Non-Equilibrium Thermodynamics and Stochastic Dynamics of a Bistable Catalytic Surface Reaction



Abstract
1. Introduction
2. The Bistable Catalytic Reaction Model
3. Theoretical Framework
3.1. Mean-Field Stochastic Description
Stochastic Entropy Production Rate
3.2. Deterministic Mean-Field Description
3.3. Macroscopic Entropy Production Rate
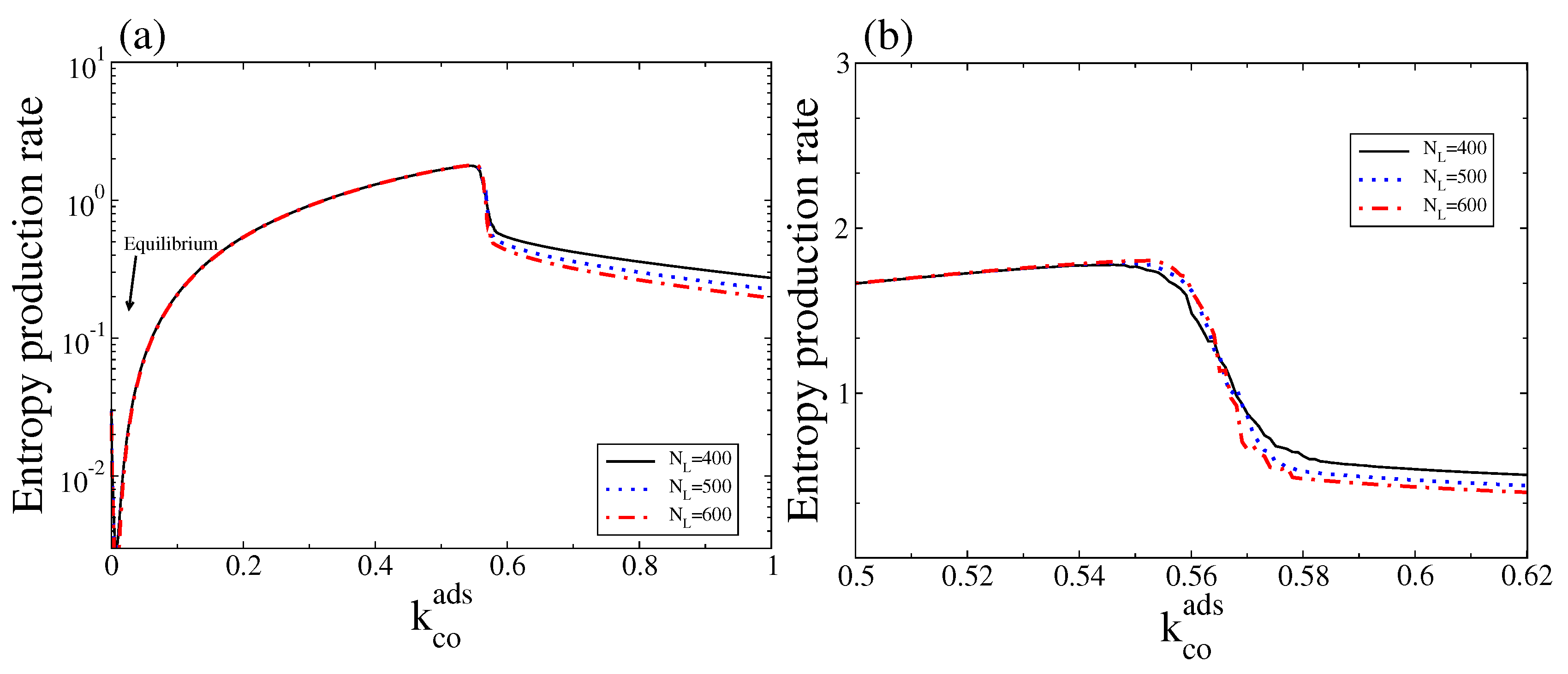
4. Results and Discussion
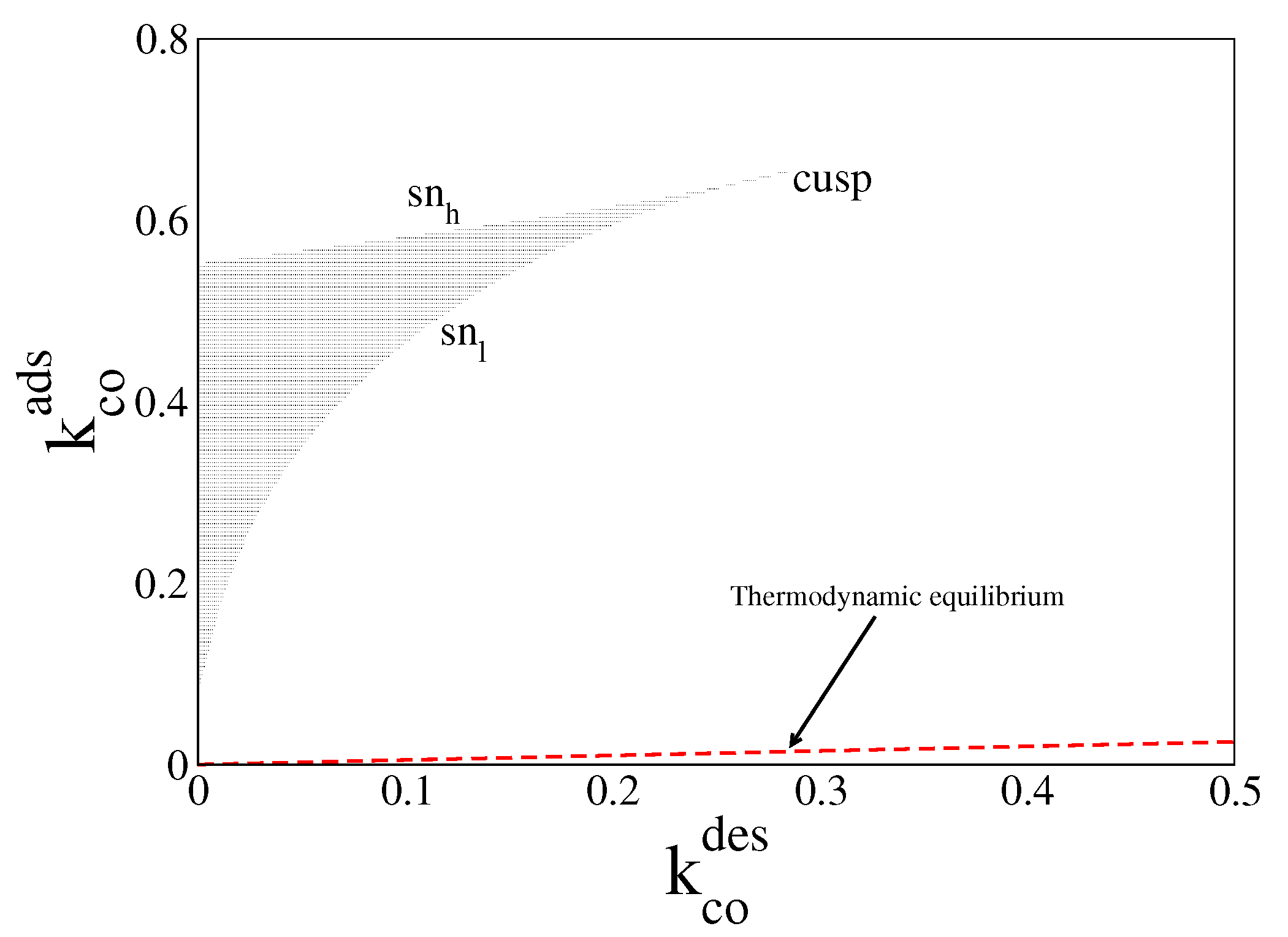
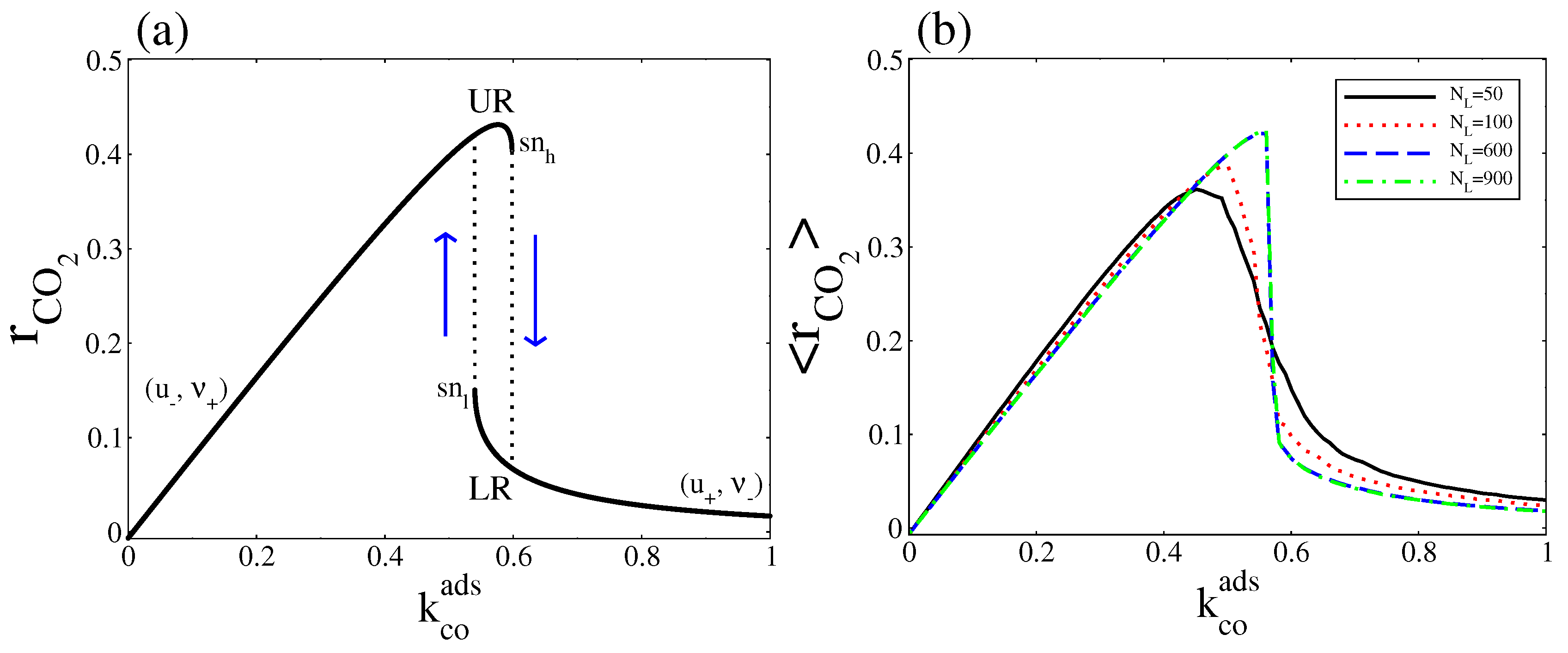
4.1. Deterministic Analysis
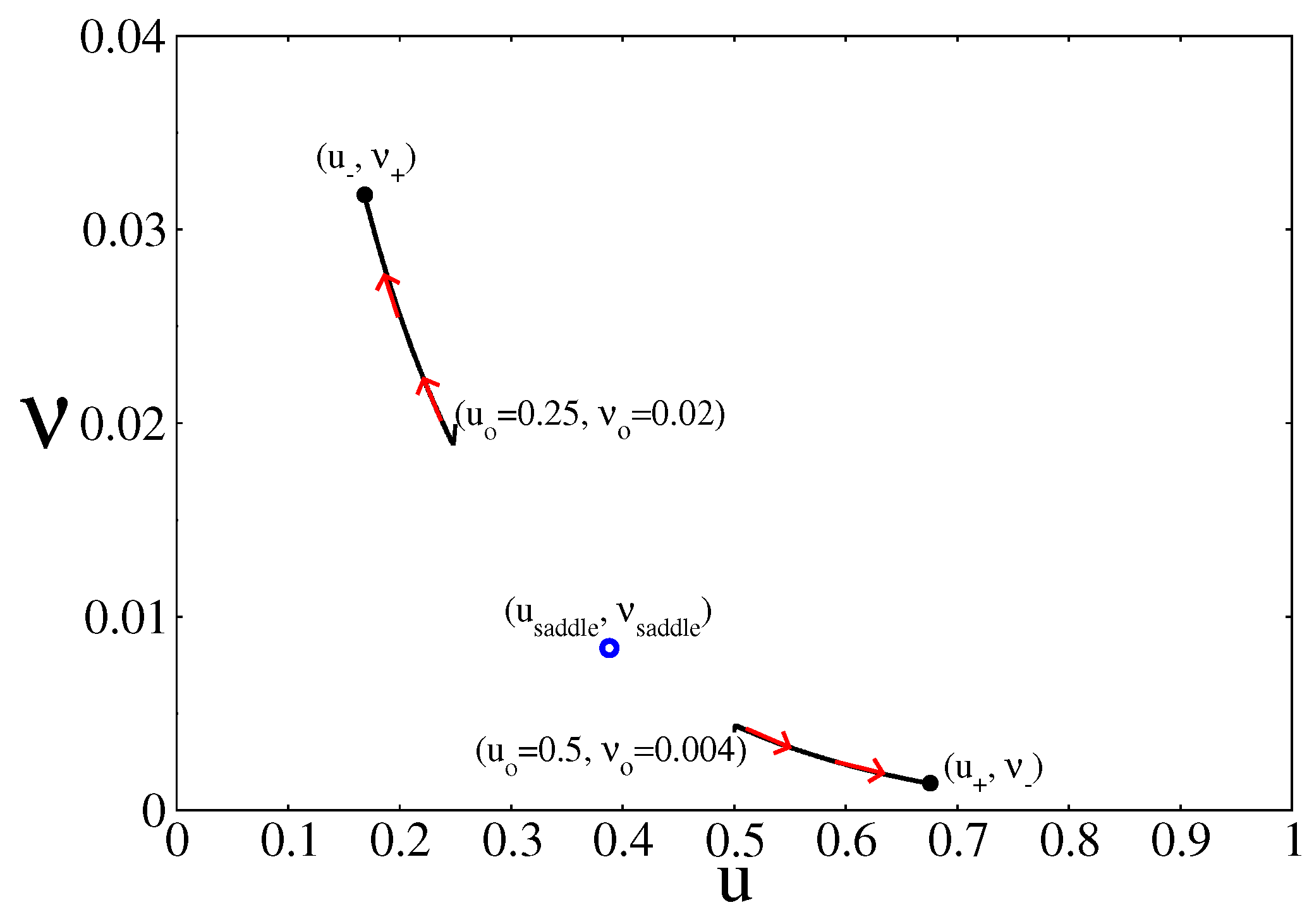
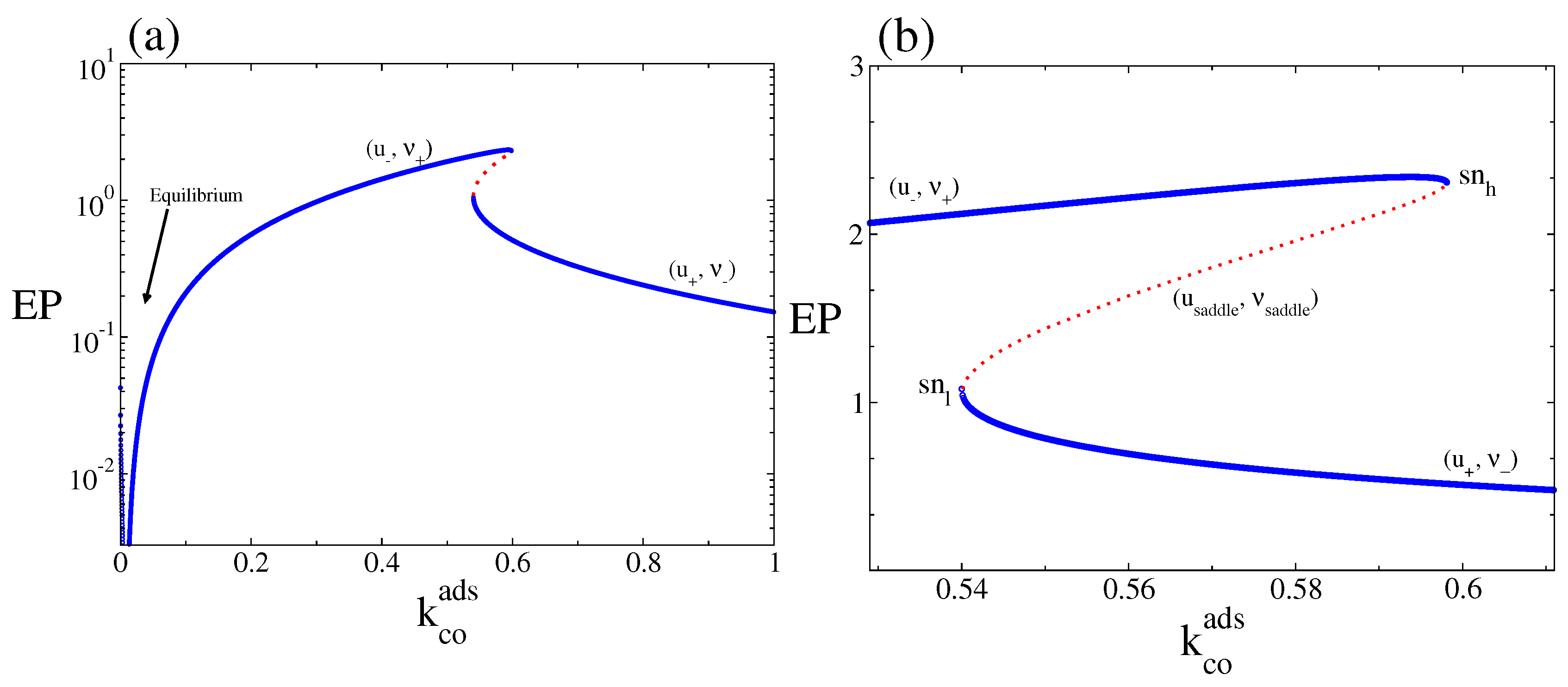
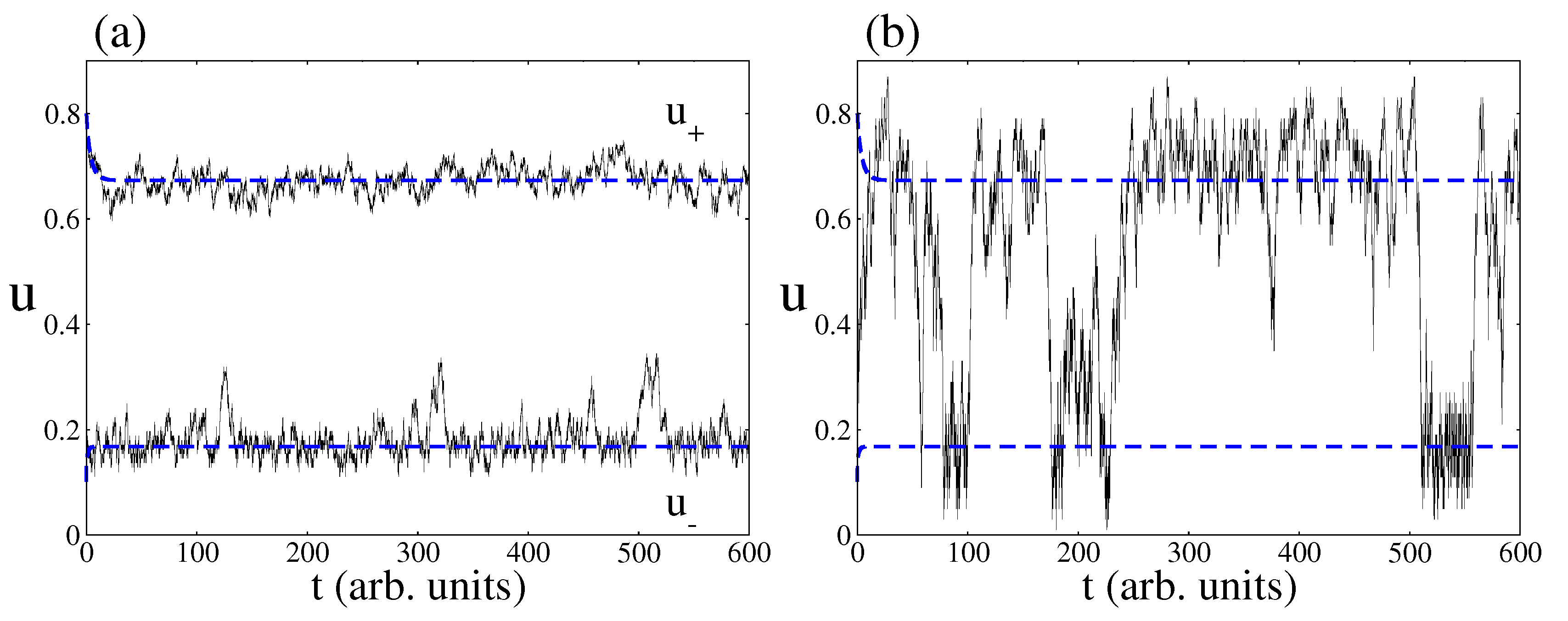
4.2. Stochastic Analysis
5. Overall CO2 Production Rate
6. Summary and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| NESS | Non-equilibrium steady state |
| ODEs | Ordinary differential equations |
| CO | Carbon monoxide |
| CO2 | Carbon dioxide |
| O2 | Oxygen |
| LH | Langmuir-Hinshelwood |
| CME | Chemical master equations |
| 2D | two dimensional |
| EP | Macroscopic entropy production rate |
| sn | Saddle node |
| UR | Upper rate |
| LR | Lower rate |
| MinEPP | Minimium entropy production principle |
| MaxEPP | Maximium entropy production principle |
Appendix A. Transition Rates and Detailed Balance
Appendix A.1. CO(gas) Adsorption and CO(ads) Desorption.
Appendix A.2. Dissociative O2(gas) Adsorption and Associative O(ads) Desorption.
Appendix A.3. CO(ads)+O(ads) Reaction and CO2(gas) Dissociative Adsorption
References
