
The Neural Gut–Brain Axis of Pathological Protein Aggregation in Parkinson’s Disease and Its Counterpart in Peroral Prion Infections


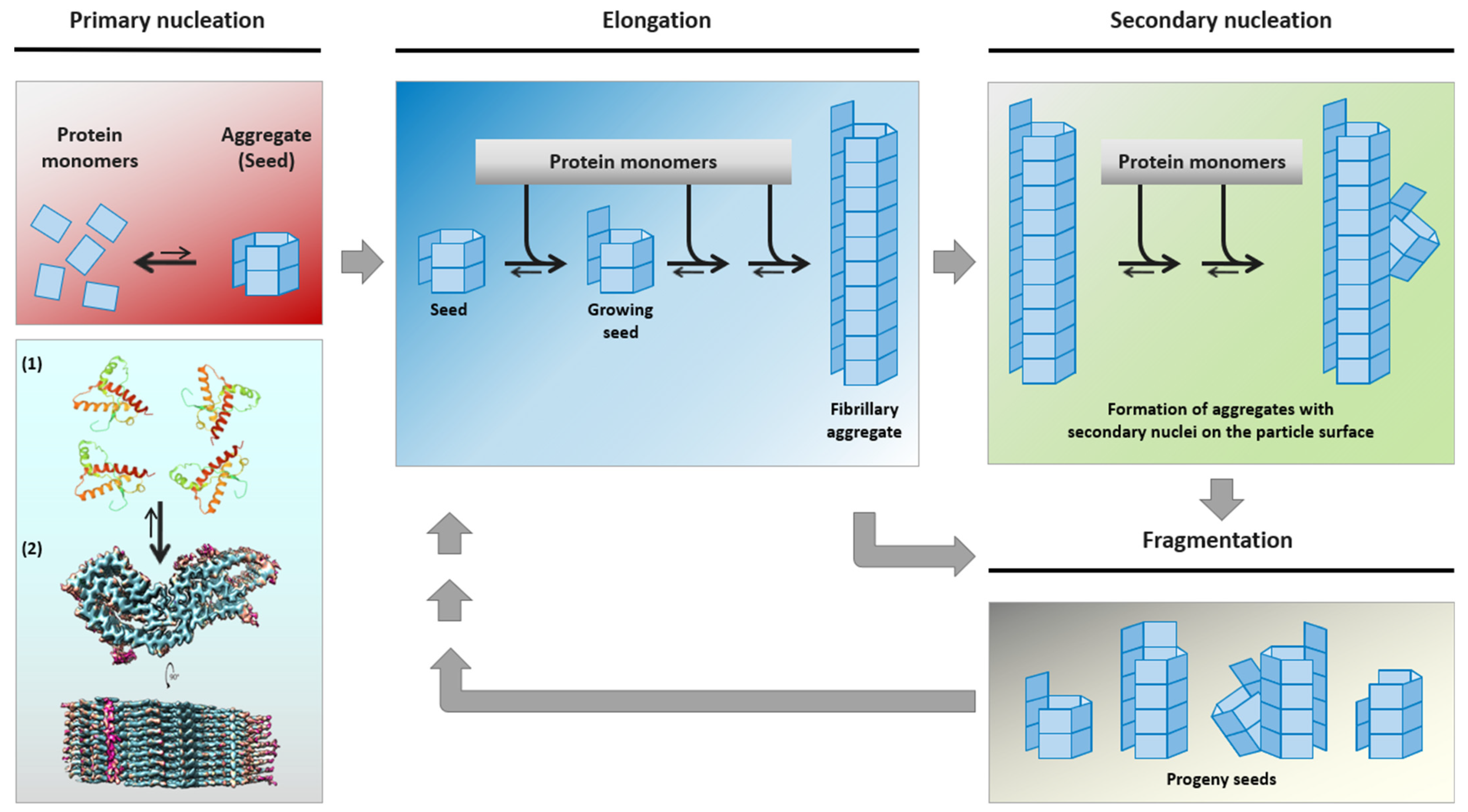{kind=link}
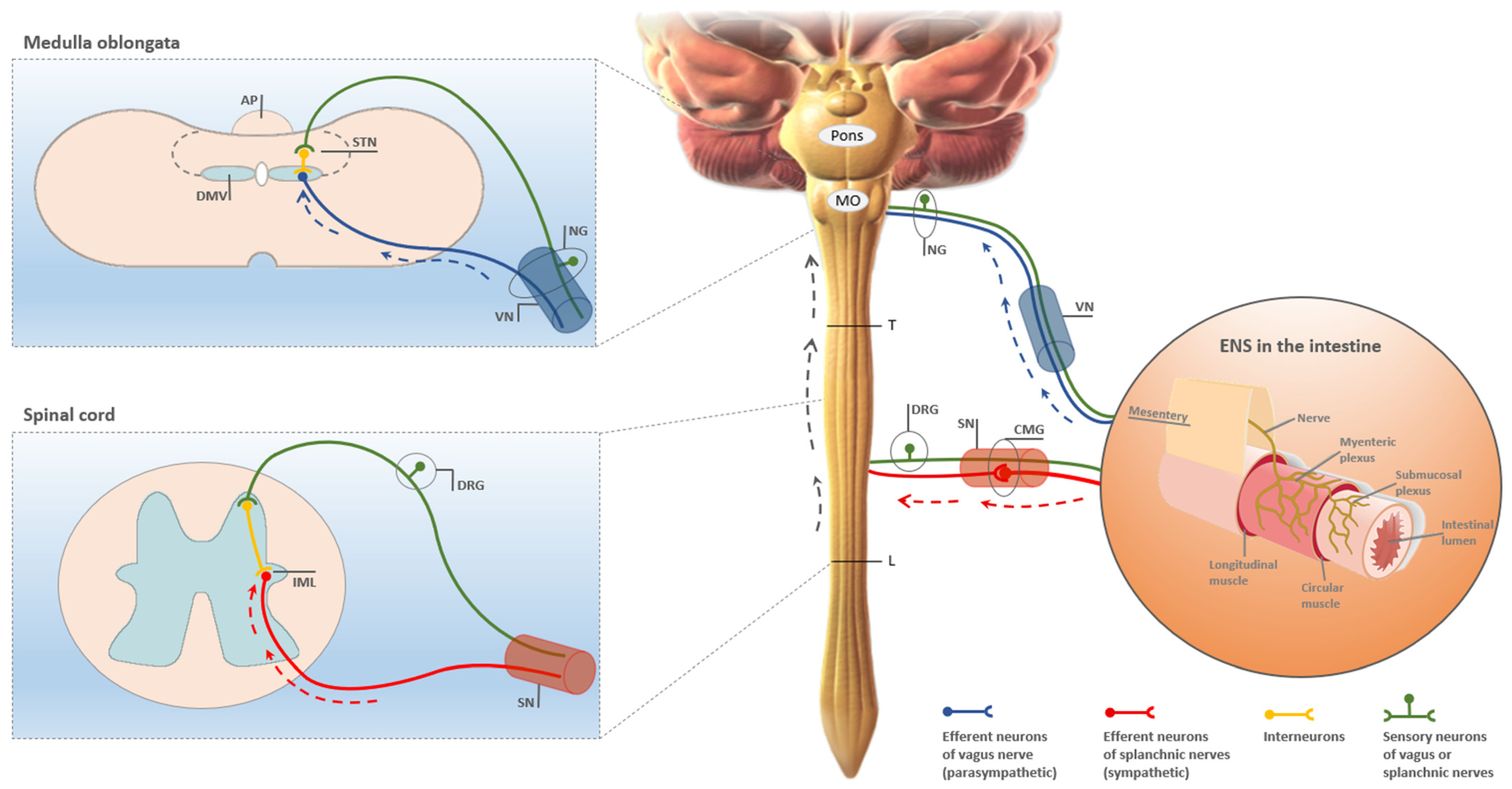{kind=link}
Abstract
1. Introduction
2. The Established Neural Gut–Brain Axis in Peroral Prion Infections
3. Haematogenous Neuroinvasion of the Brain in Peroral Prion Infections
4. The Postulated Neural Gut–Brain Axis in Parkinson’s Disease (PD)
5. Is There a Neural Gut–Brain Axis in PD? Pros, Cons and Insights from Peroral Prion Infections
5.1. Studies Based on Human Tissues and Epidemiological Data
5.2. Studies in Animals
6. Conclusions
6.1. Does the Concept of a Neural Gut–Brain Axis of Pathological Protein Aggregation Hold in PD and Which Alternative or Complementary Hypotheses Are Being Discussed?
6.2. Possible Future Directions of Research
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dickson, D.W. Parkinson’s disease and parkinsonism: Neuropathology. Cold Spring Harb. Perspect. Med. 2012, 2, a009258. [Google Scholar] [CrossRef]
- Schulz-Schaeffer, W.J. The synaptic pathology of alpha-synuclein aggregation in dementia with Lewy bodies, Parkinson’s disease and Parkinson’s disease dementia. Acta Neuropathol. 2010, 120, 131–143. [Google Scholar] [CrossRef]
- Come, J.H.; Fraser, P.E.; Lansbury, P.T., Jr. A kinetic model for amyloid formation in the prion diseases: Importance of seeding. Proc. Natl. Acad. Sci. USA 1993, 90, 5959–5963. [Google Scholar] [CrossRef] [PubMed]
- Beekes, M.; Thomzig, A.; Schulz-Schaeffer, W.J.; Burger, R. Is there a risk of prion-like disease transmission by Alzheimer- or Parkinson-associated protein particles? Acta Neuropathol. 2014, 128, 463–476. [Google Scholar] [CrossRef] [PubMed]
- Oueslati, A.; Ximerakis, M.; Vekrellis, K. Protein Transmission, Seeding and Degradation: Key Steps for α-Synuclein Prion-Like Propagation. Exp. Neurobiol. 2014, 23, 324–336. [Google Scholar] [CrossRef]
- Melki, R. Alpha-synuclein and the prion hypothesis in Parkinson’s disease. Rev. Neurol. 2018, 174, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Baral, P.K.; Swayampakula, M.; Aguzzi, A.; James, M.N. X-ray structural and molecular dynamical studies of the globular domains of cow, deer, elk and Syrian hamster prion proteins. J. Struct. Biol. 2015, 192, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Kraus, A.; Hoyt, F.; Schwartz, C.L.; Hansen, B.; Hughson, A.G.; Artikis, E.; Race, B.; Caughey, B. Structure of an infectious mammalian prion. bioRxiv 2021. [Google Scholar] [CrossRef]
- Meisl, G.; Rajah, L.; Cohen, S.A.I.; Pfammatter, M.; Šarić, A.; Hellstrand, E.; Buell, A.K.; Aguzzi, A.; Linse, S.; Vendruscolo, M.; et al. Scaling behaviour and rate-determining steps in filamentous self-assembly. Chem. Sci. 2017, 8, 7087–7097. [Google Scholar] [CrossRef] [PubMed]
- Antony, P.M.; Diederich, N.J.; Krüger, R.; Balling, R. The hallmarks of Parkinson’s disease. FEBS J. 2013, 280, 5981–5993. [Google Scholar] [CrossRef]
