
A Combined Experimental and Theoretical Study of Nitrofuran Antibiotics: Crystal Structures, DFT Computations, Sublimation and Solution Thermodynamics

 , , ,
, , ,  and
and 
Abstract
1. Introduction
2. Results and Discussion
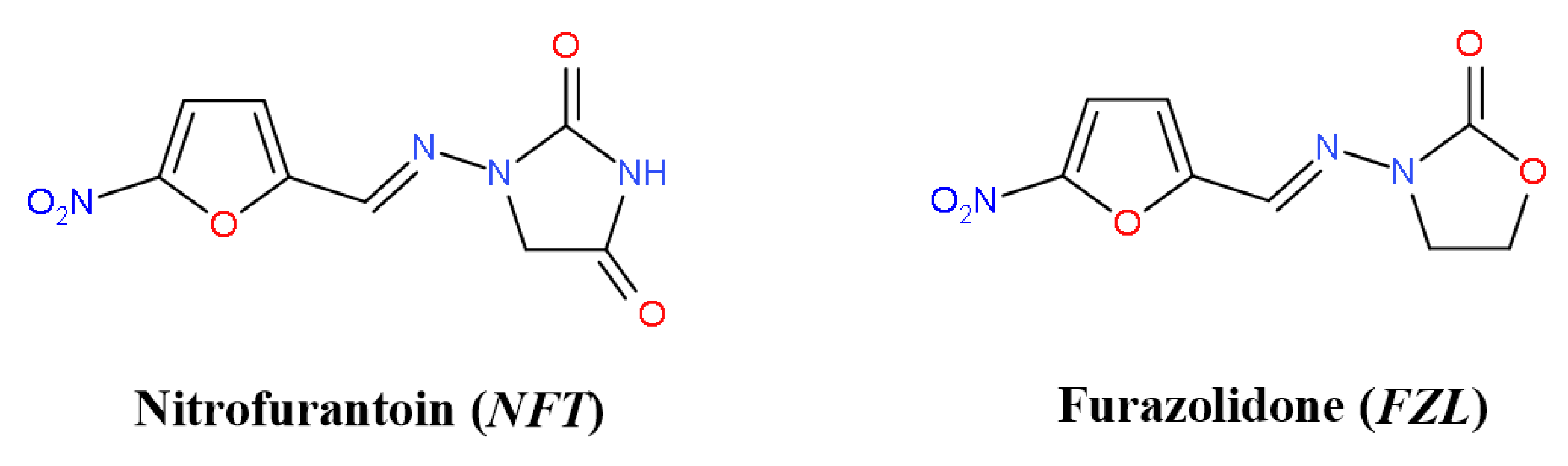
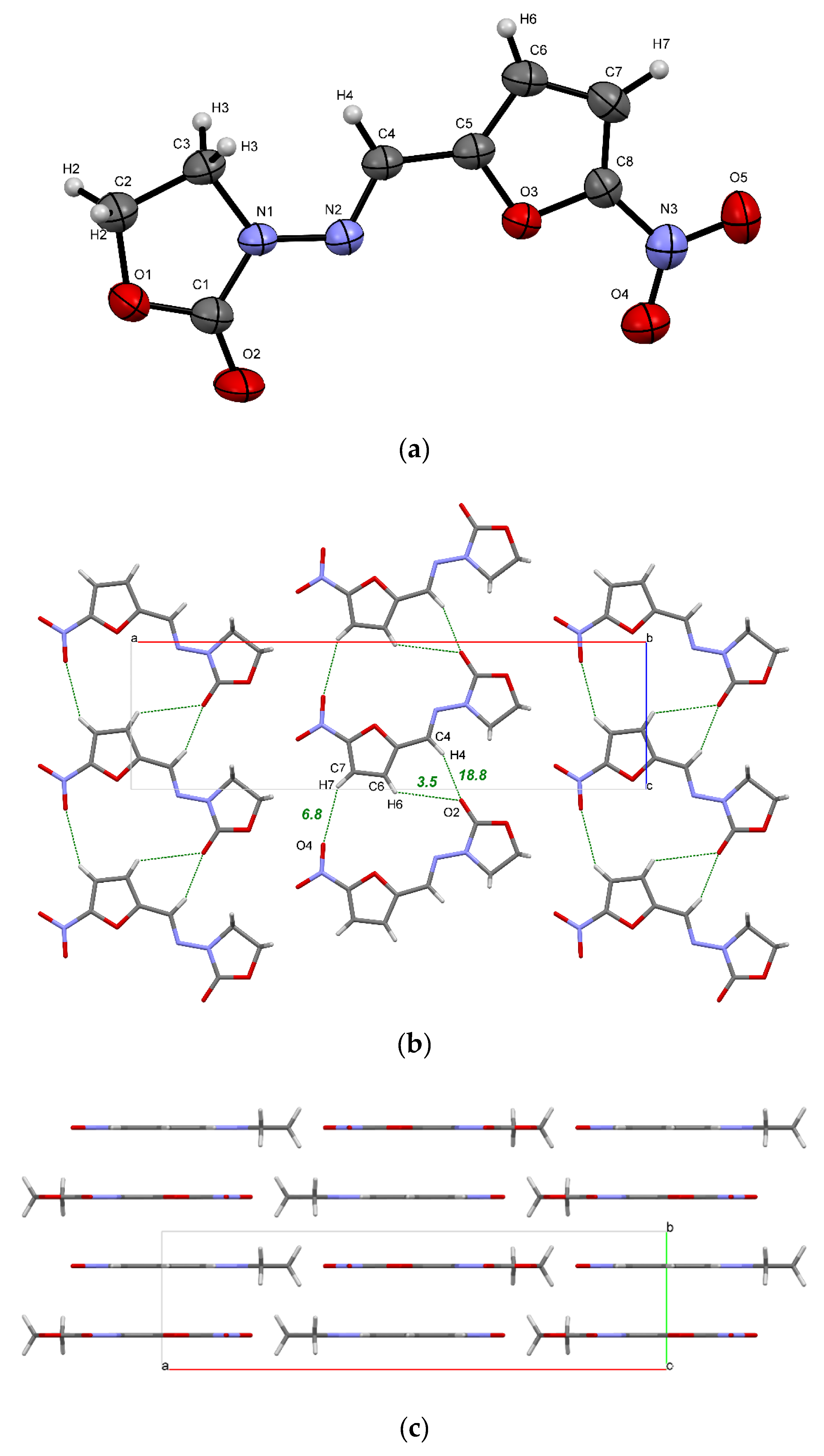
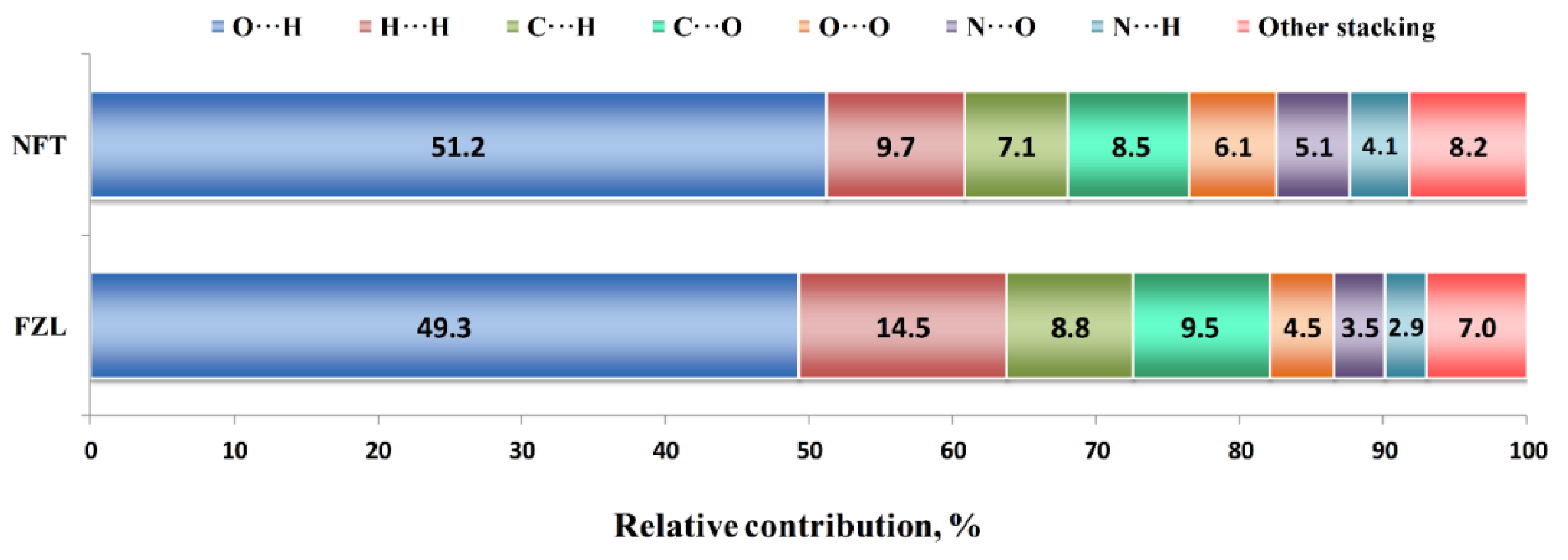
2.1. Crystal Structure Analysis
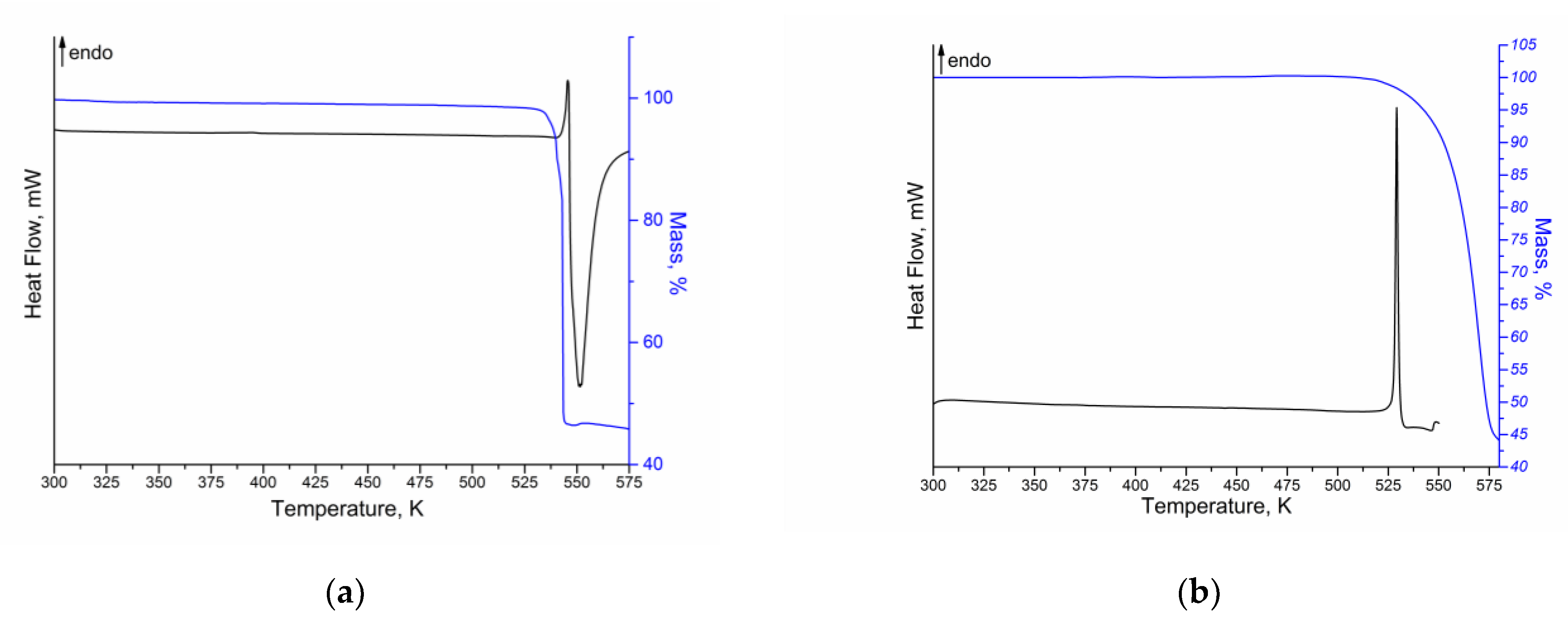
2.2. Thermal Study of the Compounds
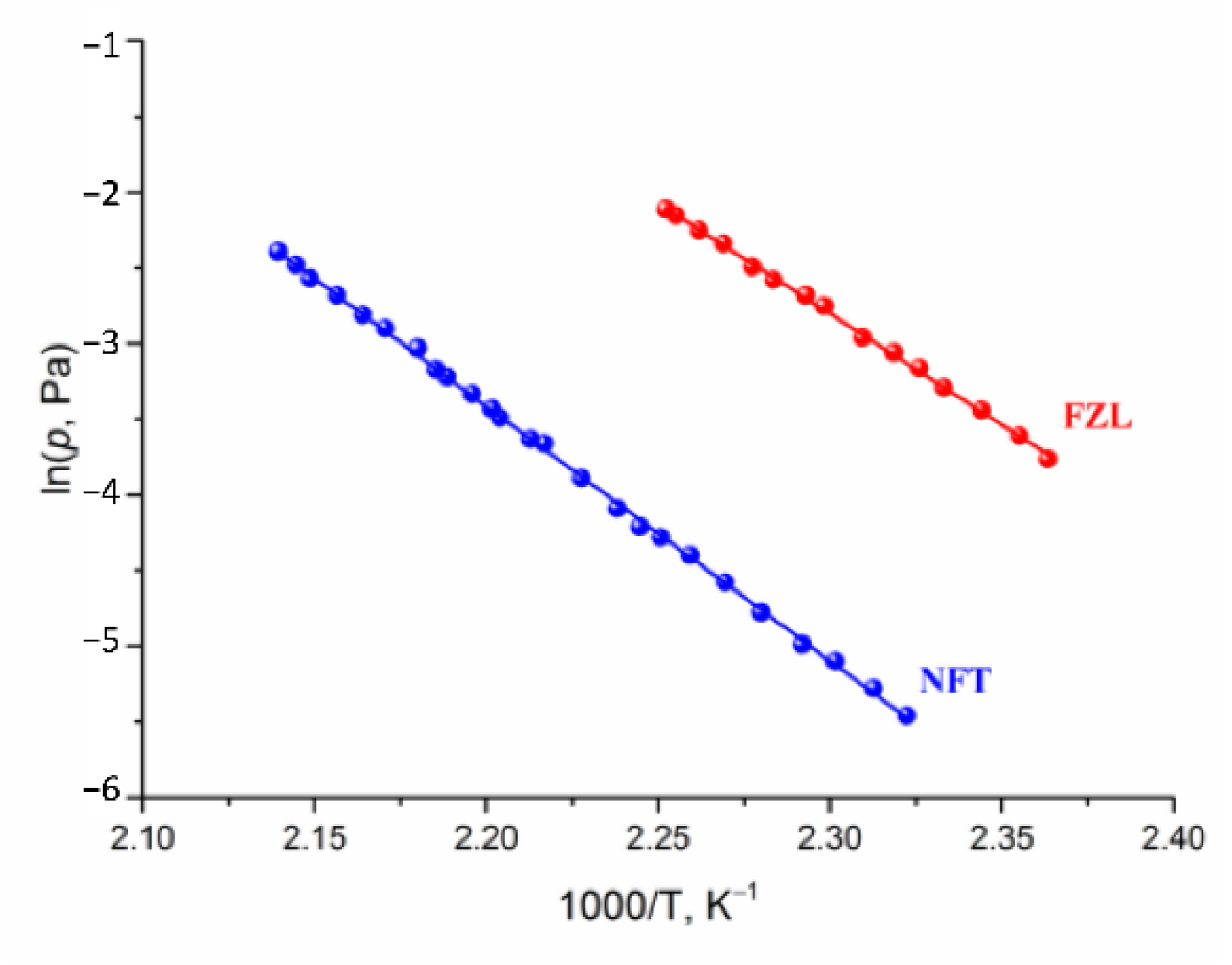
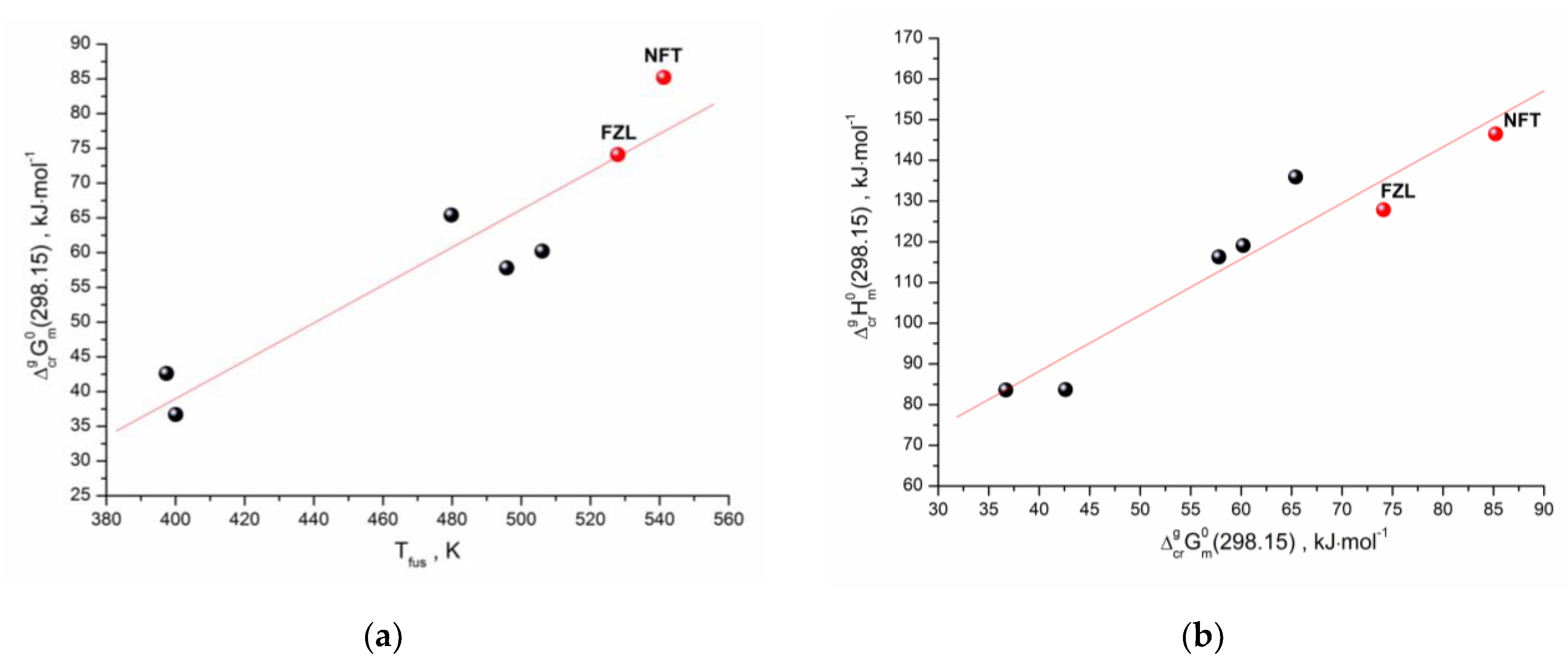
2.3. Vapor Pressure and Sublimation Thermodynamics
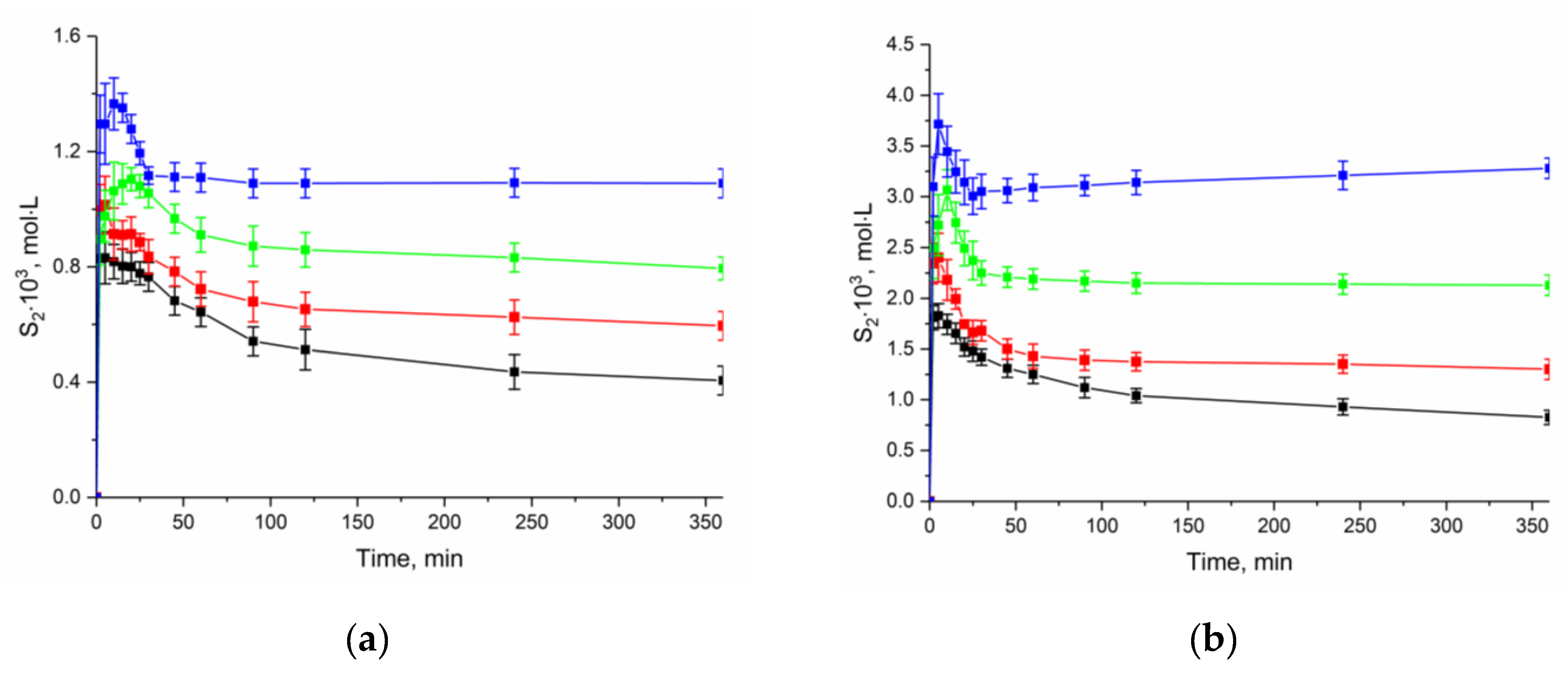
2.4. Solubility Study
3. Experimental Section
3.1. Materials
3.2. Powder X-Ray Diffraction (PXRD)
3.3. Differential Scanning Calorimetry (DSC)
3.4. DSC/TG/Mass-Spectrometry
3.5. Vapor Pressure Measurements
3.6. Single Crystal X-ray Diffraction (SCXRD)
3.7. Solubility Experiment
3.7.1. Equilibrium Solubility Determination
3.7.2. Dissolution Study
3.8. Computation Details
3.8.1. Solid-State DFT Calculations and Energy of Intermolecular Interactions
3.8.2. Van der Waals Volumes Calculation
3.8.3. Tanimoto Similarity Indices (Clusterization)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hutchings, M.I.; Truman, A.W.; Wilkinson, B. Antibiotics: Past, present and future. Curr. Opin. Microbiol. 2019, 51, 72–80. [Google Scholar] [CrossRef]
