
Combined LXR and RXR Agonist Therapy Increases ABCA1 Protein Expression and Enhances ApoAI-Mediated Cholesterol Efflux in Cultured Endothelial Cells

Abstract
1. Introduction
2. Results
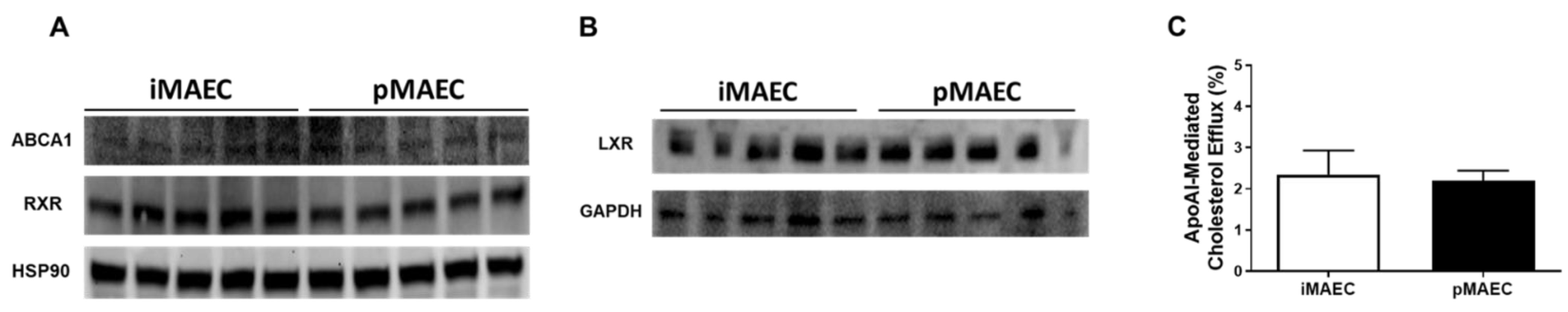
2.1. iMAEC Express ABCA1, LXR, and RXR Proteins, and Are Capable of Effluxing Cholesterol to ApoAI
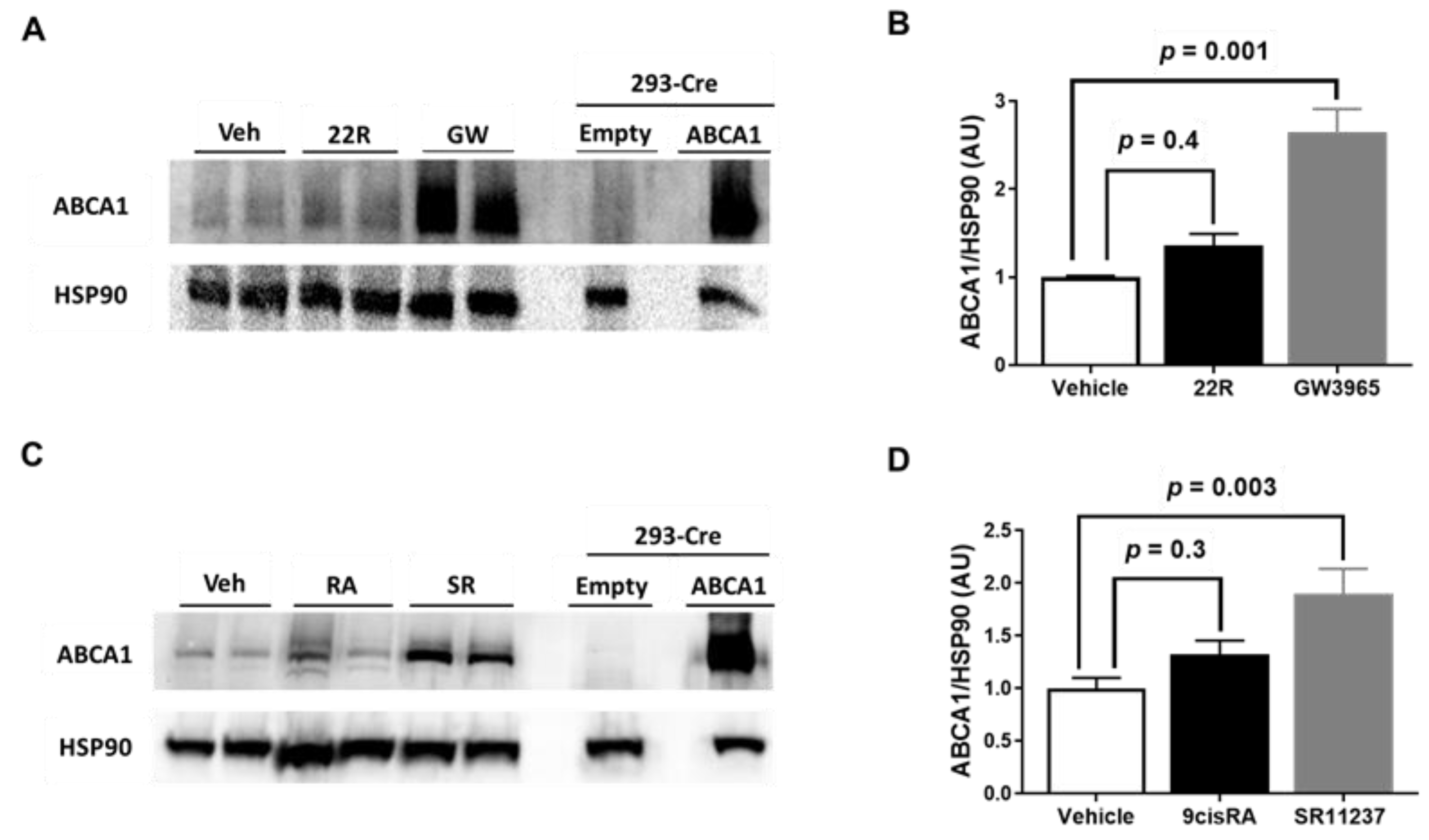
2.2. Synthetic LXR and RXR Agonists, but Not Endogenous LXR and RXR Agonists, Increase ABCA1 Protein Expression in iMAEC
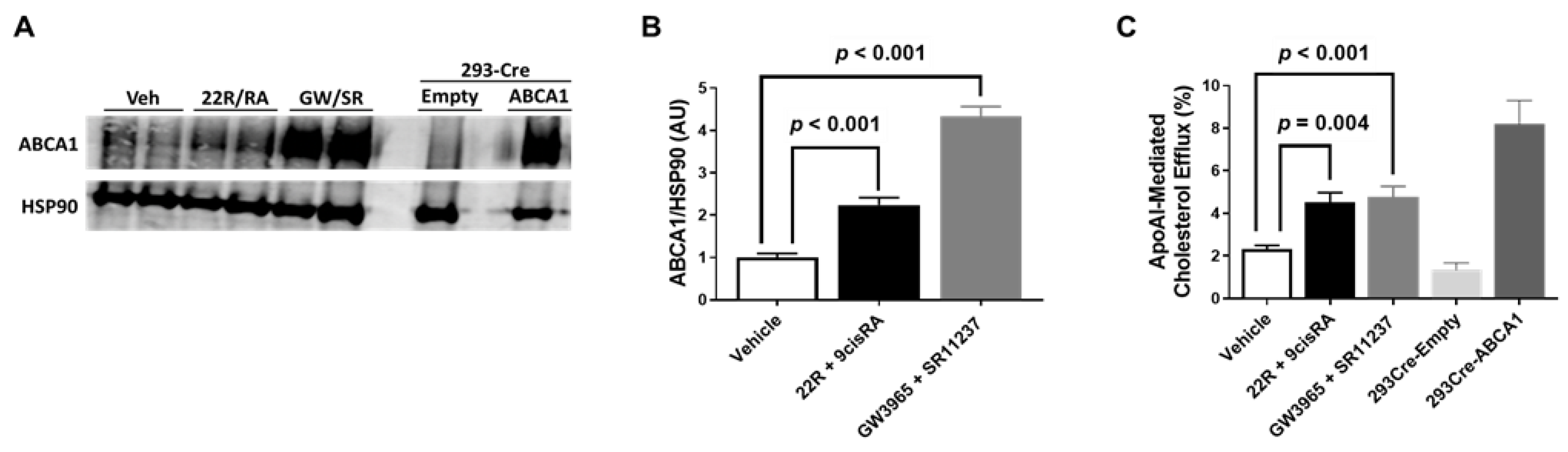
2.3. Combining LXR/RXR Agonist Treatments Increases Both ABCA1 Protein and ApoAI-Mediated Cholesterol Efflux in iMAEC
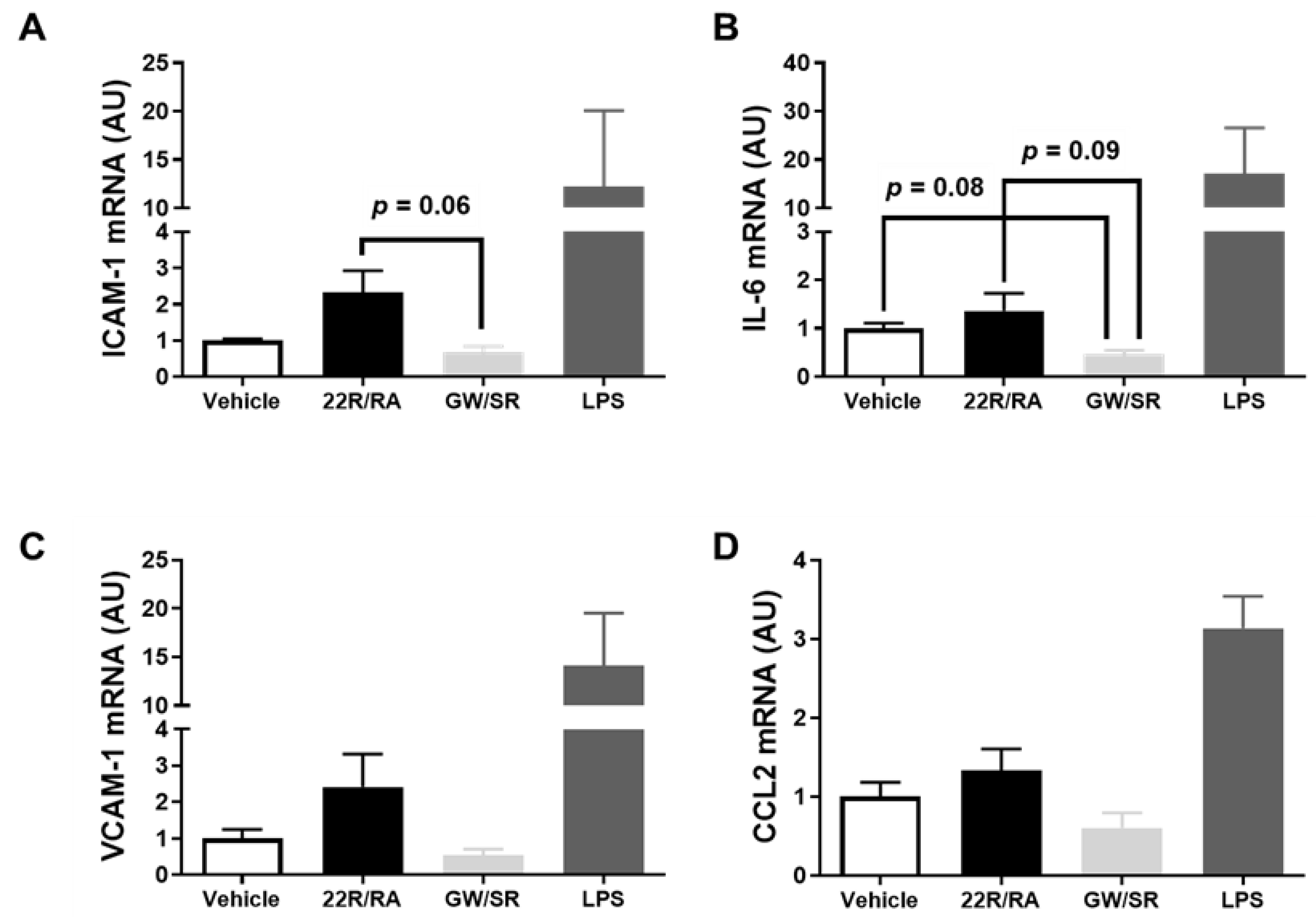
2.4. Combined LXR/RXR Agonist Therapy in iMAEC Fails to Alter Pro-Inflammatory Gene Expression
3. Discussion
4. Materials and Methods
4.1. Cell Culture Maintenance
4.2. Treating Cultured Cells
4.3. Immunoblotting
4.4. Cholesterol Efflux Assays
4.5. RT-qPCR
4.6. Statistical Analyses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ablain, J.; Durand, E.M.; Yang, S.; Zhou, Y.; Zon, L.I. A CRISPR/Cas9 vector system for tissue-specific gene disruption in zebrafish. Dev. Cell 2015, 32, 756–764. [Google Scholar] [CrossRef]
- Pahwa, R.; Jialal, I. Atherosclerosis. In StatPearls (Atherosclerosis); StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Endo, A. A historical perspective on the discovery of statins. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2010, 86, 484–493. [Google Scholar] [CrossRef]
- Davies, J.T.; Delfino, S.F.; Feinberg, C.E.; Johnson, M.F.; Nappi, V.L.; Olinger, J.T.; Schwab, A.P.; Swanson, H.I. Current and Emerging Uses of Statins in Clinical Therapeutics: A Review. Lipid Insights 2016, 9, 13–29. [Google Scholar] [CrossRef]
- DuBroff, R.; de Lorgeril, M. Cholesterol confusion and statin controversy. World J. Cardiol. 2015, 7, 404–409. [Google Scholar] [CrossRef] [PubMed]
- Hajar, R. Statins: Past and present. Heart Views 2011, 12, 121–127. [Google Scholar] [CrossRef]
