipidated Peptidomimetics with Improved Antimicrobial Activity. ACS Med. Chem. Lett. 2012, 3, 683–686. [Google Scholar] [CrossRef]
- Chu-Kung, A.F.; Nguyen, R.; Bozzelli, K.N.; Tirrell, M. Chain length dependence of antimicrobial peptide–fatty acid conjugate activity. J. Colloid Interface Sci. 2010, 345, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Veronese, F.M.; Mero, A. The Impact of PEGylation on Biological Therapies. BioDrugs 2008, 22, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Konno, K.; Hisada, M.; Fontana, R.; Lorenzi, C.C.; Naoki, H.; Itagaki, Y.; Miwa, A.; Kawai, N.; Nakata, Y.; Yasuhara, T.; et al. Anoplin, a novel antimicrobial peptide from the venom of the solitary wasp Anoplius samariensis. Biochim. Biophys. Acta (BBA)—Protein Struct. Mol. Enzym. 2001, 1550, 70–80. [Google Scholar] [CrossRef]
- Rinaldi, A.C.; Mangoni, M.L.; Rufo, A.; Luzi, C.; Barra, D.; Zhao, H.; Kinnunen, P.K.; Bozzi, A.; Di Giulio, A.; Simmaco, M. Temporin L: Antimicrobial, haemolytic and cytotoxic activities, and effects on membrane permeabilization in lipid vesicles. Biochem. J. 2002, 368, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Chamorro, C.; Boerman, M.A.; Arnusch, C.J.; Breukink, E.; Pieters, R.J. Enhancing membrane disruption by targeting and multivalent presentation of antimicrobial peptides. Biochim. Biophys. Acta (BBA)—Biomembr. 2012, 1818, 2171–2174. [Google Scholar] [CrossRef]
- Xiao, M.; Jasensky, J.; Gerszberg, J.; Chen, J.; Tian, J.; Lin, T.; Lu, T.; Lahann, J.; Chen, Z. Chemically Immobilized Antimicrobial Peptide on Polymer and Self-Assembled Monolayer Substrates. Langmuir 2018, 34, 12889–12896. [Google Scholar] [CrossRef]
- Kim, J.-M.; Jang, S.-J.; Yang, M.-H.; Cho, H.-J.; Lee, K.-H. Characterization of Antibacterial Activity and Synergistic Effect of Cationic Antibacterial Peptide-resin Conjugates. Bull. Korean Chem. Soc. 2011, 32, 3928–3932. [Google Scholar] [CrossRef]
- Tam, J.P.; Lu, Y.-A.; Yang, J.-L. Antimicrobial dendrimeric peptides. Eur. J. Biol. Chem. 2002, 269, 923–932. [Google Scholar] [CrossRef]
- Reymond, J.-L.; Darbre, T. Peptide and glycopeptide dendrimer apple trees as enzyme models and for biomedical applications. Org. Biomol. Chem. 2012, 10, 1483–1492. [Google Scholar] [CrossRef]
- Lequeux, I.; Ducasse, E.; Jouenne, T.; Thebault, P. Addition of antimicrobial properties to hyaluronic acid by grafting of antimicrobial peptide. Eur. Polym. J. 2014, 51, 182–190. [Google Scholar] [CrossRef]
- Majumdar, I.D.; Weber, H.C. Biology of mammalian bombesin-like peptides and their receptors. Curr. Opin. Endocrinol. Diabetes Obes. 2011, 18, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Su, T.; Yang, H.; Fan, Q.; Jia, D.; Tao, Z.; Wan, L.; Lu, X. Enhancing the circulating half-life and the antitumor effects of a tumor-selective cytotoxic peptide by exploiting endogenous serum albumin as a drug carrier. Int. J. Pharm. 2016, 499, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Mohid, S.A.; Bhunia, A. Combining Antimicrobial Peptides with Nanotechnology: An Emerging Field in Theranostics. Curr. Protein Pept. Sci. 2020, 21, 413–428. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhang, M.; Li, B.; Chen, D.; Dong, X.; Wang, Y.; Gu, Y. Versatile antimicrobial peptide-based ZnO quantum dots for in vivo bacteria diagnosis and treatment with high specificity. Biomaterials 2015, 53, 532–544. [Google Scholar] [CrossRef] [PubMed]
- Galdiero, E.; Siciliano, A.; Maselli, V.; Gesuele, R.; Guida, M.; Fulgione, D.; Galdiero, S.; Lombardi, L.; Falanga, A. An integrated study on antimicrobial activity and ecotoxicity of quantum dots and quantum dots coated with the antimicrobial peptide indolicidin. Int. J. Nanomed. 2016, 11, 4199–4211. [Google Scholar] [CrossRef]