- Huttner, A.; Verhaegh, E.M.; Harbarth, S.; Muller, A.E.; Theuretzbacher, U.; Mouton, J.W. Nitrofurantoin revisited: A systematic review and meta-analysis of controlled trials. J. Antimicrob. Chemother. 2015, 70, 2456–2464. [Google Scholar] [CrossRef]
- World Health Organization. World Health Organization Model List of Essential Medicines: 21st List 2019; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- Guay, D.R. An Update on the Role of Nitrofurans in the Management of Urinary Tract Infections. Drugs 2001, 61, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Davila, M.J. Role of Old Antibiotics in the Era of Antibiotic Resistance. Highlighted Nitrofurantoin for the Treatment of Lower Urinary Tract Infections. Antibiotics 2014, 3, 39–48. [Google Scholar] [CrossRef]
- Hurdle, J.G.; Lee, R.B.; Budha, N.R.; Carson, E.I.; Qi, J.; Scherman, M.S.; Cho, S.H.; McNeil, M.R.; Lenaerts, A.J.; Franzblau, S.G.; et al. A microbiological assessment of novel nitrofuranylamides as anti-tuberculosis agents. J. Antimicrob. Chemother. 2008, 62, 1037–1045. [Google Scholar] [CrossRef]
- Jiang, X.; Sun, L.; Qiu, J.J.; Sun, X.; Li, S.; Wang, X.; So, C.W.E.; Dong, S. A Novel Application of Furazolidone: Anti-Leukemic Activity in Acute Myeloid Leukemia. PLoS ONE 2013, 8, e72335. [Google Scholar] [CrossRef] [PubMed]
- Sarvi, S.; Crispin, R.; Lu, Y.; Zeng, L.; Hurley, T.D.; Houston, D.R.; von Kriegsheim, A.; Chen, C.-H.; Mochly-Rosen, D.; Ranzani, M.; et al. ALDH1 Bio-activates Nifuroxazide to Eradicate ALDHHigh Melanoma-Initiating Cells. Cell Chem. Biol. 2018, 25, 1456–1469.e6. [Google Scholar] [CrossRef]