- Tabas, I. Macrophage apoptosis in atherosclerosis: Consequences on plaque progression and the role of endoplasmic reticulum stress. Antioxid. Redox Signal. 2009, 11, 2333–2339. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; Bornfeldt, K.E. Macrophage Phenotype and Function in Different Stages of Atherosclerosis. Circ. Res. 2016, 118, 653–667. [Google Scholar] [CrossRef]
- Tabas, I.; Garcia-Cardena, G.; Owens, G.K. Recent insights into the cellular biology of atherosclerosis. J. Cell Biol. 2015, 209, 13–22. [Google Scholar] [CrossRef]
- Baumer, Y.; McCurdy, S.; Weatherby, T.M.; Mehta, N.N.; Halbherr, S.; Halbherr, P.; Yamazaki, N.; Boisvert, W.A. Hyperlipidemia-induced cholesterol crystal production by endothelial cells promotes atherogenesis. Nat. Commun. 2017, 8, 1129. [Google Scholar] [CrossRef]
- Mundi, S.; Massaro, M.; Scoditti, E.; Carluccio, M.A.; van Hinsbergh, V.W.M.; Iruela-Arispe, M.L.; De Caterina, R. Endothelial permeability, LDL deposition, and cardiovascular risk factors-a review. Cardiovasc. Res. 2018, 114, 35–52. [Google Scholar] [CrossRef] [PubMed]
- Zelcer, N.; Tontonoz, P. Liver X receptors as integrators of metabolic and inflammatory signaling. J. Clin. Investig. 2006, 116, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Joseph, S.B.; McKilligin, E.; Pei, L.; Watson, M.A.; Collins, A.R.; Laffitte, B.A.; Chen, M.; Noh, G.; Goodman, J.; Hagger, G.N.; et al. Synthetic LXR ligand inhibits the development of atherosclerosis in mice. Proc. Natl. Acad. Sci. USA 2002, 99, 7604–7609. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.I.; Xia, Z. The retinoid X receptors and their ligands. Biochim. Biophys. Acta 2012, 1821, 21–56. [Google Scholar] [CrossRef] [PubMed]
- Wouters, E.; de Wit, N.M.; Vanmol, J.; van der Pol, S.M.A.; van Het Hof, B.; Sommer, D.; Loix, M.; Geerts, D.; Gustafsson, J.A.; Steffensen, K.R.; et al. Liver X Receptor Alpha Is Important in Maintaining Blood-Brain Barrier Function. Front. Immunol. 2019, 10, 1811. [Google Scholar] [CrossRef]