- Liddle, R.A. Parkinson’s disease from the gut. Brain Res. 2018, 1693, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, E.; Kluge, A.; Böttner, M.; Zunke, F.; Cossais, F.; Berg, D.; Arnold, P. Alpha Synuclein Connects the Gut-Brain Axis in Parkinson’s Disease Patients–A View on Clinical Aspects, Cellular Pathology and Analytical Methodology. Front. Cell Dev. Biol. 2020, 8, 573696. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.Y.; Chan, P.; Stoessl, A.J. The underlying mechanism of prodromal PD: Insights from the parasympathetic nervous system and the olfactory system. Transl. Neurodegener. 2017, 6, 4. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Braak, H.; Rub, U.; Gai, W.P.; Del Tredici, K. Idiopathic Parkinson’s disease: Possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J. Neural Transm. 2003, 110, 517–536. [Google Scholar] [CrossRef]
- Hawkes, C.H.; Del Tredici, K.; Braak, H. Parkinson’s disease: A dual-hit hypothesis. Neuropathol. Appl. Neurobiol. 2007, 33, 599–614. [Google Scholar] [CrossRef]
- Hawkes, C.H.; Del Tredici, K.; Braak, H. Parkinson’s disease: The dual hit theory revisited. Ann. N. Y. Acad. Sci. 2009, 1170, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.; Cervenakova, L. A prion lexicon (out of control). Lancet 2005, 365, 122. [Google Scholar] [CrossRef]
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. The prion diseases. Brain Pathol. 1998, 8, 499–513. [Google Scholar] [CrossRef]
- Tyson, T.; Steiner, J.A.; Brundin, P. Sorting out release, uptake and processing of alpha-synuclein during prion-like spread of pathology. J. Neurochem. 2016, 139 (Suppl. S1), 275–289. [Google Scholar] [CrossRef]
- Grozdanov, V.; Danzer, K.M. Release and uptake of pathologic alpha-synuclein. Cell Tissue Res. 2018, 373, 175–182. [Google Scholar] [CrossRef]
- Volpicelli-Daley, L.; Brundin, P. Prion-like propagation of pathology in Parkinson disease. Handb. Clin. Neurol. 2018, 153, 321–335. [Google Scholar] [CrossRef] [PubMed]
- Karpowicz, R.J., Jr.; Trojanowski, J.Q.; Lee, V.M. Transmission of α-synuclein seeds in neurodegenerative disease: Recent developments. Lab. Investig. 2019, 99, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Steiner, J.A.; Quansah, E.; Brundin, P. The concept of alpha-synuclein as a prion-like protein: Ten years after. Cell Tissue Res. 2018, 373, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Killinger, B.A.; Kordower, J.H. Spreading of alpha-synuclein–relevant or epiphenomenon? J. Neurochem. 2019, 150, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Lionnet, A.; Leclair-Visonneau, L.; Neunlist, M.; Murayama, S.; Takao, M.; Adler, C.H.; Derkinderen, P.; Beach, T.G. Does Parkinson’s disease start in the gut? Acta Neuropathol. 2018, 135, 1–12. [Google Scholar] [CrossRef]
- Surmeier, D.J.; Obeso, J.A.; Halliday, G.M. Parkinson’s Disease Is Not Simply a Prion Disorder. J. Neurosci. 2017, 37, 9799–9807. [Google Scholar] [CrossRef] [PubMed]
- Scheperjans, F.; Derkinderen, P.; Borghammer, P. The Gut and Parkinson’s Disease: Hype or Hope? J. Parkinsons Dis. 2018, 8, S31–S39. [Google Scholar] [CrossRef]
- Kimberlin, R.H.; Walker, C.A. Pathogenesis of scrapie in mice after intragastric infection. Virus Res. 1989, 12, 213–220. [Google Scholar] [CrossRef]
- Beekes, M.; Baldauf, E.; Diringer, H. Sequential appearance and accumulation of pathognomonic markers in the central nervous system of hamsters orally infected with scrapie. J. Gen. Virol. 1996, 77, 1925–1934. [Google Scholar] [CrossRef]
- McBride, P.A.; Schulz-Schaeffer, W.J.; Donaldson, M.; Bruce, M.; Diringer, H.; Kretzschmar, H.A.; Beekes, M. Early spread of scrapie from the gastrointestinal tract to the central nervous system involves autonomic fibers of the splanchnic and vagus nerves. J. Virol. 2001, 75, 9320–9327. [Google Scholar] [CrossRef]