- Gao, W.; Hu, C.-M.J.; Fang, R.H.; Zhang, L. Liposome-like nanostructures for drug delivery. J. Mater. Chem. B 2013, 1, 6569–6585. [Google Scholar] [CrossRef]
- Makowski, M.; Silva, Í.C.; Pais do Amaral, C.; Gonçalves, S.; Santos, N.C. Advances in Lipid and Metal Nanoparticles for Antimicrobial Peptide Delivery. Pharmaceutics 2019, 11, 588. [Google Scholar] [CrossRef] [PubMed]
- Bonam, S.R.; Wang, F.; Muller, S. Lysosomes as a therapeutic target. Nat. Rev. Drug Discov. 2019, 18, 923–948. [Google Scholar] [CrossRef]
- Almeida, B.; Nag, O.K.; Rogers, K.E.; Delehanty, J.B. Recent Progress in Bioconjugation Strategies for Liposome-Mediated Drug Delivery. Molecules 2020, 25, 5672. [Google Scholar] [CrossRef]
- Loutet, S.A.; Valvano, M.A. Extreme Antimicrobial Peptide and Polymyxin B Resistance in the Genus Burkholderia. Front. Microbiol. 2011, 2, 159. [Google Scholar] [CrossRef]
- Desai, T.R.; Tyrrell, G.J.; Ng, T.; Finlay, W.H. In Vitro Evaluation of Nebulization Properties, Antimicrobial Activity, and Regional Airway Surface Liquid Concentration of Liposomal Polymyxin B Sulfate. Pharm. Res. 2003, 20, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Haisma, E.M.; De Breij, A.; Chan, H.; Van Dissel, J.T.; Drijfhout, J.W.; Hiemstra, P.; El Ghalbzouri, A.; Nibbering, P.H. LL-37-Derived Peptides Eradicate Multidrug-Resistant Staphylococcus aureus from Thermally Wounded Human Skin Equivalents. Antimicrob. Agents Chemother. 2014, 58, 4411–4419. [Google Scholar] [CrossRef] [PubMed]
- Haisma, E.M.; Göblyös, A.; Ravensbergen, B.; Adriaans, A.E.; Cordfunke, R.A.; Schrumpf, J.; Limpens, R.; Schimmel, K.J.M.; Hartigh, J.D.; Hiemstra, P.; et al. Antimicrobial Peptide P60.4Ac-Containing Creams and Gel for Eradication of Methicillin-Resistant Staphylococcus aureus from Cultured Skin and Airway Epithelial Surfaces. Antimicrob. Agents Chemother. 2016, 60, 4063–4072. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.U.; Pirzadeh, M.; Förster, C.Y.; Shityakov, S.; Shariati, M.A. Role of Milk-Derived Antibacterial Peptides in Modern Food Biotechnology: Their Synthesis, Applications and Future Perspectives. Biomolecules 2018, 8, 110. [Google Scholar] [CrossRef]
- Mohan, A.; McClements, D.J.; Udenigwe, C.C. Encapsulation of bioactive whey peptides in soy lecithin-derived nanoliposomes: Influence of peptide molecular weight. Food Chem. 2016, 213, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Bruno, B.J.; Miller, G.D.; Lim, C.S. Basics and recent advances in peptide and protein drug delivery. Ther. Deliv. 2013, 4, 1443–1467. [Google Scholar] [CrossRef]
- Mizukami, S.; Hosoda, M.; Satake, T.; Okada, S.; Hori, Y.; Furuta, T.; Kikuchi, K. Photocontrolled Compound Release System Using Caged Antimicrobial Peptide. J. Am. Chem. Soc. 2010, 132, 9524–9525. [Google Scholar] [CrossRef]
- Malmsten, M. Inorganic nanomaterials as delivery systems for proteins, peptides, DNA, and siRNA. Curr. Opin. Colloid Interface Sci. 2013, 18, 468–480. [Google Scholar] [CrossRef]
- Malmsten, M. Nanomaterials as Antimicrobial Agents. In Handbook of Nanomaterials Properties; Bhushan, B., Luo, D., Schricker, S.R., Sigmund, W., Zauscher, S., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 1053–1075. [Google Scholar]
- Hajipour, M.J.; Fromm, K.M.; Ashkarran, A.A.; de Aberasturi, D.J.; de Larramendi, I.R.; Rojo, T.; Serpooshan, V.; Parak, W.J.; Mahmoudi, M. Antibacterial properties of nanoparticles. Trends Biotechnol. 2012, 30, 499–511. [Google Scholar] [CrossRef]