- Oram, J.F. Molecular basis of cholesterol homeostasis: Lessons from Tangier disease and ABCA1. Trends Mol. Med. 2002, 8, 168–173. [Google Scholar] [CrossRef]
- Oram, J.F.; Lawn, R.M. ABCA1. The gatekeeper for eliminating excess tissue cholesterol. J. Lipid Res. 2001, 42, 1173–1179. [Google Scholar] [CrossRef]
- Oram, J.F.; Vaughan, A.M. ABCA1-mediated transport of cellular cholesterol and phospholipids to HDL apolipoproteins. Curr. Opin. Lipidol. 2000, 11, 253–260. [Google Scholar] [CrossRef]
- Oram, J.F.; Heinecke, J.W. ATP-binding cassette transporter A1: A cell cholesterol exporter that protects against cardiovascular disease. Physiol. Rev. 2005, 85, 1343–1372. [Google Scholar] [CrossRef]
- Cavelier, C.; Rohrer, L.; von Eckardstein, A. ATP-Binding cassette transporter A1 modulates apolipoprotein A-I transcytosis through aortic endothelial cells. Circ. Res. 2006, 99, 1060–1066. [Google Scholar] [CrossRef]
- Liao, H.; Langmann, T.; Schmitz, G.; Zhu, Y. Native LDL upregulation of ATP-binding cassette transporter-1 in human vascular endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 127–132. [Google Scholar] [CrossRef]
- Hayashi, T.; Kotani, H.; Yamaguchi, T.; Taguchi, K.; Iida, M.; Ina, K.; Maeda, M.; Kuzuya, M.; Hattori, Y.; Ignarro, L.J. Endothelial cellular senescence is inhibited by liver X receptor activation with an additional mechanism for its atheroprotection in diabetes. Proc. Natl. Acad. Sci. USA 2014, 111, 1168–1173. [Google Scholar] [CrossRef]
- Morello, F.; Saglio, E.; Noghero, A.; Schiavone, D.; Williams, T.A.; Verhovez, A.; Bussolino, F.; Veglio, F.; Mulatero, P. LXR-activating oxysterols induce the expression of inflammatory markers in endothelial cells through LXR-independent mechanisms. Atherosclerosis 2009, 207, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Kolseth, I.B.; Agren, J.; Sundvold-Gjerstad, V.; Lyngstadaas, S.P.; Wang, J.E.; Dahle, M.K. 9-cis retinoic acid inhibits inflammatory responses of adherent monocytes and increases their ability to induce classical monocyte migration. J. Innate Immun. 2012, 4, 176–186. [Google Scholar] [CrossRef]