- Baldauf, E.; Beekes, M.; Diringer, H. Evidence for an alternative direct route of access for the scrapie agent to the brain bypassing the spinal cord. J. Gen. Virol. 1997, 78, 1187–1197. [Google Scholar] [CrossRef]
- Beekes, M.; McBride, P.A. Early accumulation of pathological PrP in the enteric nervous system and gut-associated lymphoid tissue of hamsters orally infected with scrapie. Neurosci. Lett. 2000, 278, 181–184. [Google Scholar] [CrossRef]
- Beekes, M.; McBride, P.A.; Baldauf, E. Cerebral targeting indicates vagal spread of infection in hamsters fed with scrapie. J. Gen. Virol. 1998, 79, 601–607. [Google Scholar] [CrossRef]
- McBride, P.A.; Beekes, M. Pathological PrP is abundant in sympathetic and sensory ganglia of hamsters fed with scrapie. Neurosci. Lett. 1999, 265, 135–138. [Google Scholar] [CrossRef]
- McKinley, M.P.; Bolton, D.C.; Prusiner, S.B. A protease-resistant protein is a structural component of the scrapie prion. Cell 1983, 35, 57–62. [Google Scholar] [CrossRef]
- Gabizon, R.; McKinley, M.P.; Prusiner, S.B. Purified prion proteins and scrapie infectivity copartition into liposomes. Proc. Natl. Acad. Sci. USA 1987, 84, 4017–4021. [Google Scholar] [CrossRef]
- Bolton, D.C.; Rudelli, R.D.; Currie, J.R.; Bendheim, P.E. Copurification of Sp33-37 and scrapie agent from hamster brain prior to detectable histopathology and clinical disease. J. Gen. Virol. 1991, 72, 2905–2913. [Google Scholar] [CrossRef]
- Rubenstein, R.; Merz, P.A.; Kascsak, R.J.; Scalici, C.L.; Papini, M.C.; Carp, R.I.; Kimberlin, R.H. Scrapie-infected spleens: Analysis of infectivity, scrapie-associated fibrils, and protease-resistant proteins. J. Infect. Dis. 1991, 164, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Jendroska, K.; Heinzel, F.P.; Torchia, M.; Stowring, L.; Kretzschmar, H.A.; Kon, A.; Stern, A.; Prusiner, S.B.; DeArmond, S.J. Proteinase-resistant prion protein accumulation in Syrian hamster brain correlates with regional pathology and scrapie infectivity. Neurology 1991, 41, 1482–1490. [Google Scholar] [CrossRef] [PubMed]
- Ironside, J.W.; McCardle, L.; Horsburgh, A.; Lim, Z.; Head, M.W. Pathological diagnosis of variant Creutzfeldt-Jakob disease. APMIS 2002, 110, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Wadsworth, J.D.; Joiner, S.; Hill, A.F.; Campbell, T.A.; Desbruslais, M.; Luthert, P.J.; Collinge, J. Tissue distribution of protease resistant prion protein in variant Creutzfeldt-Jakob disease using a highly sensitive immunoblotting assay. Lancet 2001, 358, 171–180. [Google Scholar] [CrossRef]
- Schulz-Schaeffer, W.J.; Tschoke, S.; Kranefuss, N.; Drose, W.; Hause-Reitner, D.; Giese, A.; Groschup, M.H.; Kretzschmar, H.A. The paraffin-embedded tissue blot detects PrP(Sc) early in the incubation time in prion diseases. Am. J. Pathol. 2000, 156, 51–56. [Google Scholar] [CrossRef]
- Groschup, M.H.; Beekes, M.; McBride, P.A.; Hardt, M.; Hainfellner, J.A.; Budka, H. Deposition of disease-associated prion protein involves the peripheral nervous system in experimental scrapie. Acta Neuropathol. 1999, 98, 453–457. [Google Scholar] [CrossRef]
- Beekes, M.; McBride, P.A. The spread of prions through the body in naturally acquired transmissbile spongiform encephalopathies. FEBS J. 2007, 264, 588–605. [Google Scholar] [CrossRef]
- Krüger, D.; Thomzig, A.; Lenz, G.; Kampf, K.; McBride, P.; Beekes, M. Faecal shedding, alimentary clearance and intestinal spread of prions in hamsters fed with scrapie. Vet. Res. 2009, 40, 4. [Google Scholar] [CrossRef] [PubMed]