- Vivero-Escoto, J.L.; Slowing, I.; Trewyn, B.G.; Lin, V.S.-Y. Mesoporous Silica Nanoparticles for Intracellular Controlled Drug Delivery. Small 2010, 6, 1952–1967. [Google Scholar] [CrossRef] [PubMed]
- Gultepe, E.; Nagesha, D.; Sridhar, S.; Amiji, M. Nanoporous inorganic membranes or coatings for sustained drug delivery in implantable devices. Adv. Drug Deliv. Rev. 2010, 62, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Fadeel, B.; Garcia-Bennett, A.E. Better safe than sorry: Understanding the toxicological properties of inorganic nanoparticles manufactured for biomedical applications. Adv. Drug Deliv. Rev. 2010, 62, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Braun, K.; Pochert, A.; Lindén, M.; Davoudi, M.; Schmidtchen, A.; Nordström, R.; Malmsten, M. Membrane interactions of mesoporous silica nanoparticles as carriers of antimicrobial peptides. J. Colloid Interface Sci. 2016, 475, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo-Barba, I.; Vallet-Regí, M.; Kupferschmidt, N.; Terasaki, O.; Schmidtchen, A.; Malmsten, M. Incorporation of antimicrobial compounds in mesoporous silica film monolith. Biomaterials 2009, 30, 5729–5736. [Google Scholar] [CrossRef]
- Bitar, A.; Ahmad, N.M.; Fessi, H.; Elaissari, A. Silica-based nanoparticles for biomedical applications. Drug Discov. Today 2012, 17, 1147–1154. [Google Scholar] [CrossRef]
- Li, L.-L.; Wang, H. Enzyme-Coated Mesoporous Silica Nanoparticles as Efficient Antibacterial Agents In Vivo. Adv. Healthc. Mater. 2013, 2, 1351–1360. [Google Scholar] [CrossRef]
- Roy, P.; Berger, S.; Schmuki, P. TiO2 Nanotubes: Synthesis and Applications. Angew. Chem. Int. Ed. 2011, 50, 2904–2939. [Google Scholar] [CrossRef]
- Ma, M.; Kazemzadeh-Narbat, M.; Hui, Y.; Lu, S.; Ding, C.; Chen, D.D.Y.; Hancock, R.E.W.; Wang, R. Local delivery of antimicrobial peptides using self-organized TiO2 nanotube arrays for peri-implant infections. J. Biomed. Mater. Res. Part A 2012, 100A, 278–285. [Google Scholar] [CrossRef]
- Mody, V.V.; Siwale, R.; Singh, A.K.; Mody, H.R. Introduction to metallic nanoparticles. J. Pharm. Bioallied Sci. 2010, 2, 282–289. [Google Scholar] [CrossRef]
- Rai, M.; Ingle, A.P.; Gupta, I.; Brandelli, A. Bioactivity of noble metal nanoparticles decorated with biopolymers and their application in drug delivery. Int. J. Pharm. 2015, 496, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Peng, M.; He, Y.; Yeung, E.S. Functionalized fluorescent gold nanodots: Synthesis and application for Pb2+ sensing. Chem. Commun. 2011, 47, 11981–11983. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-Y.; Chang, H.-Y.; Lu, J.-K.; Huang, Y.-C.; Harroun, S.G.; Tseng, Y.-T.; Li, Y.-J.; Huang, C.-C.; Chang, H.-T. Self-Assembly of Antimicrobial Peptides on Gold Nanodots: Against Multidrug-Resistant Bacteria and Wound-Healing Application. Adv. Funct. Mater. 2015, 25, 7189–7199. [Google Scholar] [CrossRef]
- Bogdanovic, G.; Djordjevic, A. Carbon nanomaterials: Biologically active fullerene derivatives. Srp. Arh. Celok. Lek. 2016, 144, 222–231. [Google Scholar] [CrossRef]
- Bechinger, B.; Zasloff, M.; Opella, S. Structure and Dynamics of the Antibiotic Peptide PGLa in Membranes by Solution and Solid-State Nuclear Magnetic Resonance Spectroscopy. Biophys. J. 1998, 74, 981–987. [Google Scholar] [CrossRef]
- Nellore, B.P.V.; Kanchanapally, R.; Pedraza, F.; Sinha, S.S.; Pramanik, A.; Hamme, A.T.; Arslan, Z.; Sardar, D.; Ray, P.C. Bio-Conjugated CNT-Bridged 3D Porous Graphene Oxide Membrane for Highly Efficient Disinfection of Pathogenic Bacteria and Removal of Toxic Metals from Water. ACS Appl. Mater. Interfaces 2015, 7, 19210–19218. [Google Scholar] [CrossRef]