- Wilkinson, S.R.; Bot, C.; Kelly, J.M.; Hall, B.S. Trypanocidal activity of nitroaromatic prodrugs: Current treatments and future perspectives. Curr. Top. Med. Chem. 2011, 11, 2072–2084. [Google Scholar] [CrossRef]
- Perlovich, G.L.; Surov, A.O.; Bauer-Brandl, A. Thermodynamic properties of flufenamic and niflumic acids--specific and non-specific interactions in solution and in crystal lattices, mechanism of solvation, partitioning and distribution. J. Pharm. Biomed. Anal. 2007, 45, 679–687. [Google Scholar] [CrossRef]
- Ahrenberg, M.; Brinckmann, M.; Schmelzer, J.W.P.; Beck, M.; Schmidt, C.; Keßler, O.; Kragl, U.; Verevkin, S.P.; Schick, C. Determination of volatility of ionic liquids at the nanoscale by means of ultra-fast scanning calorimetry. Phys. Chem. Chem. Phys. 2014, 16, 2971–2980. [Google Scholar] [CrossRef] [PubMed]
- Abdelaziz, A.; Zaitsau, D.H.; Buzyurov, A.; Minakov, A.A.; Verevkin, S.P.; Schick, C. Fast scanning calorimetry: Sublimation thermodynamics of low volatile and thermally unstable compounds. Thermochim. Acta 2019, 676, 249–262. [Google Scholar] [CrossRef]
- Drozd, K.V.; Manin, A.N.; Voronin, A.P.; Perlovich, G.L. Sublimation thermodynamics of pyrazinoic, dipicolinic and quinolinic acids: Experiment and theoretical prediction. J. Chem. Thermodyn. 2021, 155, 106369. [Google Scholar] [CrossRef]
