
Compression-Complexity Measures for Analysis and Classification of Coronaviruses

 ,
, 
Abstract
1. Introduction
- Identifying various genes that constitute the genome.
- Identifying the origin of the genome sequence.
- Understanding the information content present in the coding and non-coding regions.
- Reconstructing the phylogenetic tree to study evolutionary patterns.
- Automatic classification and identification of unknown genome sequences.
2. Genomic Sequences and Comparison
2.1. Genome and Gene
2.2. Genome Sequence Comparison
- Alignment-free methods: These are computationally less intensive methods that consider the genome sequences as character strings and use distance-based methods involving frequency and distribution of bases [8,9,10,11,12]. Our focus in this paper is on alignment-free methodology, especially on using compression-complexity measures for sequence comparisons.
3. Materials and Methods
3.1. Genome Sequences Used in This Study
3.1.1. Mammalian Sequences
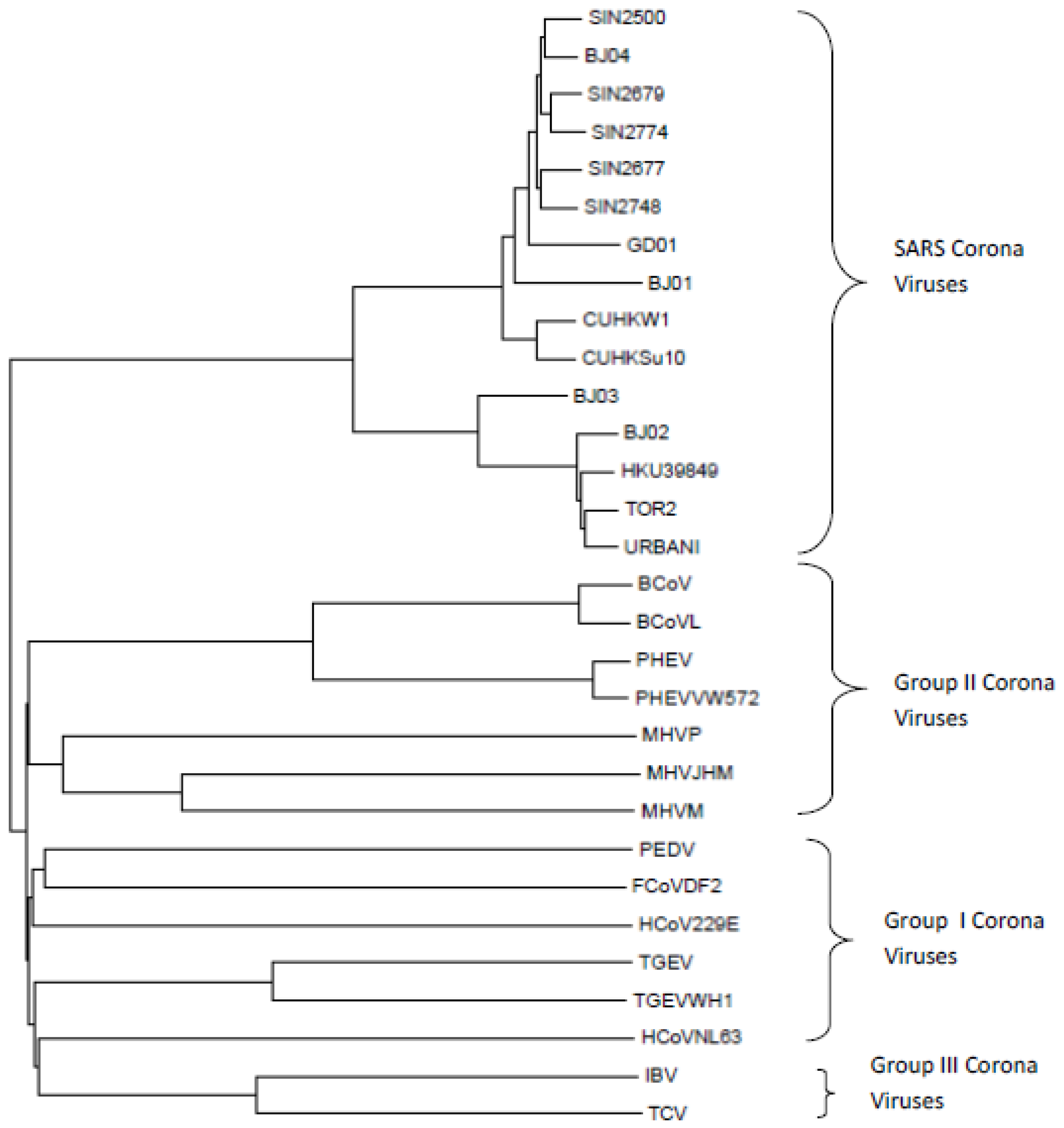
3.1.2. Coronaviruses (SARS-CoV-1)
- 15 SARS-CoV-1 coronaviruses
- 15 Non-SARS-CoV-1 coronaviruses belonging to Groups I, II and III coronaviruses
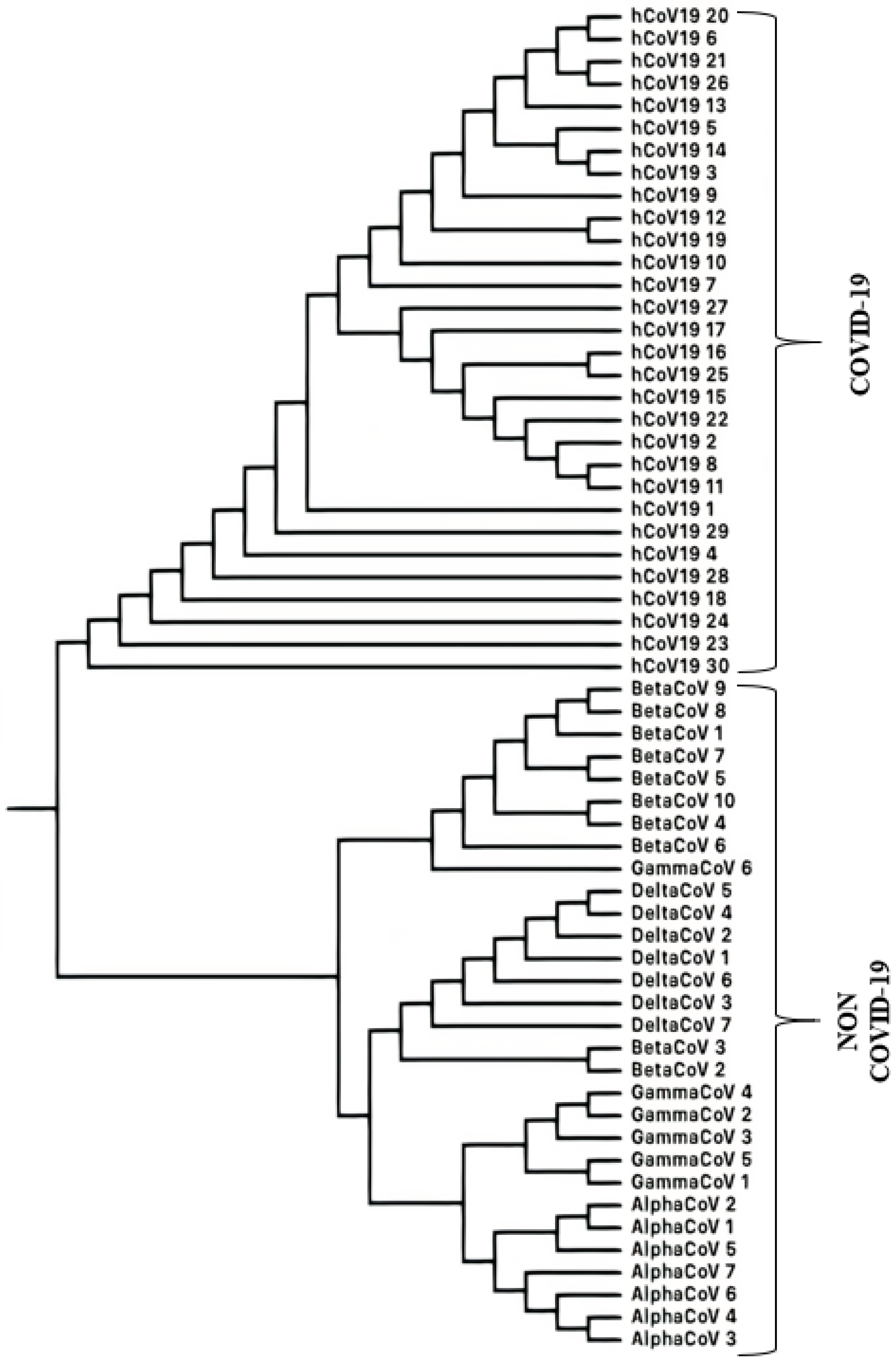
3.1.3. SARS-CoV-2 (COVID-19 Causing Corona Viruses)
3.2. Mathematical and Computational Methods Used in This Study
3.2.1. Compression Complexity Measures: Lempel–Ziv (LZ) and Effort-to-Compress (ETC)
3.2.2. Distance Measure
3.2.3. Machine Learning Algorithms Used in the Study
4. Results and Discussion
Classification of SARS-CoV-2 Sequences
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ETC | Effort-to-Compress complexity |
| LZC | Lempel ziv complexity |
| LSVM | Linear Support Vector Machine |
| QSVM | Quadratic Support Vector Machine |
| LD | Linear Discriminant |
| FKNN | Fine K-Nearest Neighbors |
References
- Lebatteux, D.; Remita, A.M.; Diallo, A.B. Toward an alignment-free method for feature extraction and accurate classification of viral sequences. J. Comput. Biol. 2019, 26, 519–535. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xue, X.; Xie, X. An alignment-free measure based on physicochemical properties of amino acids for protein sequence comparison. Comput. Biol. Chem. 2019, 80, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Lesk, A. Introduction to genomics; Oxford University Press: Oxford, UK, 2012. [Google Scholar]