- Subbalakshmi, C.; Sitaram, N. Mechanism of antimicrobial action of indolicidin. FEMS Microbiol. Lett. 1998, 160, 91–96. [Google Scholar] [CrossRef]
- Sur, A.; Pradhan, B.; Banerjee, A.; Aich, P. Immune Activation Efficacy of Indolicidin Is Enhanced upon Conjugation with Carbon Nanotubes and Gold Nanoparticles. PLOS ONE 2015, 10, e0123905. [Google Scholar] [CrossRef]
- Zielińska, A.; Carreiró, F.; Oliveira, A.M.; Neves, A.; Pires, B.; Venkatesh, D.N.; Durazzo, A.; Lucarini, M.; Eder, P.; Silva, A.M.; et al. Polymeric Nanoparticles: Production, Characterization, Toxicology and Ecotoxicology. Molecules 2020, 25, 3731. [Google Scholar] [CrossRef]
- D’Angelo, I.; Casciaro, B.; Miro, A.; Quaglia, F.; Mangoni, M.L.; Ungaro, F. Overcoming barriers in Pseudomonas aeruginosa lung infections: Engineered nanoparticles for local delivery of a cationic antimicrobial peptide. Colloids Surf. B Biointerfaces 2015, 135, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Lian, Z.; Ji, T. Functional peptide-based drug delivery systems. J. Mater. Chem. B 2020, 8, 6517–6529. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.-K.; Kim, C.; Seo, C.H.; Park, Y. The therapeutic applications of antimicrobial peptides (AMPs): A patent review. J. Microbiol. 2017, 55, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kościuczuk, E.M.; Lisowski, P.; Jarczak, J.; Strzałkowska, N.; Jóźwik, A.; Horbańczuk, J.O.; Krzyżewski, J.; Zwierzchowski, L.; Bagnicka, E. Cathelicidins: Family of antimicrobial peptides. A review. Mol. Biol. Rep. 2012, 39, 10957–10970. [Google Scholar] [CrossRef] [PubMed]
- Seo, M.-D.; Won, H.-S.; Kim, J.-H.; Mishig-Ochir, T.; Lee, B.-J. Antimicrobial Peptides for Therapeutic Applications: A Review. Molecules 2012, 17, 12276–12286. [Google Scholar] [CrossRef]
- Giuliani, A.; Pirri, G.; Nicoletto, S. Antimicrobial peptides: An overview of a promising class of therapeutics. Open Life Sci. 2007, 2, 1–33. [Google Scholar] [CrossRef]
- Chen, C.H.; Lu, T.K. Development and Challenges of Antimicrobial Peptides for Therapeutic Applications. Antibiotics 2020, 9, 24. [Google Scholar] [CrossRef]
- Law, V.; Knox, C.; Djoumbou, Y.; Jewison, T.; Guo, A.C.; Liu, Y.; Maciejewski, A.; Arndt, D.; Wilson, M.; Neveu, V.; et al. DrugBank 4.0: Shedding new light on drug metabolism. Nucleic Acids Res. 2014, 42, D1091–D1097. [Google Scholar] [CrossRef]
- Howard, B. Bacitracin. In xPharm: The Comprehensive Pharmacology Reference; Enna, S.J., Bylund, D.B., Eds.; Elsevier: New York, NY, USA, 2007; pp. 1–4. [Google Scholar]
- O’Donnell, J.A.; Gelone, S.P.; Safdar, A. 37—Topical Antibacterials. In Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases, 8th ed.; Bennett, J.E., Dolin, R., Blaser, M.J., Eds.; W.B. Saunders: Philadelphia, PA, USA, 2015; pp. 452–462. [Google Scholar]
- Kumar, P. 33—Pharmacology of Specific Drug Groups: Antibiotic Therapy. In Pharmacology and Therapeutics for Dentistry, 7th ed.; Dowd, F.J., Johnson, B.S., Mariotti, A.J., Eds.; Mosby: Maryland Heights, MO, USA, 2017; pp. 457–487. [Google Scholar]
- Parente, D.M.; Laplante, K.L. 145—Glycopeptides. In Infectious Diseases, 4th ed.; Cohen, J., Powderly, W.G., Opal, S.M., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 1249–1255. [Google Scholar]
- Murray, B.E.; Arias, C.A.; Nannini, E.C. 30—Glycopeptides (Vancomycin and Teicoplanin), Streptogramins (Quinupristin-Dalfopristin), Lipopeptides (Daptomycin), and Lipoglycopeptides (Telavancin). In Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases, 8th ed.; Bennett, J.E., Dolin, R., Blaser, M.J., Eds.; W.B. Saunders: Philadelphia, PA, USA, 2015; pp. 377–400. [Google Scholar]
