
On the Configurational Entropy of Nanoscale Solutions for More Accurate Surface and Bulk Nano-Thermodynamic Calculations

Abstract
:1. Introduction
2. The Exact Equation for the Integral Molar Configurational Entropy of Solutions
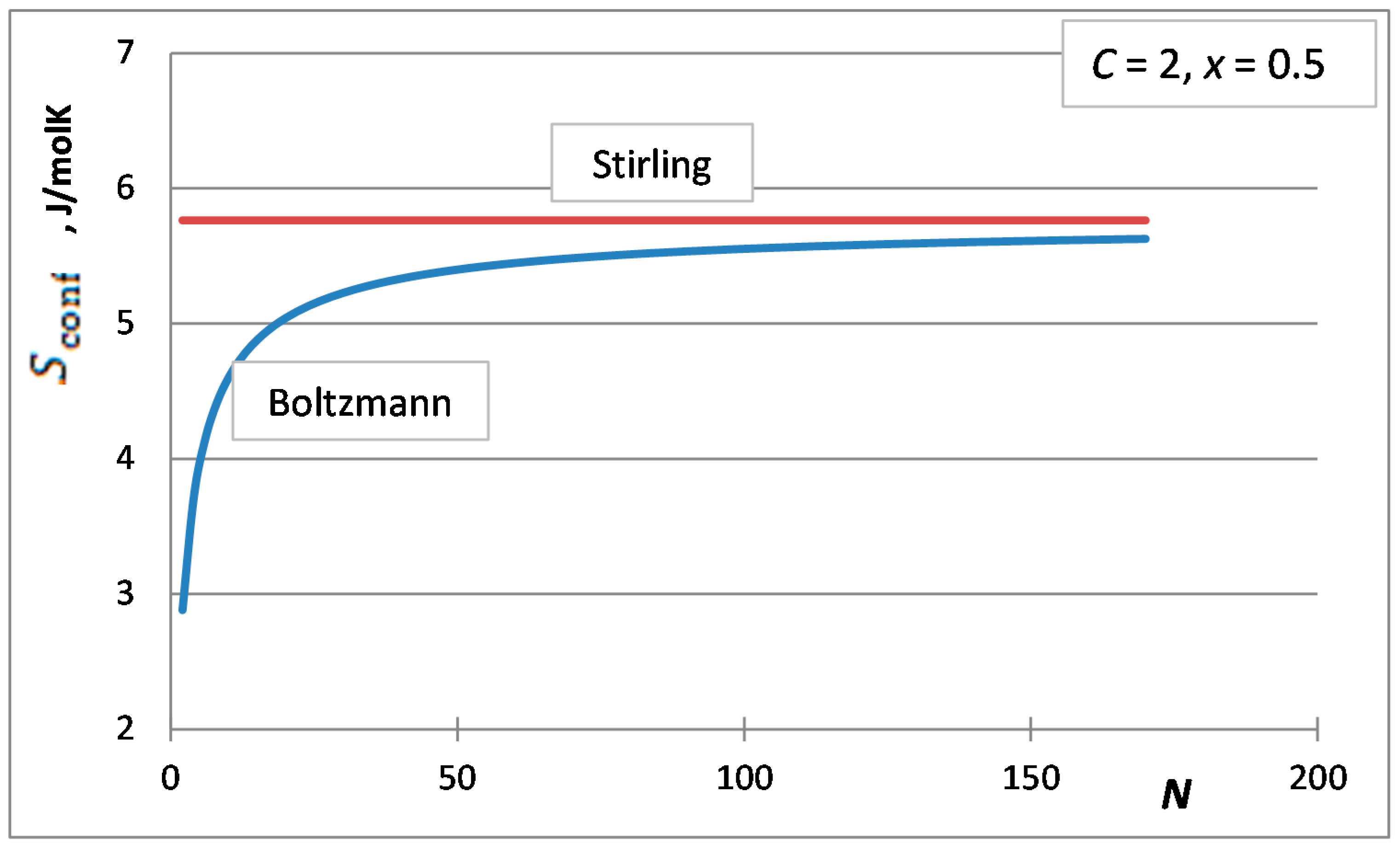
3. The Stirling Approximation for the Integral Molar Configurational Entropy of Solutions