- Kondepudi, D.; Prigogine, I. Modern Thermodynamics: From Heat Engines to Dissipative Structures; Wiley: New York, NY, USA, 1998. [Google Scholar]
- Kondepudi, D.; Petrosky, T.; Pojman, J.A. Dissipative structures and irreversibility in nature: Celebrating 100th birth anniversary of Ilya Prigogine (1917–2003). Chaos 2017, 21, 104501. [Google Scholar] [CrossRef] [PubMed]
- Endres, R.G. Entropy production selects non-equilibrium states in multistable systems. Sci. Rep. 2017, 7, 14437. [Google Scholar] [CrossRef] [PubMed]
- Martyushev, L.M. The maximum entropy production principle: Two questions. Phil. Trans. R. Soc. B Rep. 2010, 365, 1333. [Google Scholar] [CrossRef] [PubMed]
- Jaynes, E.T. The minimum entropy production principle. Annu. Rev. Phys. Chem. 1980, 31, 579–601. [Google Scholar] [CrossRef]
- Kawazura, Y.; Yoshida, Z. Entropy production rate in a flux-driven self-organising system. Phys. Rev. E 2010, 82, 066403. [Google Scholar] [CrossRef] [PubMed]
- Vellela, M.; Quian, H. Stochastic dynamics and non-equilibrium thermodynamics of a bistable chemical system: the Schlögl model revisited. J. R. Soc. Interface 2009, 6, 925–940. [Google Scholar] [CrossRef] [PubMed]
- Nicoli, C.; Nicolis, G. Stability, complexity and the maximum dissipation conjecture. Q. J. R. Meteorol. Soc. 2010, 136, 1161–1169. [Google Scholar] [CrossRef]
- Dewar, R.C. Maximum entropy production and the fluctuation theorem. J. Phys. A Math. Gen. 2005, 38, L371–L381. [Google Scholar] [CrossRef]