- Covaci, O.-I.; Mitran, R.-A.; Buhalteanu, L.; Dumitrescu, D.G.; Shova, S.; Manta, C.-M. Bringing new life into old drugs: A case study on nifuroxazide polymorphism. CrystEngComm 2017, 19, 3584–3591. [Google Scholar] [CrossRef]
- Alhalaweh, A.; George, S.; Basavoju, S.; Childs, S.L.; Rizvi, S.A.A.; Velaga, S.P. Pharmaceutical cocrystals of nitrofurantoin: Screening, characterization and crystal structure analysis. CrystEngComm 2012, 14, 5078–5088. [Google Scholar] [CrossRef]
- Vangala, V.R.; Chow, P.S.; Tan, R.B.H. Co-Crystals and Co-Crystal Hydrates of the Antibiotic Nitrofurantoin: Structural Studies and Physicochemical Properties. Cryst. Growth Des. 2012, 12, 5925–5938. [Google Scholar] [CrossRef]
- Pienaar, E.W.; Caira, M.R.; Lötter, A.P. Polymorphs of nitrofurantoin. 2. Preparation and X-ray crystal structures of two anhydrous forms of nitrofurantoin. J. Crystallogr. Spectrosc. Res. 1993, 23, 785–790. [Google Scholar] [CrossRef]
- Bandoli, G.; Ongaro, A.; Lotto, F.; Rossi, M. Crystal data of Nitrofurantoin C 8 H 6 N 4 O 5. Powder Diffr. 1993, 8, 107–108. [Google Scholar] [CrossRef]
- Wang, H.; Xiao, H.; Liu, N.; Zhang, B.; Shi, Q. Three New Compounds Derived from Nitrofurantoin: X-Ray Structures and Hirshfeld Surface Analyses. Open J. Inorg. Chem. 2015, 05, 63–73. [Google Scholar] [CrossRef]
- Pienaar, E.W.; Caira, M.R.; Lötter, A.P. Polymorphs of nitrofurantoin. I. Preparation and X-ray crystal structures of two monohydrated forms of nitrofurantoin. J. Crystallogr. Spectrosc. Res. 1993, 23, 739–744. [Google Scholar] [CrossRef]