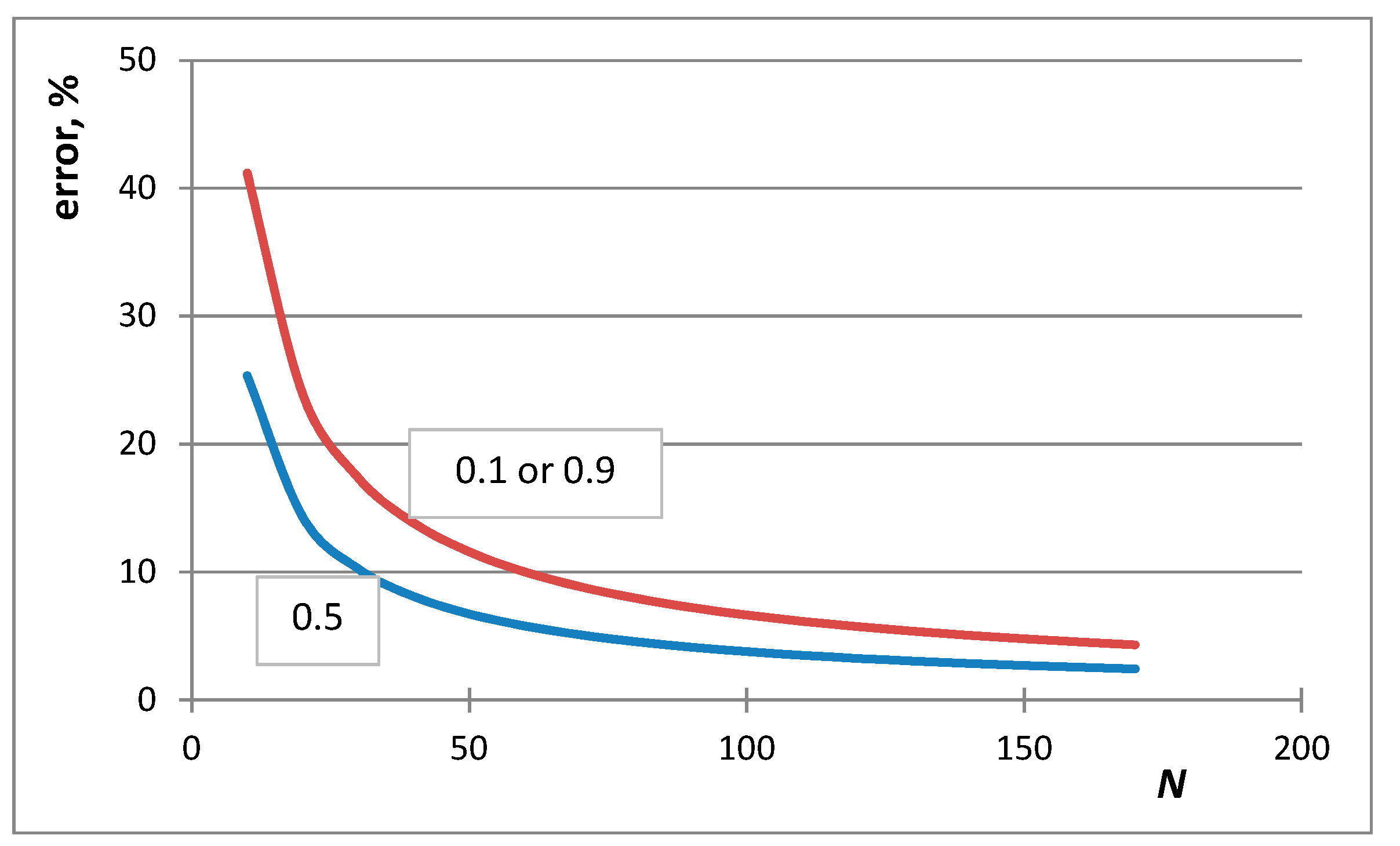
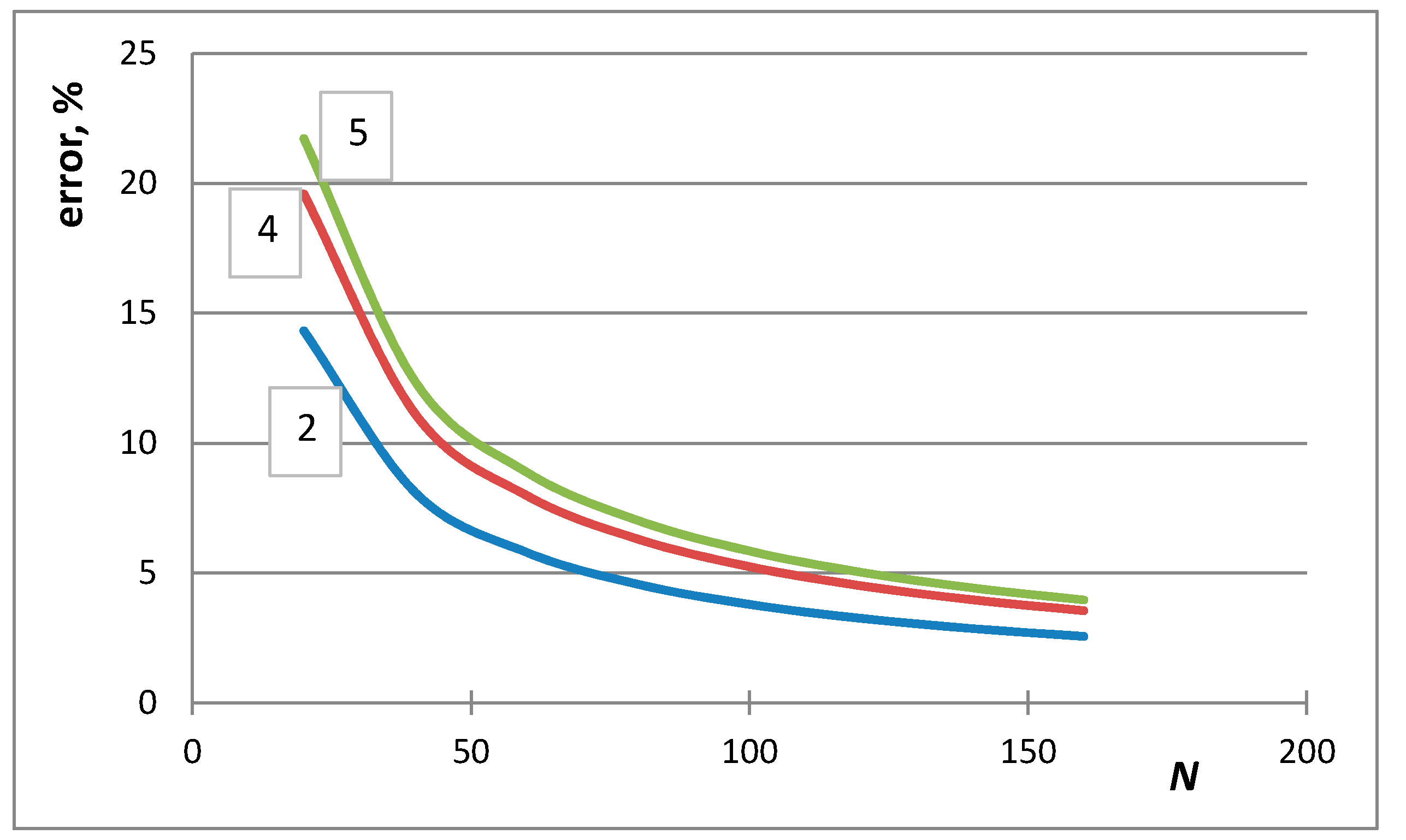
4. Selection of the Optimum Approximation to Replace the Stirling Approximation
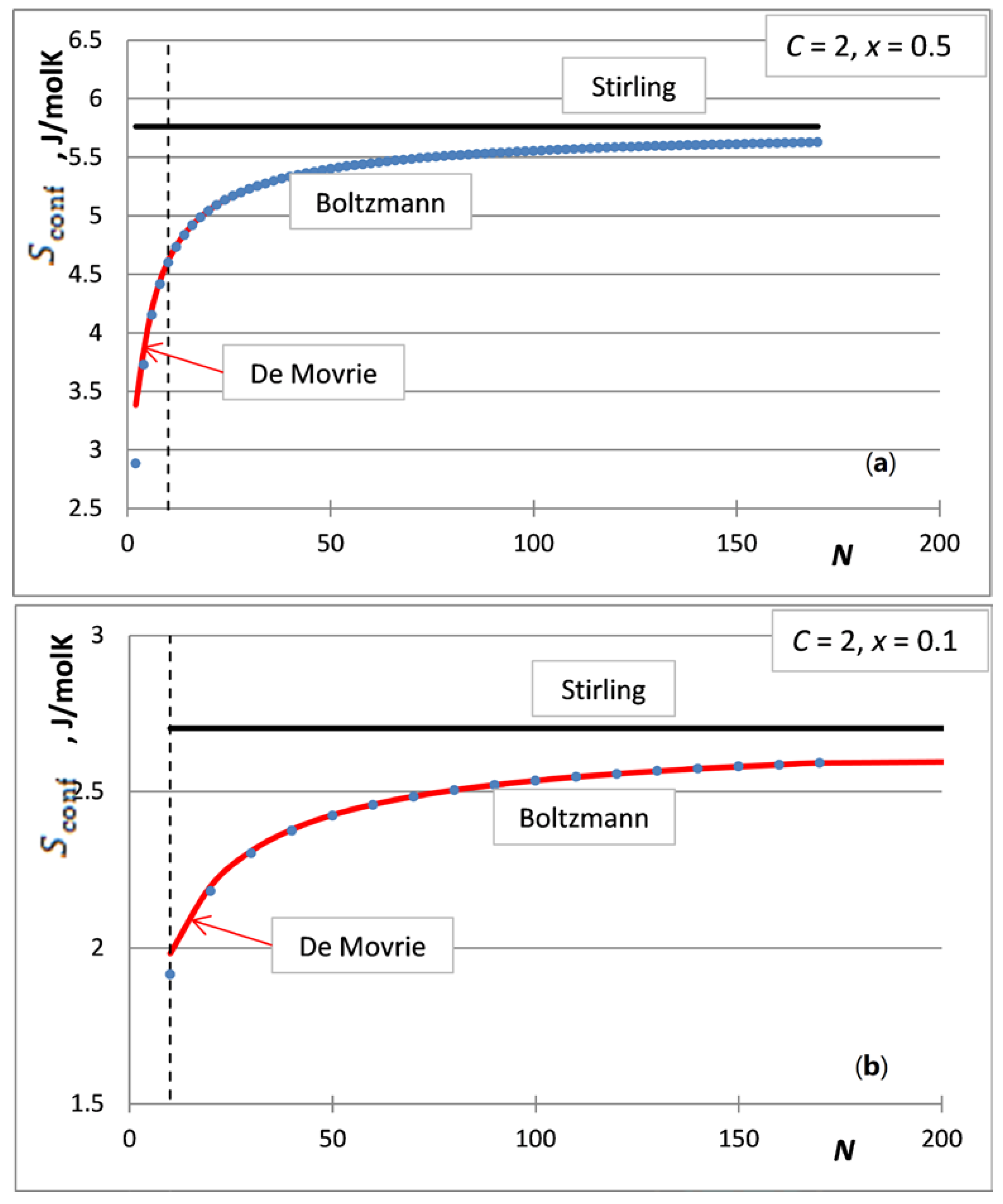
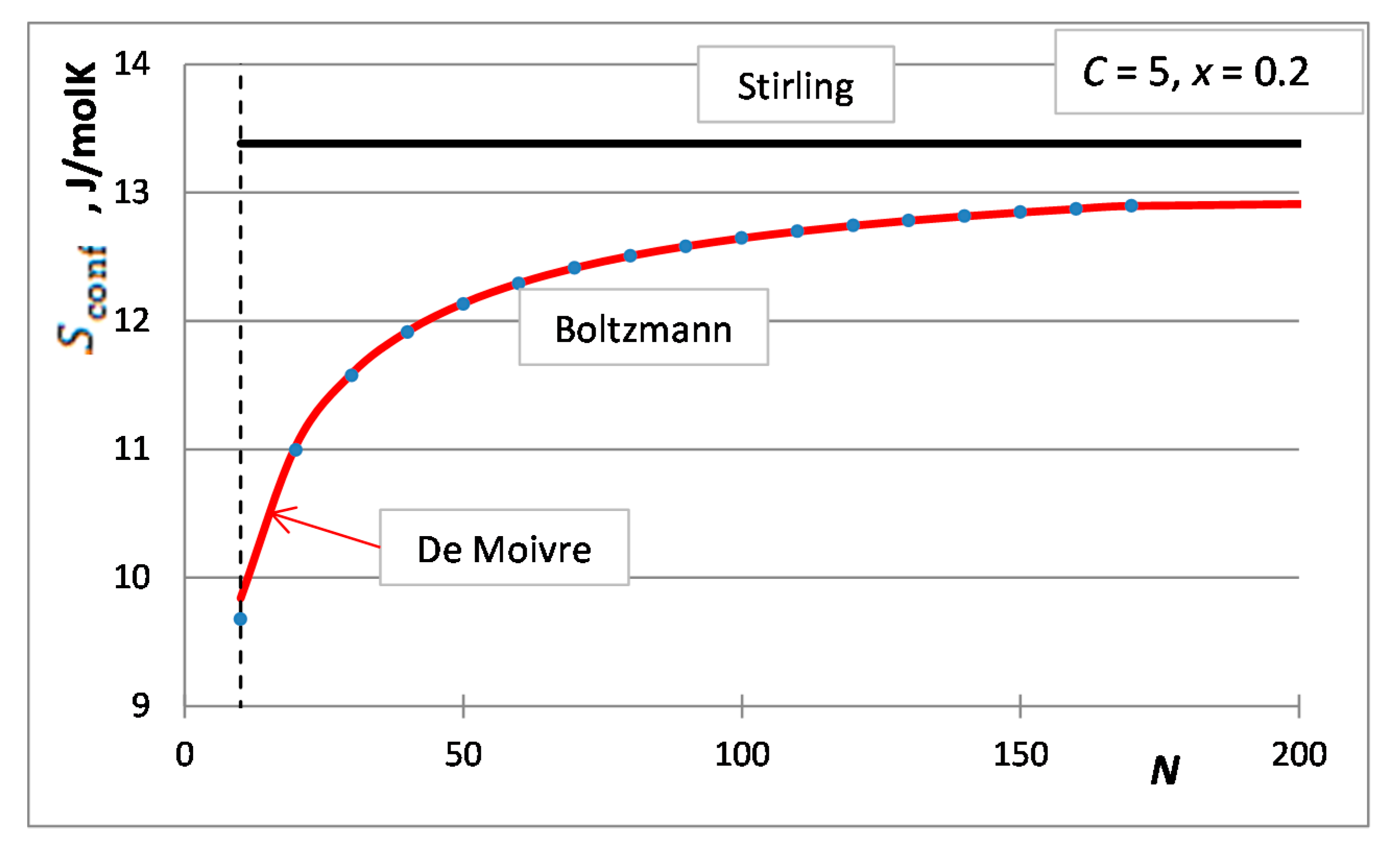
5. The Integral Configurational Entropy of Nano-Solutions in the de Moivre Approximation
6. The Partial Configurational Entropies of Components of Nano-Solutions in the de Moivre Approximation
7. Situations When the de Moivre Approximation Must Be Used
- The first case is the bulk calculations below the size of the solution phase, when the integral entropies calculated by the Stirling approximation lead to more than 0.1% relative error compared to the de Moivre approximation (in this case, this error cannot be compared to the exact Boltzmann equation, as it cannot be calculated for large numbers). For example, the difference between the Stirling approximation and the de Moivre approximation becomes smaller than 0.1% for Figure 4a,b and Figure 5 at , , and , respectively. Thus, the de Moivre approximation should be used whenever the number of atoms is below 15,000. Let us take the average molar volume of a metal (10 cm3/mol), leading to the volume of the phase 2.5 × 10−25 m3. If it is a cubic phase, its side length equals 6.3 nm. For molecular solutions, this critical size will be larger, as the molecules are larger than the atoms.