- Grinstein, G.; Linsker, R. Comments on a derivation and application of the ‘Maximum entropy production’ principle. J. Phys. A Math. Gen. 2007, 40, 9717–9720. [Google Scholar] [CrossRef]
- Nicoli, C.; Nicolis, G. Comment on the connection between stability and entropy production. Q. J. R. Meteorol. Soc. 2003, 129, 3501–3505. [Google Scholar] [CrossRef]
- Dewar, R.C.; Lineweaver, C.; Niven, R.K.; Regenauer-Lieb, K. Beyond the Second Law— Entropy Poduction and Non-Equilibrium Systems; Springer: Berlin/Heidelberg, Germany, 2014; Chapter 1. [Google Scholar]
- Niven, R.K. Simultaneous extrema in the entropy production for steady-state fluid flow in parallel pipe. J. Non-Equil. Thermodyn. 2010, 35, 347–378. [Google Scholar] [CrossRef]
- Luo, J.; van de Broeck, C.; Nicolis, G. Stability Criteria and Fluctuations around Nonequilibrium States. Z. Phys. B Condens. Matter. 1984, 56, 165–170. [Google Scholar]
- Feynman, R.P.; Leighton, R.B.; Sands, M.L. Feynman Lectures on Physics; Pearson/Addison-Wesley: San Francisco, CA, USA, 2006; Volume 2, Chapter 19. [Google Scholar]
- Paltridge, G.W. The steady state format of global climate. Quart. J. Royal Meteorol. Soc. 1978, 104, 927–945. [Google Scholar] [CrossRef]
- Martyushev, L.M.; Seleznev, V.D. Maximum entropy production principle in physics, chemistry and biology. Phys. Rep. 2006, 426, 1–45. [Google Scholar] [CrossRef]
- Paltridge, G.W.; Farquhar, G.D.; Cuntz, M. Maximum entropy production, cloud feedback, and climate change. Geophys. Res. Lett. 2007, 34, L14708. [Google Scholar] [CrossRef]