- Manzano, V.M.; Munoz, J.C.; Jimenez, J.R.; Puyol, M.R.; Puyol, D.R.; Kitamura, M.; Cazana, F.J. Human renal mesangial cells are a target for the anti-inflammatory action of 9-cis retinoic acid. Br. J. Pharmacol. 2000, 131, 1673–1683. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Drew, P.D. 9-Cis-retinoic acid suppresses inflammatory responses of microglia and astrocytes. J. Neuroimmunol. 2006, 171, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. Inflammation in atherosclerosis. Nature 2002, 420, 868–874. [Google Scholar] [CrossRef]
- Libby, P. Inflammation in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2045–2051. [Google Scholar] [CrossRef]
- Ni, C.W.; Kumar, S.; Ankeny, C.J.; Jo, H. Development of immortalized mouse aortic endothelial cell lines. Vasc. Cell 2014, 6, 7. [Google Scholar] [CrossRef]
- Stamatikos, A.; Dronadula, N.; Ng, P.; Palmer, D.; Knight, E.; Wacker, B.K.; Tang, C.; Kim, F.; Dichek, D.A. ABCA1 Overexpression in Endothelial Cells In Vitro Enhances ApoAI-Mediated Cholesterol Efflux and Decreases Inflammation. Hum. Gene Ther. 2019, 30, 236–248. [Google Scholar] [CrossRef]
- Chai, D.; Wang, B.; Shen, L.; Pu, J.; Zhang, X.K.; He, B. RXR agonists inhibit high-glucose-induced oxidative stress by repressing PKC activity in human endothelial cells. Free Radic. Biol. Med. 2008, 44, 1334–1347. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Yuhanna, I.S.; Umetani, J.; Lee, W.R.; Korach, K.S.; Shaul, P.W.; Umetani, M. LXRbeta/estrogen receptor-alpha signaling in lipid rafts preserves endothelial integrity. J. Clin. Investig. 2013, 123, 3488–3497. [Google Scholar] [CrossRef] [PubMed]
- Vaisman, B.L.; Demosky, S.J.; Stonik, J.A.; Ghias, M.; Knapper, C.L.; Sampson, M.L.; Dai, C.; Levine, S.J.; Remaley, A.T. Endothelial expression of human ABCA1 in mice increases plasma HDL cholesterol and reduces diet-induced atherosclerosis. J. Lipid Res. 2012, 53, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Stamatikos, A.; Knight, E.; Vojtech, L.; Bi, L.; Wacker, B.K.; Tang, C.; Dichek, D.A. Exosome-Mediated Transfer of Anti-miR-33a-5p from Transduced Endothelial Cells Enhances Macrophage and Vascular Smooth Muscle Cell Cholesterol Efflux. Hum. Gene Ther. 2020, 31, 219–232. [Google Scholar] [CrossRef]
- Stamatikos, A.D.; da Silva, R.P.; Lewis, J.T.; Douglas, D.N.; Kneteman, N.M.; Jacobs, R.L.; Paton, C.M. Tissue Specific Effects of Dietary Carbohydrates and Obesity on ChREBPalpha and ChREBPbeta Expression. Lipids 2016, 51, 95–104. [Google Scholar] [CrossRef]
- Esobi, I.C.; Barksdale, C.; Heard-Tate, C.; Reigers Powell, R.; Bruce, T.F.; Stamatikos, A. MOVAS Cells: A Versatile Cell Line for Studying Vascular Smooth Muscle Cell Cholesterol Metabolism. Lipids 2021, 56, 413–422. [Google Scholar] [CrossRef]
- Zannis, V.I.; Chroni, A.; Krieger, M. Role of apoA-I, ABCA1, LCAT, and SR-BI in the biogenesis of HDL. J. Mol. Med. 2006, 84, 276–294. [Google Scholar] [CrossRef]
- Albers, M.; Blume, B.; Schlueter, T.; Wright, M.B.; Kober, I.; Kremoser, C.; Deuschle, U.; Koegl, M. A novel principle for partial agonism of liver X receptor ligands. Competitive recruitment of activators and repressors. J. Biol. Chem. 2006, 281, 4920–4930. [Google Scholar] [CrossRef] [PubMed]
- Aravindhan, K.; Webb, C.L.; Jaye, M.; Ghosh, A.; Willette, R.N.; DiNardo, N.J.; Jucker, B.M. Assessing the effects of LXR agonists on cellular cholesterol handling: A stable isotope tracer study. J. Lipid Res. 2006, 47, 1250–1260. [Google Scholar] [CrossRef]
- Mitro, N.; Vargas, L.; Romeo, R.; Koder, A.; Saez, E. T0901317 is a potent PXR ligand: Implications for the biology ascribed to LXR. FEBS Lett. 2007, 581, 1721–1726. [Google Scholar] [CrossRef]
- Benoit, G.; Altucci, L.; Flexor, M.; Ruchaud, S.; Lillehaug, J.; Raffelsberger, W.; Gronemeyer, H.; Lanotte, M. RAR-independent RXR signaling induces t(15;17) leukemia cell maturation. EMBO J. 1999, 18, 7011–7018. [Google Scholar] [CrossRef]