- The second case is when surface or interface equilibrium calculations are performed, being routinely used in nano-equilibrium calculations. In this case, the de Moivre approximation should be used whenever the surface/interface region contains less than 15,000 atoms. Suppose a single layer of atoms along the surface and the average molar surface area of 45,000 m2/mol. Then, 15,000 atoms will cover a 1.2 × 10−15 m2 surface area. If this phase is cubic, its side length is 13.7 nm. So, we can conclude that when the diameter of a nano-phase is below 15 nm, the de Moivre approximation should be used instead of the Stirling approximation. For molecular solutions, this critical size will be larger, as the molecules are larger than the atoms.
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gibbs, J.W. On the Equilibrium of Heterogeneous Substances. Am. J. Sci. 1878, 96, 441–458. [Google Scholar] [CrossRef]
- Thomson, W. On the Equilibrium of Vapour at a Curved Surface of Liquid. Proc. R. Soc. Edinb. 1872, 7, 63–68. [Google Scholar] [CrossRef]
- Ostwald, W. Über die vermeintliche Isomerie des roten und gelben Quecksilberoxyds und die Oberflächenspannung fester Körper. Z. Phys. Chem. 1900, 34, 495–503. (In German) [Google Scholar] [CrossRef]
- Pawlow, P. Über die Abhängigkeit des Schmelzpunktes von der Oberflächenenergie eines Festen Körpers. Z. Phys. Chem. 1908, 55, 545–548. (In German) [Google Scholar] [CrossRef]
- Reiss, H.; Wilson, I.B. The Effect of Surface on Melting Point. J. Colloid Sci. 1948, 3, 551–561. [Google Scholar] [CrossRef]
- Couchman, P.R.; Jesser, W.A. Thermodynamic theory of size dependence of melting temperature in metals. Nature 1977, 269, 481–483. [Google Scholar] [CrossRef]
- Samsonov, V.M.; Malkov, O.A. Thermodynamic model of crystallization and melting of small particles. Cent. Eur. J. Phys. 2004, 2, 90–103. [Google Scholar] [CrossRef]
- Lee, J.H.; Tanaka, T.; Lee, J.; Mori, H. Effect of substrates on the melting temperature of gold nanoparticles. Calphad 2007, 31, 105–111. [Google Scholar] [CrossRef]
- Kaptay, G. On the size and shape dependence of the solubility of nano-particles in solutions. Int. J. Pharm. 2012, 430, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Junkaew, A.; Ham, B.; Zhang, X.; Arróyave, R. Tailoring the formation of metastable Mg through interfacial engineering: A phase stability analysis. Calphad 2014, 45, 145–150. [Google Scholar] [CrossRef]
- Guenther, G.; Guillon, O. Models of size-dependent nanoparticle melting tested on gold. J. Mater. Sci. 2014, 49, 7915–7932. [Google Scholar] [CrossRef]
- Kaptay, G.; Janczak-Rusch, J.; Pigozzi, G.; Jeurgens, L.P.H. Theoretical Analysis of Melting Point Depression of Pure Metals in Different Initial Configurations. J. Mater. Eng. Perform. 2014, 23, 1600–1607. [Google Scholar] [CrossRef]
- Zhu, J.; Fu, Q.; Xue, Y.; Cui, Z. Comparison of different models of melting transformation. J. Mater. Sci. 2016, 51, 2262–2269. [Google Scholar] [CrossRef]
- Arabczyk, W.; Ekiert, E.A.; Pelka, R. Hysteresis phenomenon in a reaction system of nanocrystalline iron and a mixture of ammonia and hydrogen. Phys. Chem. Chem. Phys. 2016, 18, 25796–25800. [Google Scholar] [CrossRef] [PubMed]