- Scholar, E. Vancomycin. In xPharm: The Comprehensive Pharmacology Reference; Enna, S.J., Bylund, D.B., Eds.; Elsevier: New York, NY, USA, 2007; pp. 1–6. [Google Scholar]
- Sáez-Llorens, X.; McCracken, G.H. Chapter 37—Clinical Pharmacology of Antibacterial Agents. In Infectious Diseases of the Fetus and Newborn Infant, 6th ed.; Remington, J.S., Klein, J.O., Wilson, C.B., Baker, C.J., Eds.; W.B. Saunders: Philadelphia, PA, USA, 2006; pp. 1223–1267. [Google Scholar]
- Maddison, J.E.; Watson, A.D.J.; Elliott, J. Chapter 8—Antibacterial drugs. In Small Animal Clinical Pharmacology, 2nd ed.; Maddison, J.E., Page, S.W., Church, D.B., Eds.; W.B. Saunders: Edinburgh, UK, 2008; pp. 148–185. [Google Scholar]
- Daum, R.S. 115—Staphylococcus aureus. In Principles and Practice of Pediatric Infectious Diseases, 5th ed.; Long, S.S., Prober, C.G., Fischer, M., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 692–706. [Google Scholar]
- Oritavancin. In Meyler’s Side Effects of Drugs, 16th ed.; Aronson, J.K., Ed.; Elsevier: Oxford, UK, 2016; p. 391. [Google Scholar]
- Padberg, S. 2.6—Anti-infective Agents. In Drugs during Pregnancy and Lactation, 3rd ed.; Schaefer, C., Peters, P., Miller, R.K., Eds.; Academic Press: San Diego, CA, USA, 2015; pp. 115–176. [Google Scholar]
- Chen, A.Y.; Zervos, M.J.; Vazquez, J.A. Dalbavancin: A novel antimicrobial. Int. J. Clin. Pract. 2007, 61, 853–863. [Google Scholar] [CrossRef]
- Bork, J.T.; Heil, E.L.; Berry, S.; Lopes, E.; Davé, R.; Gilliam, B.L.; Amoroso, A. Dalbavancin Use in Vulnerable Patients Receiving Outpatient Parenteral Antibiotic Therapy for Invasive Gram-Positive Infections. Infect. Dis. Ther. 2019, 8, 171–184. [Google Scholar] [CrossRef]
- Scholar, E. Daptomycin. In xPharm: The Comprehensive Pharmacology Reference; Enna, S.J., Bylund, D.B., Eds.; Elsevier: New York, NY, USA, 2009; pp. 1–5. [Google Scholar]
- Tascini, C.; Sbrana, F.; Sozio, E.; Pino, B.D.; Bertolino, G.; Ripoli, A.; Pallotto, C.; Emdin, M.; Sampietro, T. Statins during daptomycin therapy: To give or not to give? Minerva Anestesiol. 2019, 85, 689–690. [Google Scholar] [CrossRef]
- Van Bambeke, F.; Mingeot-Leclercq, M.-P.; Glupczynski, Y.; Tulkens, P.M. 137—Mechanisms of Action. In Infectious Diseases, 4th ed.; Cohen, J., Powderly, W.G., Opal, S.M., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 1162–1180. [Google Scholar]
- Barie, P.S.; Eachempati, S.R.; Shapiro, M.J. Chapter 97—Antibacterial therapy: The old, the new, and the future. In Current Therapy of Trauma and Surgical Critical Care; Asensio, J.A., Trunkey, D.D., Eds.; Mosby: Philadelphia, PA, USA, 2008; pp. 688–701. [Google Scholar]
- Groman, R.P. Chapter 200—Miscellaneous Antibiotics. In Small Animal Critical Care Medicine; Silverstein, D.C., Hopper, K., Eds.; W.B. Saunders: St. Louis, MO, USA, 2009; pp. 845–849. [Google Scholar]
- Carr, E.A. Chapter 177—Systemic Inflammatory Response Syndrome. In Robinson’s Current Therapy in Equine Medicine, 7th ed.; Sprayberry, K.A., Robinson, N.E., Eds.; W.B. Saunders: St. Louis, MO, USA, 2015; pp. 741–745. [Google Scholar]
- Scholar, E. Polymyxin B. In xPharm: The Comprehensive Pharmacology Reference; Enna, S.J., Bylund, D.B., Eds.; Elsevier: New York, NY, USA, 2007; pp. 1–4. [Google Scholar]
- Papich, M.G. Polymyxin B Sulfate. In Saunders Handbook of Veterinary Drugs, 4th ed.; Papich, M.G., Ed.; W.B. Saunders: St. Louis, MO, USA, 2016; pp. 647–649. [Google Scholar]