- Pearson, W.R. An introduction to sequence similarity (“homology”) searching. Curr. Protoc. Bioinform. 2013, 42, 3.1.1–3.1.8. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.K.; Niyogi, R.; Misra, M. A framework for alignment-free methods to perform similarity analysis of biological sequence. In Proceedings of the Sixth International Conference on Contemporary Computing (IC3), Noida, India, 8–10 August 2013; pp. 337–342. [Google Scholar]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Zielezinski, A.; Vinga, S.; Almeida, J.; Karlowski, W.M. Alignment-free sequence comparison: Benefits, applications, and tools. Genome Biol. 2017, 18, 186. [Google Scholar] [CrossRef]
- Xia, X. Distance-Based Phylogenetic Methods. In Bioinformatics and the Cell; Springer: Berlin/Heidelberg, Germany, 2018; pp. 343–379. [Google Scholar]
- Zielezinski, A.; Girgis, H.Z.; Bernard, G.; Leimeister, C.A.; Tang, K.; Dencker, T.; Lau, A.K.; Röhling, S.; Choi, J.J.; Waterman, M.S.; et al. Benchmarking of alignment-free sequence comparison methods. Genome Biol. 2019, 20, 144. [Google Scholar] [CrossRef]
- Monge, R.E.; Crespo, J.L. Analysis of data complexity in human dna for gene-containing zone prediction. Entropy 2015, 17, 1673–1689. [Google Scholar] [CrossRef]
- Dehghanzadeh, H.; Ghaderi-Zefrehei, M.; Mirhoseini, S.Z.; Esmaeilkhaniyan, S.; Haruna, I.L.; Najafabadi, H.A. A new DNA sequence entropy-based Kullback–Leibler algorithm for gene clustering. J. Appl. Genet. 2020, 61, 231–238. [Google Scholar] [CrossRef]
- Otu, H.H.; Sayood, K. A new sequence distance measure for phylogenetic tree construction. Bioinformatics 2003, 19, 2122–2130. [Google Scholar] [CrossRef]
- Grumbach, S.; Tahi, F. A new challenge for compression algorithms: Genetic sequences. Inf. Process. Manag. 1994, 30, 875–886. [Google Scholar] [CrossRef]