- Kleidon, A. Non-equilibrium thermodynamics and maximum entropy production in the Earth system: applications and implications. Naturwissenschaften 2009, 96, 635–677. [Google Scholar] [CrossRef] [PubMed]
- Ertl, G. Reactions at surfaces: From atoms to complexity (Nobel lecture). Angew. Chem. Int. Ed. 2008, 47, 3524–3535. [Google Scholar] [CrossRef] [PubMed]
- Imbihl, R.; Ertl, G. Oscillatory kinetics in heterogeneous catalysis. Chem. Rev. 1995, 95, 697–733. [Google Scholar] [CrossRef]
- Berdau, M.; Yelenin, G.G.; Karpowicz, A.; Ehsasi, M.; Christmann, K.; Block, J.H. Macroscopic and mesoscopic characterization of a bistable reaction system: CO oxidation on Pt(111) surface. J. Chem. Phys. 1999, 110, 11551. [Google Scholar] [CrossRef]
- Bär, M.; Zülicke, C.; Eiswirth, M.; Ertl, G. Theoretical modeling of spatiotemporal self-organization in a surface catalyzed reaction exhibiting bistable kinetics. J. Chem. Phys. 1992, 96, 8595. [Google Scholar] [CrossRef]
- Grosfils, P.; Gaspard, P.; Visart de Bocarmé, T. The role of fluctuations in bistability and oscillations during the H2 + O2 reaction on nanosized rhodium crystals. J. Chem. Phys. 2015, 143, 064705. [Google Scholar] [CrossRef] [PubMed]
- Suchorski, Y.; Beben, J.; James, E.W.; Evans, J.W.; Liu, D.-J.; Imbihl, R. Fluctuation-Induced Transitions in a Bistable Surface Reaction: Catalytic CO Oxidation on a Pt Field Emitter Tip. Phys. Rev. Lett. 1999, 82, 1907. [Google Scholar] [CrossRef]