- Mabbott, N.A.; MacPherson, G.G. Prions and their lethal journey to the brain. Nat. Rev. Microbiol. 2006, 4, 201–211. [Google Scholar] [CrossRef]
- van Keulen, L.J.; Schreuder, B.E.; Vromans, M.E.; Langeveld, J.P.; Smits, M.A. Pathogenesis of natural scrapie in sheep. Arch. Virol. Suppl. 2000, 16, 57–71. [Google Scholar] [CrossRef]
- van Keulen, L.J.; Schreuder, B.E.; Vromans, M.E.; Langeveld, J.P.; Smits, M.A. Scrapie-associated prion protein in the gastrointestinal tract of sheep with natural scrapie. J. Comp. Pathol. 1999, 121, 55–63. [Google Scholar] [CrossRef]
- van Keulen, L.J.; Vromans, M.E.; van Zijderveld, F.G. Early and late pathogenesis of natural scrapie infection in sheep. APMIS 2002, 110, 23–32. [Google Scholar] [CrossRef]
- Sigurdson, C.J.; Spraker, T.R.; Miller, M.W.; Oesch, B.; Hoover, E.A. PrP(CWD) in the myenteric plexus, vagosympathetic trunk and endocrine glands of deer with chronic wasting disease. J. Gen. Virol. 2001, 82, 2327–2334. [Google Scholar] [CrossRef]
- Fox, K.A.; Jewell, J.E.; Williams, E.S.; Miller, M.W. Patterns of PrPCWD accumulation during the course of chronic wasting disease infection in orally inoculated mule deer (Odocoileus hemionus). J. Gen. Virol. 2006, 87, 3451–3461. [Google Scholar] [CrossRef]
- Hoffmann, C.; Ziegler, U.; Buschmann, A.; Weber, A.; Kupfer, L.; Oelschlegel, A.; Hammerschmidt, B.; Groschup, M.H. Prions spread via the autonomic nervous system from the gut to the central nervous system in cattle incubating bovine spongiform encephalopathy. J. Gen. Virol. 2007, 88, 1048–1055. [Google Scholar] [CrossRef] [PubMed]
- Kaatz, M.; Fast, C.; Ziegler, U.; Balkema-Buschmann, A.; Hammerschmidt, B.; Keller, M.; Oelschlegel, A.; McIntyre, L.; Groschup, M.H. Spread of classic BSE prions from the gut via the peripheral nervous system to the brain. Am. J. Pathol. 2012, 181, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Holznagel, E.; Yutzy, B.; Kruip, C.; Bierke, P.; Schulz-Schaeffer, W.; Löwer, J. Foodborne-Transmitted Prions From the Brain of Cows With Bovine Spongiform Encephalopathy Ascend in Afferent Neurons to the Simian Central Nervous System and Spread to Tonsils and Spleen at a Late Stage of the Incubation Period. J. Infect. Dis. 2015, 212, 1459–1468. [Google Scholar] [CrossRef]
- Haik, S.; Faucheux, B.A.; Sazdovitch, V.; Privat, N.; Kemeny, J.L.; Perret-Liaudet, A.; Hauw, J.J. The sympathetic nervous system is involved in variant Creutzfeldt-Jakob disease. Nat. Med. 2003, 9, 1121–1123. [Google Scholar] [CrossRef]
- Ironside, J.W. Pathology of variant Creutzfeldt-Jakob disease. Arch. Virol. Suppl. 2000, 143–151. [Google Scholar] [CrossRef]
- Thomzig, A.; Kratzel, C.; Lenz, G.; Kruger, D.; Beekes, M. Widespread PrPSc accumulation in muscles of hamsters orally infected with scrapie. EMBO Rep. 2003, 4, 530–533. [Google Scholar] [CrossRef] [PubMed]
- Thomzig, A.; Schulz-Schaeffer, W.; Kratzel, C.; Mai, J.; Beekes, M. Preclinical deposition of pathological prion protein PrPSc in muscles of hamsters orally exposed to scrapie. J. Clin. Investig. 2004, 113, 1465–1472. [Google Scholar] [CrossRef][Green Version]
- Thomzig, A.; Schulz-Schaeffer, W.; Wrede, A.; Wemheuer, W.; Brenig, B.; Kratzel, C.; Lemmer, K.; Beekes, M. Accumulation of pathological prion protein PrPSc in the skin of animals with experimental and natural scrapie. PLoS Pathog. 2007, 3, e66. [Google Scholar] [CrossRef]