- Cunha, C.B.; Opal, S.M. 42—How do I optimize antibiotic use in critical illness. In Evidence-Based Practice of Critical Care, 3rd ed.; Deutschman, C.S., Neligan, P.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 291–298. [Google Scholar]
- Ahn, J.M.; Kassees, K.; Lee, T.K.; Manandhar, B.; Yousif, A.M. 6.03—Strategy and Tactics for Designing Analogs: Biochemical Characterization of the Large Molecules. In Comprehensive Medicinal Chemistry III; Chackalamannil, S., Rotella, D., Ward, S.E., Eds.; Elsevier: Oxford, UK, 2017; pp. 66–115. [Google Scholar]
- Sauberan, J.B.; Bradley, J.S. 292—Antimicrobial Agents. In Principles and Practice of Pediatric Infectious Diseases, 5th ed.; Long, S.S., Prober, C.G., Fischer, M., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 1499–1531. [Google Scholar]
- Biswas, S.; Brunel, J.-M.; Dubus, J.-C.; Reynaud-Gaubert, M.; Rolain, J.-M. Colistin: An update on the antibiotic of the 21st century. Expert Rev. Anti-Infect. Ther. 2012, 10, 917–934. [Google Scholar] [CrossRef]
- Yahav, D.; Farbman, L.; Leibovici, L.; Paul, M. Colistin: New lessons on an old antibiotic. Clin. Microbiol. Infect. 2012, 18, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Pavithrra, G.; Rajasekaran, R. Gramicidin Peptide to Combat Antibiotic Resistance: A Review. Int. J. Pept. Res. Ther. 2020, 26, 191–199. [Google Scholar] [CrossRef]
- Stein, W.D.; Litman, T. Chapter 3—Ion Channels across Cell Membranes. In Channels, Carriers, and Pumps, 2nd ed.; Stein, W.D., Litman, T., Eds.; Elsevier: London, UK, 2015; pp. 81–130. [Google Scholar]
- Kamal, S.M. Chapter 6—Hepatitis C Treatment in the Era of Direct-Acting Antiviral Agents: Challenges in Developing Countries. In Hepatitis C in Developing Countries, Kamal, S.M., Ed.; Academic Press: Cambridge, MA, USA, 2018; pp. 209–246. [Google Scholar]
- Firsov, A.A.; Smirnova, M.V.; Lubenko, I.Y.; Vostrov, S.N.; Portnoy, Y.; Zinner, S.H. Testing the mutant selection window hypothesis with Staphylococcus aureus exposed to daptomycin and vancomycin in an in vitro dynamic model. J. Antimicrob. Chemother. 2006, 58, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- Couet, W.; Grégoire, N.; Gobin, P.; Saulnier, P.J.; Frasca, D.; Marchand, S.; Mimoz, O. Pharmacokinetics of Colistin and Colistimethate Sodium After a Single 80-mg Intravenous Dose of CMS in Young Healthy Volunteers. Clin. Pharmacol. Ther. 2011, 89, 875–879. [Google Scholar] [CrossRef]
- Woodford, N. Chapter 20—Glycopeptides. In Antibiotic and Chemotherapy, 9th ed.; Finch, R.G., Greenwood, D., Norrby, S.R., Whitley, R.J., Eds.; W.B. Saunders: London, UK, 2010; pp. 265–271. [Google Scholar]
- Moise, P.A.; Sakoulas, G. Chapter 140—Glycopeptides. In Infectious Diseases, 3rd ed.; Cohen, J., Opal, S.M., Powderly, W.G., Eds.; Mosby: London, UK, 2010; pp. 1399–1406. [Google Scholar]
- Matthews, S.J.; Lancaster, J.W. Telaprevir: A Hepatitis C NS3/4A Protease Inhibitor. Clin. Ther. 2012, 34, 1857–1882. [Google Scholar] [CrossRef] [PubMed]
- Gentile, I.; Viola, C.; Borgia, F.; Castaldo, G.; Borgia, G. Telaprevir: A Promising Protease Inhibitor for the Treatment of Hepatitis C Virus Infection. Curr. Med. Chem. 2009, 16, 1115–1121. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pawlotsky, J.-M. Chapter Five—Hepatitis C Virus: Standard-of-Care Treatment. In Advances in Pharmacology, De Clercq, E., Ed.; Academic Press: Cambridge, MA, USA, 2013; Volume 67, pp. 169–215. [Google Scholar]
- Miller, M.D.; Hazuda, D.J. HIV resistance to the fusion inhibitor enfuvirtide: Mechanisms and clinical implications. Drug Resist. Updat. 2004, 7, 89–95. [Google Scholar] [CrossRef]