- Johánek, V.; Laurin, M.; Grant, A.W.; Kasemo, C.; Henry, C.R.; Libuda, J. Fluctuations and bistabilities on catalyst nanoparticles. Science 2004, 304, 1639. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.-J.; Evans, J.W. Fluctuations and bistability in a “hybrid” atomistic model for CO oxidation on nanofacets: An effective potential analysis. J. Chem. Phys. 2002, 117, 7319. [Google Scholar] [CrossRef]
- Pineda, M.; Imbihl, R.; Schimansky-Geier, L.; Zülicke, C. Theoretical analysis of internal fluctuations and bistability in CO oxidation on nanoscale surfaces. J. Chem. Phys. 2006, 124, 044701. [Google Scholar] [CrossRef] [PubMed]
- Chorkendorff, I.; Niemantsverdriet, H. Concepts of Modern Catalysis and Kinetics; Wiley-VCH: Weinheim, German, 2003. [Google Scholar]
- Engel, T.; Ertl, G. Elementary steps in the catalytic oxidation of carbon monoxide on platinum metals. Adv. Catal. 1979, 28, 1–78. [Google Scholar]
- Stamatakis, M.; Vlachos, D.G. Equivalence of on-lattice stochastic chemical kinetics with the well-mixed chemical master equation in the limit of fast diffusion. Comput. Chem. Eng. 2011, 35, 2602–2610. [Google Scholar] [CrossRef]
- Jansen, A.P.J. An Introduction to Monte Carlo Simulations of Surface Reactions; Springer: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Gillespie, D.T. Exact stochastic simulation of coupled chemical reactions. J. Phys. Chem. 1977, 81, 2340–2361. [Google Scholar] [CrossRef]
- Gaspard, P. Fluctuation theorem for nonequilibrium reactions. J. Chem. Phys. 2004, 120, 8898. [Google Scholar] [CrossRef] [PubMed]