- Varr, J.; Delahaye, J.P.; Rivals, E. Transformation distances: A family of dissimilarity measures based on movements of segments. Bioinformatics 1999, 15, 194–202. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, M.; Badger, J.H.; Chen, X.; Kwong, S.; Kearney, P.; Zhang, H. An information-based sequence distance and its application to whole mitochondrial genome phylogeny. Bioinformatics 2001, 17, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Kwong, S.; Li, M. A compression algorithm for DNA sequences and its applications in genome comparison. In Proceedings of the Fourth Annual International Conference on Computational Molecular Biology, Tokyo, Japan, 8–11 April 2000; ACM: New York, NY, USA, 2000; p. 107. [Google Scholar]
- Ming, L.; Vitányi, P.M. Kolmogorov complexity and its applications. Algorithms Complex. 2014, 1, 187. [Google Scholar]
- Cover, T.M.; Thomas, J.A. Elements of Information Theory; John Wiley & Sons: Hoboken, NJ, USA, 2012. [Google Scholar]
- Lempel, A.; Ziv, J. On the complexity of finite sequences. IEEE Trans. Inf. Theory 1976, 22, 75–81. [Google Scholar] [CrossRef]
- Liu, N.; Wang, T.m. A relative similarity measure for the similarity analysis of DNA sequences. Chem. Phys. Lett. 2005, 408, 307–311. [Google Scholar] [CrossRef]
- Zhang, Y.; Hao, J.; Zhou, C.; Chang, K. Normalized Lempel-Ziv complexity and its application in bio-sequence analysis. J. Math. Chem. 2009, 46, 1203–1212. [Google Scholar] [CrossRef]
- Li, B.; Li, Y.B.; He, H.B. LZ complexity distance of DNA sequences and its application in phylogenetic tree reconstruction. Genom. Proteomics Bioinform. 2005, 3, 206–212. [Google Scholar] [CrossRef]
- Liu, L.; Li, D.; Bai, F. A relative Lempel-Ziv complexity: Application to comparing biological sequences. Chem. Phys. Lett. 2012, 530, 107–112. [Google Scholar] [CrossRef]
- Yu, C.; He, R.L.; Yau, S.S.T. Viral genome phylogeny based on Lempel–Ziv complexity and Hausdorff distance. J. Theor. Biol. 2014, 348, 12–20. [Google Scholar] [CrossRef]
- Song, Y.J.; Cho, D.H. Classification of various genomic sequences based on distribution of repeated k-word. In Proceedings of the 2017 39th Annual International Conference of the IEEE Engineering in Medicine and Biology Society (EMBC), Jeju, Republic of Korea, 11–15 July 2017; IEEE: Piscataway, NJ, USA, 2017; pp. 3894–3897. [Google Scholar]