- Reeves, J.D.; Lee, F.-H.; Miamidian, J.L.; Jabara, C.B.; Juntilla, M.M.; Doms, R.W. Enfuvirtide Resistance Mutations: Impact on Human Immunodeficiency Virus Envelope Function, Entry Inhibitor Sensitivity, and Virus Neutralization. J. Virol. 2005, 79, 4991–4999. [Google Scholar] [CrossRef] [PubMed]
- Tsibris, A.M.N.; Hirsch, M.S. 130—Antiretroviral Therapy for Human Immunodeficiency Virus Infection. In Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases, 8th ed.; Bennett, J.E., Dolin, R., Blaser, M.J., Eds.; W.B. Saunders: Philadelphia, PA, USA, 2015; pp. 1622–1641. [Google Scholar]
- Enfuvirtide. In Meyler’s Side Effects of Drugs, 16th ed.; Aronson, J.K., Ed.; Elsevier: Oxford, UK, 2016; pp. 53–54. [Google Scholar]
- Scholar, E. Atazanivir. In xPharm: The Comprehensive Pharmacology Reference; Enna, S.J., Bylund, D.B., Eds.; Elsevier: New York, NY, USA, 2009; pp. 1–7. [Google Scholar]
- Vernazza, P.L.; Schmid, P. 29—Antiviral drugs. In Side Effects of Drugs Annual, Aronson, J.K., Ed.; Elsevier: Amsterdam, The Netherlands, 2005; Volume 28, pp. 326–341. [Google Scholar]
- Canadian Agency for Drugs and Technologies in Health. Clinical Review Report: Tesamorelin (Egrifta); Canadian Agency for Drugs and Technologies in Health: Ottawa, ON, Canada, 2016. [Google Scholar]
- Dhillon, S. Tesamorelin: A Review of its Use in the Management of HIV-Associated Lipodystrophy. Drugs 2011, 71, 1071–1091. [Google Scholar] [CrossRef]
- Dahmer, S.; Kligler, B. Chapter 19—HIV Disease and AIDS. In Integrative Medicine, 4th ed.; Rakel, D., Ed.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 180–190. [Google Scholar]
- Park, N.-H.; Shin, K.-H.; Kang, M.K. 34—Antifungal and Antiviral Agents. In Pharmacology and Therapeutics for Dentistry, 7th ed.; Dowd, F.J., Johnson, B.S., Mariotti, A.J., Eds.; Mosby: Maryland Heights, MO, USA, 2017; pp. 488–503. [Google Scholar]
- Rex, J.H.; Stevens, D.A. 39—Drugs Active against Fungi, Pneumocystis, and Microsporidia. In Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases, 8th ed.; Bennett, J.E., Dolin, R., Blaser, M.J., Eds.; W.B. Saunders: Philadelphia, PA, USA, 2015; pp. 479–494. [Google Scholar]
- Stevens, D.A. 339—Systemic Antifungal Agents. In Goldman’s Cecil Medicine, 24th ed.; Goldman, L., Schafer, A.I., Eds.; W.B. Saunders: Philadelphia, PA, USA, 2012; pp. 1971–1977. [Google Scholar]
- Guo, J.; Hu, H.; Zhao, Q.; Wang, T.; Zou, Y.; Yu, S.; Wu, Q.; Guo, Z. Synthesis and Antifungal Activities of Glycosylated Derivatives of the Cyclic Peptide Fungicide Caspofungin. ChemMedChem 2012, 7, 1496–1503. [Google Scholar] [CrossRef] [PubMed]
- Scholar, E. Micafungin. In xPharm: The Comprehensive Pharmacology Reference; Enna, S.J., Bylund, D.B., Eds.; Elsevier: New York, NY, USA, 2009; pp. 1–4. [Google Scholar]
- Pappas, P.G.; Rotstein, C.M.F.; Betts, R.F.; Nucci, M.; Talwar, D.; De Waele, J.J.; Vazquez, J.A.; Dupont, B.F.; Horn, D.L.; Ostrosky-Zeichner, L.; et al. Micafungin versus Caspofungin for Treatment of Candidemia and Other Forms of Invasive Candidiasis. Clin. Infect. Dis. 2007, 45, 883–893. [Google Scholar] [CrossRef] [PubMed]
- Anidulafungin. In Meyler’s Side Effects of Drugs, 16th ed.; Aronson, J.K., Ed.; Elsevier: Oxford, UK, 2016; pp. 498–499. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Name | Active Ingredients | Peptide | Applications |
|---|---|---|---|
| Orbactiv | Oritavancin diphosphate | Lipoglycopeptide | Treatment of Gram-positive bacteria causing complicated skin and skin structure infections (cSSSI). |