- Andrieux, D.; Gaspard, P. Fluctuation theorem and Onsager reciprocity relations. J. Chem. Phys. 2004, 121, 6167. [Google Scholar] [CrossRef] [PubMed]
- Mou, C.Y.; Luo, J.; Nicolis, G. Stochastic thermodynamics of nonequilibrium steady states in chemical reaction systems. J. Phys. Chem. 1986, 12, 7011. [Google Scholar] [CrossRef]
- Seifert, U. Stochastic thermodynamics, fluctuation theorems and molecular machines. Rep. Prog. Phys. 2012, 75, 126001. [Google Scholar] [CrossRef] [PubMed]
- Van den Broeck, C.; Esposito, M. Ensemble and trajectory thermodynamics: A brief introduction. Phys. A Stat. Mech. Appl. 2015, 418, 6–16. [Google Scholar] [CrossRef]
- Schnakenberg, J. Network theory of microscopic and macroscopic behaviour of master equation systems. Rev. Mod. Phys. 1976, 48, 571. [Google Scholar] [CrossRef]
- Tomé, T.; de Oliveira, J. Stochastic thermodynamics and entropy production of chemical reaction systems. J. Chem. Phys. 2018, 148, 224104. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, C.W. Handbook of Stochastic Methods for Physics, Chemistry, and the Natural Sciences, 2nd ed.; Springer: New York, NY, USA, 1985. [Google Scholar]
- Pineda, M.; Stamatakis, M. On the stochastic modelling of surface reactions through reflected chemical Langevin equations. Comput. Chem. Eng. 2018, 117, 145–158. [Google Scholar] [CrossRef]
- Nicolis, G.; de Decker, Y. Stochastic approach to irreversible thermodynamics. Chaos 2017, 27, 104615. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, T. The smallest chemical reaction system with bistability. BMC Syst. Biol. 2009, 3, 90. [Google Scholar] [CrossRef] [PubMed]