- Monge, R.E.; Crespo, J.L. Comparison of complexity measures for DNA sequence analysis. In Proceedings of the International Work Conference on Bio-inspired Intelligence (IWOBI), Liberia, Costa Rica, 16–18 July 2014; pp. 71–75. [Google Scholar]
- Sayood, K.; Otu, H.H.; Hinrichs, S.H. System and Method for Sequence Distance Measure for Phylogenetic Tree Construction. US Patent 8,725,419, 13 May 2014. [Google Scholar]
- Wu, T.J.; Hsieh, Y.C.; Li, L.A. Statistical measures of DNA sequence dissimilarity under Markov chain models of base composition. Biometrics 2001, 57, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Blaisdell, B.E. A measure of the similarity of sets of sequences not requiring sequence alignment. Proc. Natl. Acad. Sci. USA 1986, 83, 5155–5159. [Google Scholar] [CrossRef]
- Bzhalava, Z.; Hultin, E.; Dillner, J. Extension of the viral ecology in humans using viral profile hidden Markov models. PLoS ONE 2018, 13, e0190938. [Google Scholar] [CrossRef]
- Pham, T.D.; Zuegg, J. A probabilistic measure for alignment-free sequence comparison. Bioinformatics 2004, 20, 3455–3461. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Deng, M.; Yau, S.S.T. DNA sequence comparison by a novel probabilistic method. Inf. Sci. 2011, 181, 1484–1492. [Google Scholar] [CrossRef]
- Omari, M.; Barrus, T.W.; Sanders, M.; Negron, D. Rapid Genomic Sequence Classification Using Probabilistic Data Structures. US Patent App. 15/977,667, 15 November 2018. [Google Scholar]
- Li, Y.; He, L.; He, R.L.; Yau, S.S.T. A novel fast vector method for genetic sequence comparison. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, N.; Balasubramanian, K.; Dey, S. A new complexity measure for time series analysis and classification. Eur. Phys. J. Spec. Top. 2013, 222, 847–860. [Google Scholar] [CrossRef]
- Balasubramanian, K.; Nagaraj, N. Aging and cardiovascular complexity: Effect of the length of RR tachograms. PeerJ 2016, 4, e2755. [Google Scholar] [CrossRef]
- Nagaraj, N.; Balasubramanian, K. Dynamical complexity of short and noisy time series. Eur. Phys. J. Spec. Top. 2017, 226, 2191–2204. [Google Scholar] [CrossRef]