- Dupuis, H.; Pest, M.A.; Hadzic, E.; Vo, T.X.; Hardy, D.B.; Beier, F. Exposure to the RXR Agonist SR11237 in Early Life Causes Disturbed Skeletal Morphogenesis in a Rat Model. Int. J. Mol. Sci 2019, 20, 5198. [Google Scholar] [CrossRef]
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgozoglu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Primers 2019, 5, 56. [Google Scholar] [CrossRef] [PubMed]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte chemoattractant protein-1 (MCP-1): An overview. J. Interferon Cytokine Res. 2009, 29, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Westerterp, M.; Tsuchiya, K.; Tattersall, I.W.; Fotakis, P.; Bochem, A.E.; Molusky, M.M.; Ntonga, V.; Abramowicz, S.; Parks, J.S.; Welch, C.L.; et al. Deficiency of ATP-Binding Cassette Transporters A1 and G1 in Endothelial Cells Accelerates Atherosclerosis in Mice. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1328–1337. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Kim, J.A.; Kim, K.C.; Suh, S.H. Isolation and in vitro culture of vascular endothelial cells from mice. Korean J. Physiol. Pharmacol. 2015, 19, 35–42. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, J.M.; Chen, A.F.; Zhang, K. Isolation and Primary Culture of Mouse Aortic Endothelial Cells. J. Vis. Exp. 2016, 118, e52965. [Google Scholar] [CrossRef]
- Zhu, R.; Ou, Z.; Ruan, X.; Gong, J. Role of liver X receptors in cholesterol efflux and inflammatory signaling (review). Mol. Med. Rep. 2012, 5, 895–900. [Google Scholar] [CrossRef] [PubMed]
- Kruth, H.S. Lipoprotein cholesterol and atherosclerosis. Curr. Mol. Med. 2001, 1, 633–653. [Google Scholar] [CrossRef]
- Cha, J.Y.; Repa, J.J. The liver X receptor (LXR) and hepatic lipogenesis. The carbohydrate-response element-binding protein is a target gene of LXR. J. Biol. Chem. 2007, 282, 743–751. [Google Scholar] [CrossRef]