- Yao, Z.-F.; Wang, J.-Y.; Pei, J. Control of π–π Stacking via Crystal Engineering in Organic Conjugated Small Molecule Crystals. Cryst. Growth Des. 2018, 18, 7–15. [Google Scholar] [CrossRef]
- Caira, M.R.; Pienaar, E.W.; Lötter, A.P. Polymorphism and Pseudopolymorphism of the Antibacterial Nitrofurantoin. Mol. Cryst. Liq. Cryst. Sci. Technol. Sect. A Mol. Cryst. Liq. Cryst. 1996, 279, 241–264. [Google Scholar] [CrossRef]
- Marian, E.; Tünde, J.; Kacso, I.; Miclaus, M.O.; Bratu, I. Interactions Analysis between Furazolidone and Excipients used in Pharmaceutical Forms. Rev. Chim. Bucharest Orig. Ed. 2013, 64, 1275–1278. [Google Scholar]
- Carvalho, S.G.; Cipriano, D.F.; de Freitas, J.C.C.; Junior, M.Â.S.; Ocaris, E.R.Y.; Teles, C.B.G.; de Jesus Gouveia, A.; Rodrigues, R.P.; Zanini, M.S.; Villanova, J.C.O. Physicochemical characterization and in vitro biological evaluation of solid compounds from furazolidone-based cyclodextrins for use as leishmanicidal agents. Drug Deliv. Transl. Res. 2020, 10, 1788–1809. [Google Scholar] [CrossRef]
- Perlovich, G.L.; Ryzhakov, A.M.; Tkachev, V.V.; Hansen, L.K.; Raevsky, O.A. Sulfonamide Molecular Crystals: Structure, Sublimation Thermodynamic Characteristics, Molecular Packing, Hydrogen Bonds Networks. Cryst. Growth Des. 2013, 13, 4002–4016. [Google Scholar] [CrossRef]