- Sisó, S.; Jeffrey, M.; González, L. Neuroinvasion in sheep transmissible spongiform encephalopathies: The role of the haematogenous route. Neuropathol. Appl. Neurobiol. 2009, 35, 232–246. [Google Scholar] [CrossRef]
- González, L.; Pitarch, J.L.; Martin, S.; Thurston, L.; Moore, J.; Acín, C.; Jeffrey, M. Identical pathogenesis and neuropathological phenotype of scrapie in valine, arginine, glutamine/valine, arginine, glutamine sheep infected experimentally by the oral and conjunctival routes. J. Comp. Pathol. 2014, 150, 47–56. [Google Scholar] [CrossRef]
- Braak, H.; Ghebremedhin, E.; Rüb, U.; Bratzke, H.; Del Tredici, K. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 2004, 318, 121–134. [Google Scholar] [CrossRef]
- Lee, H.J.; Patel, S.; Lee, S.J. Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J. Neurosci. 2005, 25, 6016–6024. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Suk, J.E.; Bae, E.J.; Lee, J.H.; Paik, S.R.; Lee, S.J. Assembly-dependent endocytosis and clearance of extracellular alpha-synuclein. Int. J. Biochem. Cell Biol. 2008, 40, 1835–1849. [Google Scholar] [CrossRef] [PubMed]
- Luk, K.C.; Song, C.; O’Brien, P.; Stieber, A.; Branch, J.R.; Brunden, K.R.; Trojanowski, J.Q.; Lee, V.M. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc. Natl. Acad. Sci. USA 2009, 106, 20051–20056. [Google Scholar] [CrossRef] [PubMed]
- Volpicelli-Daley, L.A.; Luk, K.C.; Patel, T.P.; Tanik, S.A.; Riddle, D.M.; Stieber, A.; Meaney, D.F.; Trojanowski, J.Q.; Lee, V.M. Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 2011, 72, 57–71. [Google Scholar] [CrossRef]
- Phillips, R.J.; Walter, G.C.; Wilder, S.L.; Baronowsky, E.A.; Powley, T.L. Alpha-synuclein-immunopositive myenteric neurons and vagal preganglionic terminals: Autonomic pathway implicated in Parkinson’s disease? Neuroscience 2008, 153, 733–750. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Sastre, M.; Bohl, J.R.; de Vos, R.A.; Del Tredici, K. Parkinson’s disease: Lesions in dorsal horn layer I, involvement of parasympathetic and sympathetic pre- and postganglionic neurons. Acta Neuropathol. 2007, 113, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Del Tredici, K.; Braak, H. Spinal cord lesions in sporadic Parkinson’s disease. Acta Neuropathol. 2012, 124, 643–664. [Google Scholar] [CrossRef] [PubMed]
- Bloch, A.; Probst, A.; Bissig, H.; Adams, H.; Tolnay, M. Alpha-synuclein pathology of the spinal and peripheral autonomic nervous system in neurologically unimpaired elderly subjects. Neuropathol. Appl. Neurobiol. 2006, 32, 284–295. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K. Neuropathological Staging of Brain Pathology in Sporadic Parkinson’s disease: Separating the Wheat from the Chaff. J. Parkinsons Dis. 2017, 7, S71–S85. [Google Scholar] [CrossRef] [PubMed]
- Klingelhoefer, L.; Reichmann, H. Pathogenesis of Parkinson disease--the gut-brain axis and environmental factors. Nat. Rev. Neurol. 2015, 11, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.; Gershon, M.D. The bowel and beyond: The enteric nervous system in neurological disorders. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 517–528. [Google Scholar] [CrossRef]
- Braak, H.; de Vos, R.A.; Bohl, J.; Del, T.K. Gastric alpha-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci.Lett. 2006, 396, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Duda, J.E.; Giasson, B.I.; Mabon, M.E.; Lee, V.M.; Trojanowski, J.Q. Novel antibodies to synuclein show abundant striatal pathology in Lewy body diseases. Ann. Neurol. 2002, 52, 205–210. [Google Scholar] [CrossRef]