- DeFilippis, A.P.; Blaha, M.J.; Martin, S.S.; Reed, R.M.; Jones, S.R.; Nasir, K.; Blumenthal, R.S.; Budoff, M.J. Nonalcoholic fatty liver disease and serum lipoproteins: The Multi-Ethnic Study of Atherosclerosis. Atherosclerosis 2013, 227, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Loria, P.; Marchesini, G.; Nascimbeni, F.; Ballestri, S.; Maurantonio, M.; Carubbi, F.; Ratziu, V.; Lonardo, A. Cardiovascular risk, lipidemic phenotype and steatosis. A comparative analysis of cirrhotic and non-cirrhotic liver disease due to varying etiology. Atherosclerosis 2014, 232, 99–109. [Google Scholar] [CrossRef]
- Volzke, H.; Robinson, D.M.; Kleine, V.; Deutscher, R.; Hoffmann, W.; Ludemann, J.; Schminke, U.; Kessler, C.; John, U. Hepatic steatosis is associated with an increased risk of carotid atherosclerosis. World J. Gastroenterol. 2005, 11, 1848–1853. [Google Scholar] [CrossRef] [PubMed]
- Kheirolomoom, A.; Kim, C.W.; Seo, J.W.; Kumar, S.; Son, D.J.; Gagnon, M.K.; Ingham, E.S.; Ferrara, K.W.; Jo, H. Multifunctional Nanoparticles Facilitate Molecular Targeting and miRNA Delivery to Inhibit Atherosclerosis in ApoE(-/-) Mice. ACS Nano 2015, 9, 8885–8897. [Google Scholar] [CrossRef] [PubMed]
- Dosta, P.; Tamargo, I.; Ramos, V.; Kumar, S.; Kang, D.W.; Borros, S.; Jo, H. Delivery of Anti-microRNA-712 to Inflamed Endothelial Cells Using Poly(beta-amino ester) Nanoparticles Conjugated with VCAM-1 Targeting Peptide. Adv. Healthc. Mater. 2021, 10, e2001894. [Google Scholar] [CrossRef] [PubMed]
- Warboys, C.M.; Amini, N.; de Luca, A.; Evans, P.C. The role of blood flow in determining the sites of atherosclerotic plaques. F1000 Med. Rep. 2011, 3, 5. [Google Scholar] [CrossRef]
- Fessler, M.B. The challenges and promise of targeting the Liver X Receptors for treatment of inflammatory disease. Pharmacol. Ther. 2018, 181, 1–12. [Google Scholar] [CrossRef]
- Chen, L.; Anton, M.; Graham, F.L. Production and characterization of human 293 cell lines expressing the site-specific recombinase Cre. Somat. Cell Mol. Genet. 1996, 22, 477–488. [Google Scholar] [CrossRef]