- Perlovich, G.L.; Raevsky, O.A. Sublimation of Molecular Crystals: Prediction of Sublimation Functions on the Basis of HYBOT Physicochemical Descriptors and Structural Clusterization. Cryst. Growth Des. 2010, 10, 2707–2712. [Google Scholar] [CrossRef]
- Otsuka, M.; Teraoka, R.; Matsuda, Y. Rotating-Disk Dissolution Kinetics of Nitrofurantoin Anhydrate and Monohydrate at Various Temperatures. Pharm. Res. 1992, 9, 307–311. [Google Scholar] [CrossRef]
- Chen, L.-K.; Cadwallader, D.E.; Jun, H.W. Nitrofurantoin solubility in aqueous urea and creatinine solutions. J. Pharm. Sci. 1976, 65, 868–872. [Google Scholar] [CrossRef] [PubMed]
- Domańska, U.; Pobudkowska, A.; Pelczarska, A.; Żukowski, Ł. Modelling, solubility and pKa of five sparingly soluble drugs. Int. J. Pharm. 2011, 403, 115–122. [Google Scholar] [CrossRef]
- Boetker, J.P.; Rantanen, J.; Arnfast, L.; Doreth, M.; Raijada, D.; Loebmann, K.; Madsen, C.; Khan, J.; Rades, T.; Müllertz, A.; et al. Anhydrate to hydrate solid-state transformations of carbamazepine and nitrofurantoin in biorelevant media studied in situ using time-resolved synchrotron X-ray diffraction. Eur. J. Pharm. Biopharm. 2016, 100, 119–127. [Google Scholar] [CrossRef]
- Brandenburg, J.G.; Maas, T.; Grimme, S. Benchmarking DFT and semiempirical methods on structures and lattice energies for ten ice polymorphs. J. Chem. Phys. 2015, 142, 124104. [Google Scholar] [CrossRef] [PubMed]
- Engineering ToolBox, 2010. Water-Heat of Vaporization. Available online: https://www.engineeringtoolbox.com/water-properties-d_1573.html (accessed on 29 March 2021).