- Malchow, H.; Schimansky-Geier, L. Noise and Diffusion in Bistable Non-Equilibrium System; Teubner: Berlin, German, 1985. [Google Scholar]
- Ziff, R.M.; Gulari, E.; Barshad, Y. Kinetic phase transitions in an irreversible surface-reaction model. Phys. Rev. Lett. 1986, 56, 2553–2556. [Google Scholar] [CrossRef] [PubMed]
- Schmiedl, T.; Seifert, U. Stochastic thermodynamics of chemical reaction networks. J. Chem. Phys. 2007, 126, 044101. [Google Scholar] [CrossRef] [PubMed]
- Rao, T.; Xiao, T.; Hou, Z. Entropy production in a mesoscopic chemical reaction system with oscillatory and excitable dynamics. J. Chem. Phys. 2011, 134, 214112. [Google Scholar] [CrossRef] [PubMed]
- De Decker, Y. On the stochastic thermodynamics of reactive systems. Phys. Stat. Mech. Appl. 2015, 428, 178–193. [Google Scholar] [CrossRef]
- Ge, H.; Qian, H. Thermodynamic limit of a nonequilibrium steady state: Maxwell-type construction for a bistable biochemical system. Phys. Rev. Lett. 2009, 103, 148103. [Google Scholar] [CrossRef] [PubMed]
- Vlysidis, M.; Kaznesiss, Y.N. On differences between deterministic and stochastic models of chemical reactions: Scholg solved with ZI-closure. Entropy 2018, 20, 678. [Google Scholar] [CrossRef]
- Pineda, M.; Imbihl, R.; Schimansky-Geier, L. Effects of surface size on minimalistic stochastic models for the catalytic CO oxidation. Phys. Stat. Mech. Appl. 2009, 389, 1178–1188. [Google Scholar] [CrossRef]