- Marsche, G.; Frank, S.; Raynes, J.G.; Kozarsky, K.F.; Sattler, W.; Malle, E. The lipidation status of acute-phase protein serum amyloid A determines cholesterol mobilization via scavenger receptor class B, type I. Biochem. J. 2007, 402, 117–124. [Google Scholar] [CrossRef]
- Wong, D.; Dorovini-Zis, K. Expression of vascular cell adhesion molecule-1 (VCAM-1) by human brain microvessel endothelial cells in primary culture. Microvasc. Res. 1995, 49, 325–339. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Sequence (5′–3′) | |
|---|---|---|
| GAPDH | forward: | TGACCTCAACTACATGGTCTACA |
| reverse: | CTTCCCATTCTCGGCCTTG | |
| VCAM-1 | forward: | CCCGTCATTGAGGATATTGG |
| reverse: | AATCTCAGGAGCTGGTAG | |
| ICAM-1 | forward: | CAATTTCTCATGCCGCACAG |
| reverse: | AGCTGGAAGATCGAAAGTCCG | |
| CCL2 | forward: | GTCCCTGTCATGCTTCTG |
| reverse: | CTGCTGGTGATCCTCTTG | |
| IL-6 | forward: | TCTATACCACTTCACAAGTCGGA |
| reverse: | GAATTGCCATTGCACAACTCTTT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, K.; Jo, H.; Echesabal-Chen, J.; Stamatikos, A. Combined LXR and RXR Agonist Therapy Increases ABCA1 Protein Expression and Enhances ApoAI-Mediated Cholesterol Efflux in Cultured Endothelial Cells. Metabolites 2021, 11, 640. https://doi.org/10.3390/metabo11090640
Huang K, Jo H, Echesabal-Chen J, Stamatikos A. Combined LXR and RXR Agonist Therapy Increases ABCA1 Protein Expression and Enhances ApoAI-Mediated Cholesterol Efflux in Cultured Endothelial Cells. Metabolites. 2021; 11(9):640. https://doi.org/10.3390/metabo11090640
Chicago/Turabian StyleHuang, Kun, Hanjoong Jo, Jing Echesabal-Chen, and Alexis Stamatikos. 2021. "Combined LXR and RXR Agonist Therapy Increases ABCA1 Protein Expression and Enhances ApoAI-Mediated Cholesterol Efflux in Cultured Endothelial Cells" Metabolites 11, no. 9: 640. https://doi.org/10.3390/metabo11090640
APA StyleHuang, K., Jo, H., Echesabal-Chen, J., & Stamatikos, A. (2021). Combined LXR and RXR Agonist Therapy Increases ABCA1 Protein Expression and Enhances ApoAI-Mediated Cholesterol Efflux in Cultured Endothelial Cells. Metabolites, 11(9), 640. https://doi.org/10.3390/metabo11090640