- Koradia, V.; de Diego, H.L.; Elema, M.R.; Rantanen, J. Integrated approach to study the dehydration kinetics of nitrofurantoin monohydrate. J. Pharm. Sci. 2010, 99, 3966–3976. [Google Scholar] [CrossRef] [PubMed]
- Archer, D.; Rudtsch, S. Enthalpy of Fusion of Indium: A Certified Reference Material for Differential Scanning Calorimetry. J. Chem. Eng. Data 2003, 5, 1157–1163. [Google Scholar] [CrossRef]
- Zielenkiewicz, W.; Perlovich, G.L.; Wszelaka-Rylik, M. The Vapour Pressure and the Enthalpy of Sublimation: Determination by inert gas flow method. J. Therm. Anal. Calorim. 1999, 57, 225–234. [Google Scholar] [CrossRef]
- Blokhina, S.; Sharapova, A.; Ol’khovich, M.; Perlovich, G. Sublimation thermodynamics of four fluoroquinolone antimicrobial compounds. J. Chem. Thermodyn. 2017, 105, 37–43. [Google Scholar] [CrossRef]
- Clarke, E.C.W.; Glew, D.N. Evaluation of thermodynamic functions from equilibrium constants. Trans. Faraday Soc. 1966, 62, 539–547. [Google Scholar] [CrossRef]
- Chickos, J.S.; Acree, W.E. Enthalpies of Sublimation of Organic and Organometallic Compounds. 1910–2001. J. Phys. Chem. Ref. Data 2002, 31, 537–698. [Google Scholar] [CrossRef]
- Sheldrick, G. SADABS, Program for Scaling and Correction of Area Detector Data; University of Göttingen: Gottingen, Germany, 1997. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Blokhina, S.; Sharapova, A.; Ol’khovich, M.; Perlovich, G. A thermodynamic study of sublimation, dissolution and distribution processes of anti-inflammatory drug Clonixin. J. Chem. Thermodyn. 2019, 132, 281–288. [Google Scholar] [CrossRef]
- Dovesi, R.; Erba, A.; Orlando, R.; Zicovich-Wilson, C.M.; Civalleri, B.; Maschio, L.; Rérat, M.; Casassa, S.; Baima, J.; Salustro, S.; et al. Quantum-mechanical condensed matter simulations with CRYSTAL. WIREs Comput. Mol. Sci. 2018, 8, e1360. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Pascale, F.; Zicovich-Wilson, C.M.; López Gejo, F.; Civalleri, B.; Orlando, R.; Dovesi, R. The calculation of the vibrational frequencies of crystalline compounds and its implementation in the CRYSTAL code. J. Comput. Chem. 2004, 25, 888–897. [Google Scholar] [CrossRef] [PubMed]
- Gatti, C.; Casassa, S.M. TOPOND14 User’s Manual. 2013. Available online: https://iris.unito.it/handle/2318/152446#.YLb-YqERVPY (accessed on 4 June 2021).
- Mata, I.; Alkorta, I.; Espinosa, E.; Molins, E. Relationships between interaction energy, intermolecular distance and electron density properties in hydrogen bonded complexes under external electric fields. Chem. Phys. Lett. 2011, 507, 185–189. [Google Scholar] [CrossRef]
- Gavezzotti, A.; Filippini, G. Energetic aspects of crystal packing: Experiment and computer simulations. In Theoretical Aspects and Computer Modeling of the Molecular Solid State, 1st ed.; Gavezzotti, A., Ed.; John Wiley & Sons: New York, NY, USA, 1997; pp. 61–97. [Google Scholar]
- Voronin, A.P.; Perlovich, G.L.; Vener, M. V Effects of the crystal structure and thermodynamic stability on solubility of bioactive compounds: DFT study of isoniazid cocrystals. Comput. Theor. Chem. 2016, 1092, 1–11. [Google Scholar] [CrossRef]
- Pascual-Ahuir, J.L.; Silla, E. GEPOL: An improved description of molecular surfaces. I. Building the spherical surface set. J. Comput. Chem. 1990, 11, 1047–1060. [Google Scholar] [CrossRef]
- Kitaigorodsky, A.J. Molecular Crystals and Molecules, 1st ed.; Kitaigorodsky, A.J., Ed.; Academic Press: London, UK, 1973; Volume 29, 570p. [Google Scholar]
- Raevsky, O.A.; Grigor’ev, V.J.; Trepalin, S.V. HYBOT Program Package. Russ. Patent No. 990090, 26 February 1999. [Google Scholar]
- Raevsky, O.A.; Gerasimenko, V.A.; Trepalin, S.V. MOLDIVS (MOLecular DIVersity& Similarity) Program Package. Russ. Patent No. 990093, 26 February 1999. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Empirical Formula | C8H7N3O5 |
|---|---|
| Formula weight | 225.17 |
| Crystal system | Orthorhombic |
| Space group | Pnma |
| a (Å) | 22.7977(13) |
| b (Å) | 6.2422(3) |
| c (Å) | 6.5085(5) |
| Unit cell volume (Å3) | 926.21(10) |
| Z | 4 |
| Calc. density (g·cm−3) | 1.615 |
| Absorption coefficient (mm−1) | 0.137 |
| θ range (deg) | 1.79–26.00 |
| No. of measured, independent (Rint) and observed reflections | 3737, 988 (0.069), 816 |
| Final R indices [I > 2σ(I)] | R1 = 0.0416, wR2 = 0.1024 |
| Final R indices (all data) | R1 = 0.0519, wR2 = 0.1079 |