- Duda, J.E.; Giasson, B.I.; Mabon, M.E.; Miller, D.C.; Golbe, L.I.; Lee, V.M.; Trojanowski, J.Q. Concurrence of alpha-synuclein and tau brain pathology in the Contursi kindred. Acta Neuropathol. 2002, 104, 7–11. [Google Scholar] [CrossRef]
- Giasson, B.I.; Duda, J.E.; Quinn, S.M.; Zhang, B.; Trojanowski, J.Q.; Lee, V.M. Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron 2002, 34, 521–533. [Google Scholar] [CrossRef]
- Schulz-Schaeffer, W.J. Is cell death primary or secondary in the pathophysiology of idiopathic Parkinson’s disease? Biomolecules 2015, 5, 1467–1479. [Google Scholar] [CrossRef]
- Kalaitzakis, M.E.; Graeber, M.B.; Gentleman, S.M.; Pearce, R.K. The dorsal motor nucleus of the vagus is not an obligatory trigger site of Parkinson’s disease: A critical analysis of alpha-synuclein staging. Neuropathol. Appl. Neurobiol. 2008, 34, 284–295. [Google Scholar] [CrossRef]
- Jellinger, K.A. A critical evaluation of current staging of alpha-synuclein pathology in Lewy body disorders. Biochim. Biophys. Acta 2009, 1792, 730–740. [Google Scholar] [CrossRef] [PubMed]
- Halliday, G.; McCann, H.; Shepherd, C. Evaluation of the Braak hypothesis: How far can it explain the pathogenesis of Parkinson’s disease? Expert Rev. Neurother. 2012, 12, 673–686. [Google Scholar] [CrossRef] [PubMed]
- Berg, D.; Postuma, R.B.; Bloem, B.; Chan, P.; Dubois, B.; Gasser, T.; Goetz, C.G.; Halliday, G.M.; Hardy, J.; Lang, A.E.; et al. Time to redefine PD? Introductory statement of the MDS Task Force on the definition of Parkinson’s disease. Mov. Disord. 2014, 29, 454–462. [Google Scholar] [CrossRef]
- Attems, J.; Jellinger, K.A. The dorsal motor nucleus of the vagus is not an obligatory trigger site of Parkinson’s disease. Neuropathol. Appl. Neurobiol. 2008, 34, 466–467. [Google Scholar] [CrossRef]
- Jellinger, K.A. Is Braak staging valid for all types of Parkinson’s disease? J. Neural Transm. 2019, 126, 423–431. [Google Scholar] [CrossRef]
- Adler, C.H.; Beach, T.G. Neuropathological basis of nonmotor manifestations of Parkinson’s disease. Mov. Disord. 2016, 31, 1114–1119. [Google Scholar] [CrossRef]
- Borghammer, P.; Van Den Berge, N. Brain-First versus Gut-First Parkinson’s Disease: A Hypothesis. J. Parkinsons Dis. 2019, 9, S281–S295. [Google Scholar] [CrossRef]
- Beach, T.G.; Adler, C.H.; Sue, L.I.; Vedders, L.; Lue, L.; White Iii, C.L.; Akiyama, H.; Caviness, J.N.; Shill, H.A.; Sabbagh, M.N.; et al. Multi-organ distribution of phosphorylated alpha-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol. 2010, 119, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Svensson, E.; Horváth-Puhó, E.; Thomsen, R.W.; Djurhuus, J.C.; Pedersen, L.; Borghammer, P.; Sørensen, H.T. Vagotomy and subsequent risk of Parkinson’s disease. Ann. Neurol. 2015, 78, 522–529. [Google Scholar] [CrossRef]
- Liu, B.; Fang, F.; Pedersen, N.L.; Tillander, A.; Ludvigsson, J.F.; Ekbom, A.; Svenningsson, P.; Chen, H.; Wirdefeldt, K. Vagotomy and Parkinson disease: A Swedish register-based matched-cohort study. Neurology 2017, 88, 1996–2002. [Google Scholar] [CrossRef]
- Tysnes, O.B.; Kenborg, L.; Herlofson, K.; Steding-Jessen, M.; Horn, A.; Olsen, J.H.; Reichmann, H. Does vagotomy reduce the risk of Parkinson’s disease? Ann. Neurol. 2015, 78, 1011–1012. [Google Scholar] [CrossRef] [PubMed]