- Thanaj, M.; Chipperfield, A.J.; Clough, G.F. Complexity-Based Analysis of Microvascular Blood Flow in Human Skin. In Physics of Biological Oscillators: New Insights into Non-Equilibrium and Non-Autonomous Systems; Springer: Cham, Switzerland, 2021; p. 291. [Google Scholar]
- Thanaj, M.; Chipperfield, A.J.; Clough, G.F. Multiscale analysis of microvascular blood flow and oxygenation. In Proceedings of the World Congress on Medical Physics and Biomedical Engineering 2018, Prague, Czech Republic, 3–8 June 2019; Springer: Berlin/Heidelberg, Germany, 2019; pp. 195–200. [Google Scholar]
- Albahri, A.; Hamid, R.A.; Alwan, J.K.; Al-Qays, Z.; Zaidan, A.; Zaidan, B.; Albahri, A.; AlAmoodi, A.; Khlaf, J.M.; Almahdi, E.; et al. Role of biological data mining and machine learning techniques in detecting and diagnosing the novel coronavirus (COVID-19): A systematic review. J. Med. Syst. 2020, 44, 1–11. [Google Scholar] [CrossRef]
- Callejon-Leblic, M.A.; Moreno-Luna, R.; Del Cuvillo, A.; Reyes-Tejero, I.M.; Garcia-Villaran, M.A.; Santos-Peña, M.; Maza-Solano, J.M.; Martín-Jimenez, D.I.; Palacios-Garcia, J.M.; Fernandez-Velez, C.; et al. Loss of smell and taste can accurately predict COVID-19 infection: A machine-learning approach. J. Clin. Med. 2021, 10, 570. [Google Scholar] [CrossRef] [PubMed]
- Arun, S.S.; Iyer, G.N. On the Analysis of COVID19-Novel Corona Viral Disease Pandemic Spread Data Using Machine Learning Techniques. In Proceedings of the 2020 4th International Conference on Intelligent Computing and Control Systems (ICICCS), Madurai, India, 13–15 May 2020; IEEE: Piscataway, NJ, USA, 2020; pp. 1222–1227. [Google Scholar]
- Anand, R.; Sowmya, V.; Menon, V.; Gopalakrishnan, A.; Soman, K. Modified VGG deep-learning architecture for COVID-19 classification using chest radiography images. Biomed. Biotechnol. Res. J. (BBRJ) 2021, 5, 43. [Google Scholar]
- Hari Prakash, S.; Adithya Narayan, K.; Nair, G.S.; Harikumar, S. Perceiving Machine Learning Algorithms to Analyze COVID-19 Radiographs. In Proceedings of International Conference on Recent Trends in Computing; Springer: Singapore, 2022; pp. 293–305. [Google Scholar]