| Dalvance | Dalbavancin hydrochloride | Lipoglycopeptide | Treatment of Gram-positive bacteria causing complicated skin and skin structure infections (cSSSI). |
| Incivek | Telaprevir | Chemically modified peptide | Chronic hepatitis C treatment |
| Egrifta | Tesamorelin acetate | Synthetic peptide | Human immunodeficiency virus (HIV) treatment |
| Vibativ | Telavancin hydrochloride | Lipoglycopeptide | Treatment of Gram-positive bacteria causing complicated skin and skin structure infections (cSSSI). |
| Eraxis | Anidulafungin | Lipopeptide | Antifungal drug |
| Cubicin | Daptomycin | Cyclic lipopeptide | Treatment of Gram-positive bacteria causing complicated skin and skin structure infections (cSSSI). |
| Cubicin rf | Daptomycin | Cyclic lipopeptide | Treatment of Gram-positive bacteria causing complicated skin and skin structure infections (cSSSI). |
| Reyataz | Atazanavir sulfate | Azapeptide | Human immunodeficiency virus (HIV) treatment |
| Fuzeon | Enfuvirtide | Synthetic peptide | Human immunodeficiency virus (HIV) treatment |
| Cancidas | Caspofungin acetate | Cyclic lipopeptide | Antifungal drug |
| Drug | Type of Bacteria | Mechanism of Action | Use | Half-life | Route of Administration | Reference |
|---|---|---|---|---|---|---|
| Bacitracin | Gram-positive | Inhibits cell wall synthesis | Skin infections | 1.9 days | Topical Ophthalmic Intramuscular | [267] |
| Dalbavancin | Gram-positive | Inhibits cell wall synthesis | Skin infections | 6–10 days | Intravenous | [278] |
| Daptomycin | Gram-positive | Membrane lysis | Skin infections | 9 h | Intravenous | [280,296] |
| Colistin | Gram-negative | Membrane lysis | Multidrug-resistant Gram-negative infections | 3.0 ± 0.6 h | Intravenous | [297] |
| Gramicidin D | Gram-positive, some Gram-negative | Membrane poration/lysis | Skin and eye infection | Not Determined | Topical Ophthalmic | [293] |
| Oritavancin | Gram-positive | Membrane lysis and inhibits cell wall synthesis | Skin infections | Long serum half-life (393 h) | Intravenous | [276] |
| Polymyxin B | Gram-negative | Membrane lysis | Urinary tract and bloodstream infections | 11.5 h | Ophthalmic Topical Intravenous | [286,287] |
| Teicoplanin | Gram-positive | Inhibits cell wall synthesis | Serious Gram-positive infections | Long half-life of approximately 47 h | Intramuscular Intravenous | [271] |
| Telavancin | Gram-positive | Membrane lysis and inhibits cell wall synthesis | Skin infections | 8 h | Intravenous | [298,299] |
| Vancomycin | Gram-positive | Inhibits cell wall synthesis | Serious Gram-positive infections | 7 to 12 h according to age | Oral Intravenous | [271,272] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez, A.A.; Otero-González, A.; Ghattas, M.; Ständker, L. Discovery, Optimization, and Clinical Application of Natural Antimicrobial Peptides. Biomedicines 2021, 9, 1381. https://doi.org/10.3390/biomedicines9101381
Rodríguez AA, Otero-González A, Ghattas M, Ständker L. Discovery, Optimization, and Clinical Application of Natural Antimicrobial Peptides. Biomedicines. 2021; 9(10):1381. https://doi.org/10.3390/biomedicines9101381
Chicago/Turabian StyleRodríguez, Armando A., Anselmo Otero-González, Maretchia Ghattas, and Ludger Ständker. 2021. "Discovery, Optimization, and Clinical Application of Natural Antimicrobial Peptides" Biomedicines 9, no. 10: 1381. https://doi.org/10.3390/biomedicines9101381
APA StyleRodríguez, A. A., Otero-González, A., Ghattas, M., & Ständker, L. (2021). Discovery, Optimization, and Clinical Application of Natural Antimicrobial Peptides. Biomedicines, 9(10), 1381. https://doi.org/10.3390/biomedicines9101381