- Szereda-Przestaszewska, M. Retrograde degeneration within the dorsal motor vagal nucleus following bilateral vagotomy in rabbits. Acta Anat. 1985, 121, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Ling, E.A.; Shieh, J.Y.; Wen, C.Y.; Yick, T.Y.; Wong, W.C. The dorsal motor nucleus of the vagus nerve of the hamster: Ultrastructure of vagal neurons and their responses to vagotomy. J. Anat. 1987, 152, 161–172. [Google Scholar]
- Ling, E.A.; Wong, W.C.; Yick, T.Y.; Leong, S.K. Ultrastructural changes in the dorsal motor nucleus of monkey following bilateral cervical vagotomy. J. Neurocytol. 1986, 15, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Leclair-Visonneau, L.; Neunlist, M.; Derkinderen, P.; Lebouvier, T. The gut in Parkinson’s disease: Bottom-up, top-down, or neither? Neurogastroenterol. Motil. 2020, 32, e13777. [Google Scholar] [CrossRef] [PubMed]
- Holmqvist, S.; Chutna, O.; Bousset, L.; Aldrin-Kirk, P.; Li, W.; Björklund, T.; Wang, Z.Y.; Roybon, L.; Melki, R.; Li, J.Y. Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol. 2014, 128, 805–820. [Google Scholar] [CrossRef]
- Arotcarena, M.L.; Dovero, S.; Prigent, A.; Bourdenx, M.; Camus, S.; Porras, G.; Thiolat, M.L.; Tasselli, M.; Aubert, P.; Kruse, N.; et al. Bidirectional gut-to-brain and brain-to-gut propagation of synucleinopathy in non-human primates. Brain 2020, 143, 1462–1475. [Google Scholar] [CrossRef]
- Thomzig, A.; Wagenführ, K.; Pinder, P.; Joncic, M.; Schulz-Schaeffer, W.J.; Beekes, M. Transmissible α-synuclein seeding activity in brain and stomach of patients with Parkinson’s disease. Acta Neuropathol. 2021, 141, 861–879. [Google Scholar] [CrossRef]
- Recasens, A.; Dehay, B.; Bove, J.; Carballo-Carbajal, I.; Dovero, S.; Perez-Villalba, A.; Fernagut, P.O.; Blesa, J.; Parent, A.; Perier, C.; et al. Lewy body extracts from Parkinson disease brains trigger alpha-synuclein pathology and neurodegeneration in mice and monkeys. Ann. Neurol. 2014, 75, 351–362. [Google Scholar] [CrossRef]
- Prusiner, S.B.; Woerman, A.L.; Mordes, D.A.; Watts, J.C.; Rampersaud, R.; Berry, D.B.; Patel, S.; Oehler, A.; Lowe, J.K.; Kravitz, S.N.; et al. Evidence for alpha-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc. Natl. Acad. Sci. USA 2015, 112, E5308–E5317. [Google Scholar] [CrossRef] [PubMed]
- Recasens, A.; Carballo-Carbajal, I.; Parent, A.; Bové, J.; Gelpi, E.; Tolosa, E.; Vila, M. Lack of pathogenic potential of peripheral α-synuclein aggregates from Parkinson’s disease patients. Acta Neuropathol. Commun. 2018, 6, 8. [Google Scholar] [CrossRef]
- Brundin, P.; Melki, R. Prying into the Prion Hypothesis for Parkinson’s Disease. J. Neurosci. 2017, 37, 9808–9818. [Google Scholar] [CrossRef] [PubMed]
- Kujawska, M.; Jodynis-Liebert, J. What is the evidence that Parkinson’s disease is a prion disorder, which originates in the gut? Int. J. Mol. Sci. 2018, 19, 3573. [Google Scholar] [CrossRef] [PubMed]
- Breen, D.P.; Halliday, G.M.; Lang, A.E. Gut-brain axis and the spread of α-synuclein pathology: Vagal highway or dead end? Mov. Disord. 2019, 34, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Engelender, S.; Isacson, O. The threshold theory for Parkinson’s disease. Trends Neurosci. 2017, 40, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Groveman, B.R.; Foliaki, S.T.; Orru, C.D.; Zanusso, G.; Carroll, J.A.; Race, B.; Haigh, C.L. Sporadic Creutzfeldt-Jakob disease prion infection of human cerebral organoids. Acta Neuropathol. Commun. 2019, 7, 90. [Google Scholar] [CrossRef] [PubMed]