| Goodness of fit | 1.039 |
| max/min Δρ (e·Å−3) | 0.188/−0.245 |
| NFT | FZL | |
|---|---|---|
| Elatt(0K) | 139.6 | 121.9 |
| Molecules in chain | 33.8 (24.2%) | 28.5 (23.4%) |
| Chains in layer | 40.7 (29.2%) | 18.6 (15.3%) |
| Layers in crystal | 65.2 (46.7%) | 74.8 (61.3%) |
| E (N−H···O) | 27.9 (20.0%) | 0 (0%) |
| ΣE (C−H···X) | 52.9 (37.9%) | 55.4 (45.4%) |
| ΣE (π···π) | 58.8 (42.1%) | 60.0 (49.2%) |
| ΣE (H···H) | 0 (0%) | 6.6 (5.4%) |
| NFT | FZL | |
|---|---|---|
| p(298.15K), Pa | 1.19·10−10 | 1.12·10−8 |
| , kJ·mol−1 | 85.2 | 74.1 |
| , kJ·mol−1 | 139.9 ± 0.7 | 122.0 ± 1.1 |
| a, J·mol−1·K−1 | 287.8 | 286.4 |
| , kJ·mol−1 | 146.5 ± 0.7 | 127.9 ± 1.1 |
| , J·mol−1·K−1 | 205.5 ± 2.8 | 180.6 ± 4.6 |
| , kJ·mol−1 | 61.3 | 53.9 |
| b, % | 70.5 | 70.4 |
| b, % | 29.5 | 29.6 |
| cβ, % | 36.3 | 37.5 |
| Compound | T, K | Buffer pH 2.0 X2·105 (S2·103) | Buffer pH 7.4 X2·105 (S2·103) | 1-octanol X2·105 (S2·104) |
|---|---|---|---|---|
| NFT | 298.15 | 1.49 (0.83) | 3.28 (1.82) | 3.61 (2.30) |
| 303.15 | 1.82 (1.01) | 4.33 (2.40) | 4.97 (3.17) | |
| 308.15 | 2.26 (1.25) | 5.51(3.06) | 6.53 (4.16) | |
| 313.15 | 2.46 (1.37) | 6.72(3.73) | 8.26 (5.26) | |
| [NFT + H2O] | 298.15 | 0.73 (0.40) | 1.49 (0.83) | - |
| 303.15 | 1.07 (0.59) | 2.34 (1.30) | - | |
| 308.15 | 1.43 (0.79) | 3.83 (2.13) | - | |
| 313.15 | 1.96 (1.08) | 5.90 (3.28) | - | |
| FZL | 298.15 | 3.35 (1.86) | 3.31 (1.84) | 1.13 (0.72) |
| 303.15 | 4.28 (2.38) | 4.27 (2.37) | 1.70 (1.09) | |
| 308.15 | 5.40 (3.00) | 5.43 (3.02) | 2.12 (1.59) | |
| 313.15 | 7.62 (4.23) | 7.65 (4.25) | 2.76 (2.24) |
| Solvent | X2 (mol.frac.) | % | % | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| NFT | |||||||||||
| pH 2.0 | 1.49·105 | 27.5 | 26.6 ± 2.8 | −0.9 | −3.1 ± 3.7 | −57.7 | −119.9 | −62.2 | −208.6 | 65.8 | 34.2 |
| pH 7.4 | 3.28·105 | 25.6 | 37.5 ± 1.9 | 11.9 | 39.9 ± 2.7 | −59.6 | −109.0 | −49.4 | −165.6 | 68.8 | 31.2 |
| 1-octanol | 3.61·105 | 25.4 | 42.8 ± 1.7 | 17.5 | 58.5 ± 2.5 | −59.8 | −103.7 | −43.8 | −147.0 | 70.3 | 29.7 |
| [NFT+H2O] | |||||||||||
| pH 2.0 | 0.73·105 | 29.3 | 50.5 ± 1.7 | 21.1 | 70.9 ± 2.6 | - | - | - | - | - | - |
| pH 7.4 | 1.98·105 | 26.8 | 71.8 ± 1.2 | 44.3 | 148.5 ± 2.0 | - | - | - | - | - | - |
| FZL | |||||||||||
| pH 2.0 | 3.35·106 | 31.3 | 41.8 ± 3.2 | 10.5 | 35.4 ± 4.0 | −42.8 | −86.1 | −43.3 | −145.2 | 66.5 | 33.5 |
| pH 7.4 | 3.31·106 | 31.3 | 42.8 ± 2.8 | 11.5 | 38.4 ± 4.0 | −42.8 | −85.1 | −42.4 | −142.2 | 66.7 | 33.3 |
| 1-octanol | 1.13·105 | 27.7 | 49.4 ± 4.4 | 21.7 | 72.6 ± 5.2 | −46.4 | −78.5 | −32.2 | −108.0 | 70.9 | 29.1 |
| Compound | CAS Register No. | Source | Mass Fraction Purity * |
|---|---|---|---|
| Nitrofurantoin | 67-20-9 | Sigma-Aldrich | ≥0.98 |
| Furazolidone | 67-45-8 | Sigma-Aldrich | ≥0.98 |
| 1-Octanol | 111-87-5 | Sigma-Aldrich | ≥0.99 |
| Potassium dihydrogen phosphate | 7778-77-0 | Merck | ≥0.99 |
| Disodium hydrogen phosphate dodecahydrate | 10039-32-4 | Merck | ≥0.99 |
| Potassium chloride | 7447-40-7 | Aldrich | ≥0.99 |
| Hydrochloric acid 0.1 mol·dm−3 fixanal | 7647-01-0 | Aldrich | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manin, A.N.; Drozd, K.V.; Voronin, A.P.; Churakov, A.V.; Perlovich, G.L. A Combined Experimental and Theoretical Study of Nitrofuran Antibiotics: Crystal Structures, DFT Computations, Sublimation and Solution Thermodynamics. Molecules 2021, 26, 3444. https://doi.org/10.3390/molecules26113444
Manin AN, Drozd KV, Voronin AP, Churakov AV, Perlovich GL. A Combined Experimental and Theoretical Study of Nitrofuran Antibiotics: Crystal Structures, DFT Computations, Sublimation and Solution Thermodynamics. Molecules. 2021; 26(11):3444. https://doi.org/10.3390/molecules26113444
Chicago/Turabian StyleManin, Alex N., Ksenia V. Drozd, Alexander P. Voronin, Andrei V. Churakov, and German L. Perlovich. 2021. "A Combined Experimental and Theoretical Study of Nitrofuran Antibiotics: Crystal Structures, DFT Computations, Sublimation and Solution Thermodynamics" Molecules 26, no. 11: 3444. https://doi.org/10.3390/molecules26113444
APA StyleManin, A. N., Drozd, K. V., Voronin, A. P., Churakov, A. V., & Perlovich, G. L. (2021). A Combined Experimental and Theoretical Study of Nitrofuran Antibiotics: Crystal Structures, DFT Computations, Sublimation and Solution Thermodynamics. Molecules, 26(11), 3444. https://doi.org/10.3390/molecules26113444