{kind=link}
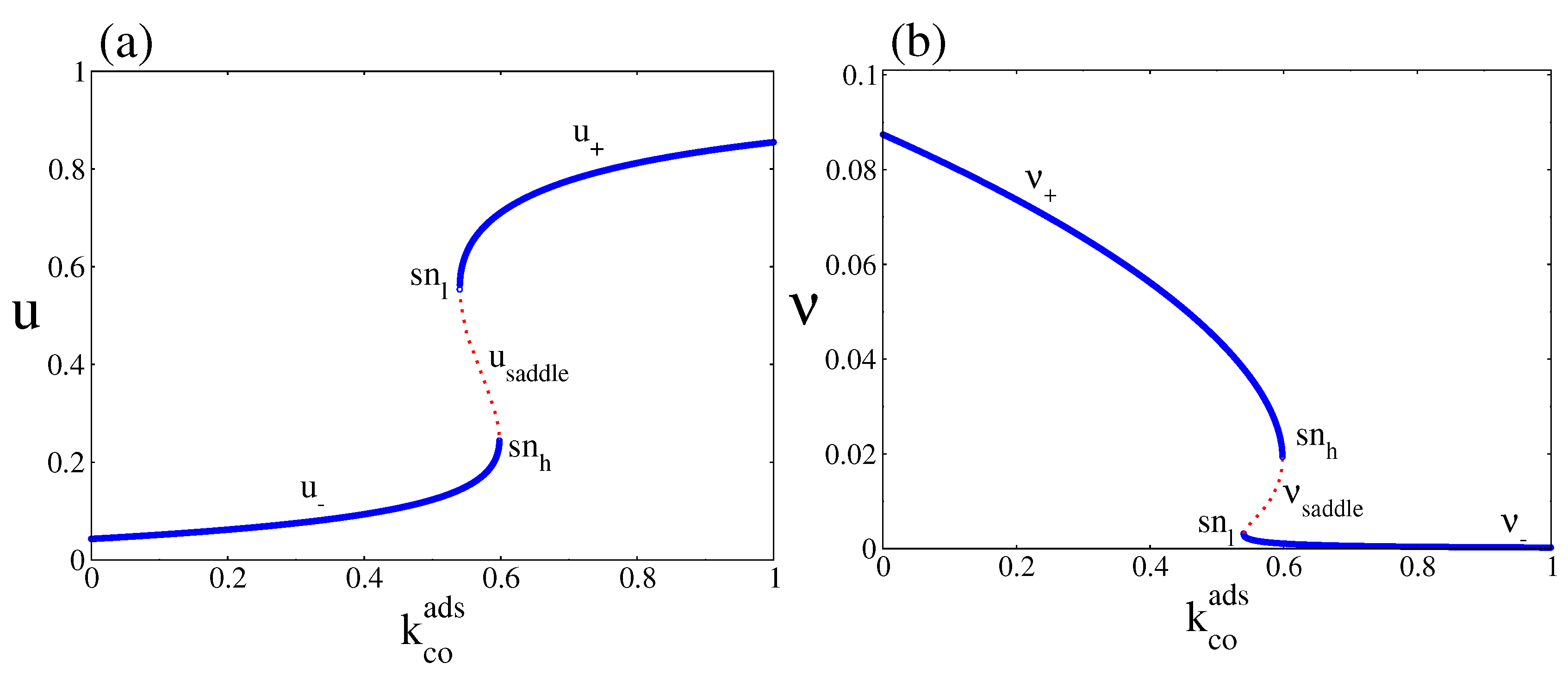{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Process | Population Change | Transition Rate |
|---|---|---|
| CO(gas) adsorption | ||
| CO(ads) desorption | ||
| O(gas) dissociative adsorption | ||
| O(ads) associative desorption | ||
| CO(gas) dissociative adsorption | ||
| CO(ads) + O(ads) reaction |
| Process | Reaction Rates or Fluxes |
|---|---|
| CO(gas) adsorption | |
| CO(ads) desorption | |
| O(gas) dissociative adsorption | |
| O(ads) associative desorption | |
| CO(gas) dissociative adsorption | |
| CO(ads) + O(ads) reaction |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pineda, M.; Stamatakis, M. Non-Equilibrium Thermodynamics and Stochastic Dynamics of a Bistable Catalytic Surface Reaction. Entropy 2018, 20, 811. https://doi.org/10.3390/e20110811
Pineda M, Stamatakis M. Non-Equilibrium Thermodynamics and Stochastic Dynamics of a Bistable Catalytic Surface Reaction. Entropy. 2018; 20(11):811. https://doi.org/10.3390/e20110811
Chicago/Turabian StylePineda, Miguel, and Michail Stamatakis. 2018. "Non-Equilibrium Thermodynamics and Stochastic Dynamics of a Bistable Catalytic Surface Reaction" Entropy 20, no. 11: 811. https://doi.org/10.3390/e20110811
APA StylePineda, M., & Stamatakis, M. (2018). Non-Equilibrium Thermodynamics and Stochastic Dynamics of a Bistable Catalytic Surface Reaction. Entropy, 20(11), 811. https://doi.org/10.3390/e20110811