- Groveman, B.R.; Walters, R.; Haigh, C.L. Using our mini-brains: Cerebral organoids as an improved cellular model for human prion disease. Neural Regen. Res. 2020, 15, 1019–1020. [Google Scholar] [CrossRef]
- Faustino Martins, J.M.; Fischer, C.; Urzi, A.; Vidal, R.; Kunz, S.; Ruffault, P.L.; Kabuss, L.; Hube, I.; Gazzerro, E.; Birchmeier, C.; et al. Self-organizing 3D human trunk neuromuscular organoids. Cell Stem Cell 2020, 26, 172–186. [Google Scholar] [CrossRef]
- Davis, S.M. Parkinson’s Disease, Smoking, and Lower Endoscopy. medRxiv 2020. [Google Scholar] [CrossRef]
- Borghammer, P.; Knudsen, K.; Fedorova, T.D.; Brooks, D.J. Imaging Parkinson’s disease below the neck. NPJ Parkinsons Dis. 2017, 3, 15. [Google Scholar] [CrossRef]
- Pinder, P.; Thomzig, A.; Schulz-Schaeffer, W.J.; Beekes, M. Alpha-synuclein seeds of Parkinson’s disease show high prion-exceeding resistance to steam sterilization. J. Hosp. Infect. 2021, 108, 25–32. [Google Scholar] [CrossRef]
- Groveman, B.R.; Orrù, C.D.; Hughson, A.G.; Raymond, L.D.; Zanusso, G.; Ghetti, B.; Campbell, K.J.; Safar, J.; Galasko, D.; Caughey, B. Rapid and ultra-sensitive quantitation of disease-associated α-synuclein seeds in brain and cerebrospinal fluid by αSyn RT-QuIC. Acta Neuropathol. Commun. 2018, 6, 7. [Google Scholar] [CrossRef]
- Wang, Z.; Becker, K.; Donadio, V.; Siedlak, S.; Yuan, J.; Rezaee, M.; Incensi, A.; Kuzkina, A.; Orrú, C.D.; Tatsuoka, C.; et al. Skin α-Synuclein Aggregation Seeding Activity as a Novel Biomarker for Parkinson Disease. JAMA Neurol. 2020, 78, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Bargar, C.; Wang, W.; Gunzler, S.A.; LeFevre, A.; Wang, Z.; Lerner, A.J.; Singh, N.; Tatsuoka, C.; Appleby, B.; Zhu, X.; et al. Streamlined alpha-synuclein RT-QuIC assay for various biospecimens in Parkinson’s disease and dementia with Lewy bodies. Acta Neuropathol. Commun. 2021, 9, 62. [Google Scholar] [CrossRef] [PubMed]
- Shahnawaz, M.; Mukherjee, A.; Pritzkow, S.; Mendez, N.; Rabadia, P.; Liu, X.; Hu, B.; Schmeichel, A.; Singer, W.; Wu, G.; et al. Discriminating α-synuclein strains in Parkinson’s disease and multiple system atrophy. Nature 2020, 578, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Manne, S.; Kondru, N.; Jin, H.; Serrano, G.E.; Anantharam, V.; Kanthasamy, A.; Adler, C.H.; Beach, T.G.; Kanthasamy, A.G. Blinded RT-QuIC Analysis of α-Synuclein Biomarker in Skin Tissue From Parkinson’s Disease Patients. Mov. Disord. 2020, 35, 2230–2239. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beekes, M. The Neural Gut–Brain Axis of Pathological Protein Aggregation in Parkinson’s Disease and Its Counterpart in Peroral Prion Infections. Viruses 2021, 13, 1394. https://doi.org/10.3390/v13071394
Beekes M. The Neural Gut–Brain Axis of Pathological Protein Aggregation in Parkinson’s Disease and Its Counterpart in Peroral Prion Infections. Viruses. 2021; 13(7):1394. https://doi.org/10.3390/v13071394
Chicago/Turabian StyleBeekes, Michael. 2021. "The Neural Gut–Brain Axis of Pathological Protein Aggregation in Parkinson’s Disease and Its Counterpart in Peroral Prion Infections" Viruses 13, no. 7: 1394. https://doi.org/10.3390/v13071394
APA StyleBeekes, M. (2021). The Neural Gut–Brain Axis of Pathological Protein Aggregation in Parkinson’s Disease and Its Counterpart in Peroral Prion Infections. Viruses, 13(7), 1394. https://doi.org/10.3390/v13071394