- Choudary, M.N.S.; Bommineni, V.B.; Tarun, G.; Reddy, G.P.; Gopakumar, G. Predicting COVID-19 Positive Cases and Analysis on the Relevance of Features using SHAP (SHapley Additive exPlanation). In Proceedings of the 2021 Second International Conference on Electronics and Sustainable Communication Systems (ICESC), Coimbatore, India, 4–6 August 2021; IEEE: Piscataway, NJ, USA, 2021; pp. 1892–1896. [Google Scholar]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| ML Methods | Accuracy | Precision | Sensitivity | Specificity | F1-Score |
|---|---|---|---|---|---|
| LSVM | 89 | 0.98 | 0.82 | 0.98 | 0.89 |
| QSVM | 90 | 1 | 0.82 | 1 | 0.90 |
| LD | 86 | 1 | 0.80 | 1 | 0.88 |
| FKNN | 92 | 0.96 | 0.89 | 0.96 | 0.92 |
| ML Methods | Accuracy | Precision | Sensitivity | Specificity | F1-Score |
|---|---|---|---|---|---|
| LSVM | 80 | 0.74 | 0.90 | 0.70 | 0.81 |
| QSVM | 83 | 0.84 | 0.74 | 0.89 | 0.79 |
| LD | 84 | 0.81 | 0.85 | 0.83 | 0.83 |
| FKNN | 88 | 0.95 | 0.79 | 0.96 | 0.86 |
| ML Methods | Accuracy | Precision | Sensitivity | Specificity | F1-Score |
|---|---|---|---|---|---|
| LSVM | 92 | 0.98 | 0.89 | 0.97 | 0.93 |
| QSVM | 95 | 1 | 0.90 | 1 | 0.95 |
| LD | 87 | 1 | 0.79 | 1 | 0.88 |
| FKNN | 98 | 1 | 0.96 | 1 | 0.98 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Munagala, N.V.T.S.; Amanchi, P.K.; Balasubramanian, K.; Panicker, A.; Nagaraj, N. Compression-Complexity Measures for Analysis and Classification of Coronaviruses. Entropy 2023, 25, 81. https://doi.org/10.3390/e25010081
Munagala NVTS, Amanchi PK, Balasubramanian K, Panicker A, Nagaraj N. Compression-Complexity Measures for Analysis and Classification of Coronaviruses. Entropy. 2023; 25(1):81. https://doi.org/10.3390/e25010081
Chicago/Turabian StyleMunagala, Naga Venkata Trinath Sai, Prem Kumar Amanchi, Karthi Balasubramanian, Athira Panicker, and Nithin Nagaraj. 2023. "Compression-Complexity Measures for Analysis and Classification of Coronaviruses" Entropy 25, no. 1: 81. https://doi.org/10.3390/e25010081
APA StyleMunagala, N. V. T. S., Amanchi, P. K., Balasubramanian, K., Panicker, A., & Nagaraj, N. (2023). Compression-Complexity Measures for Analysis and Classification of Coronaviruses. Entropy, 25(1), 81. https://doi.org/10.3390/e25010081