- Kaptay, G.; Janczak-Rusch, J.; Jeurgens, L.P.H.J. Melting Point Depression and Fast Diffusion in Nanostructured Brazing Fillers Confined between Barrier Nanolayers. Mater. Eng. Perform. 2016, 25, 3275–3284. [Google Scholar] [CrossRef]
- Weismuller, J. Alloy Effects in Nanostructures. Nanostruct. Mater. 1993, 3, 261–272. [Google Scholar] [CrossRef]
- Badmos, A.Y.; Bhadeshia, H.K. The Evolution of Solutions: A Thermodynamic Analysis of Mechanical Alloying. Metall. Mater. Trans. 1997, 28A, 2189–2194. [Google Scholar] [CrossRef]
- Jesser, W.A.; Shiflet, G.J.; Allen, G.L.; Crawford, J.L. Equilibrium phase diagrams of isolated nano-phases. Mater. Res. Innov. 1999, 2, 211–216. [Google Scholar] [CrossRef]
- Wautelet, M.; Dauchot, J.P.; Hecq, M. Phase diagrams of small particles of binary systems: A theoretical approach. Nanotechnology 2000, 11, 6–9. [Google Scholar] [CrossRef]
- Tanaka, T.; Hara, S. Thermodynamic Evaluation of Binary Phase Diagrams of Small Particle Systems. Z. Metall. 2001, 92, 467–472. [Google Scholar]
- Lee, J.G.; Mori, H. In-situ observation of alloy phase formation in nanometre-sized particles in the Sn–Bi system. Rechtsphilos. Mag. 2004, 84, 2675–2686. [Google Scholar] [CrossRef]
- Shirinayan, A.S.; Gusak, A.M. Phase diagrams of decomposing nanoalloys. Rechtsphilos. Mag. 2004, 84, 579–593. [Google Scholar] [CrossRef]
- Lee, J.; Lee, J.; Tanaka, T.; Mori, H.; Penttilá, K. Phase Diagrams of Nanometer-Sized Particles in Binary Systems. JOM 2005, 57, 56–59. [Google Scholar] [CrossRef]
- Qiao, Z.; Cao, Z.; Tanaka, T. Prediction of surface and interfacial tension based on thermodynamic data and CALPHAD approach. Rare Met. 2006, 25, 512–528. [Google Scholar] [CrossRef]
- Braidy, N.; Purdy, G.R.; Botton, G.A. Equilibrium and stability of phase-separating Au–Pt nanoparticles. Acta Mater. 2008, 56, 5972–5983. [Google Scholar] [CrossRef]
- Lee, J.; Park, J.; Tanaka, T. Effects of interaction parameters and melting points of pure metals on the phase diagrams of the binary alloy nanoparticle systems: A classical approach based on the regular solution model. Calphad 2009, 33, 377–381. [Google Scholar] [CrossRef]
- Eichhammer, Y.; Heyns, M.; Moelans, N. Calculation of phase equilibria for an alloy nanoparticle in contact with a solid nanowire. Calphad 2011, 35, 173–182. [Google Scholar] [CrossRef]
- Kaptay, G. Nano-Calphad: Extension of the Calphad method to systems with nano-phases and complexions. J. Mater. Sci. 2012, 47, 8320–8335. [Google Scholar] [CrossRef]
- Garzel, G.; Janczak-Rusch, J.; Zabdyr, L. Reassessment of the Ag–Cu phase diagram for nanosystems including particle size and shape effect. Calphad 2012, 36, 52–56. [Google Scholar] [CrossRef]
- Lee, J.; Sim, K.J. General equations of CALPHAD-type thermodynamic description for metallic nanoparticle systems. Calphad 2014, 44, 129–132. [Google Scholar] [CrossRef]
- Ghasemi, M.; Zanolli, Z.; Stankovski, M.; Johansson, J. Size- and shape-dependent phase diagram of In–Sb nano-alloys. Nanoscale 2015, 7, 17387. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, S.; Haverty, M.G.; Arróyave, R.; Goddard, W.A., III.; Shankar, S. Phase stability in nanoscale material systems: extension from bulk phase diagrams. [CrossRef] [PubMed]
- Pelegrina, J.L.; Gennari, F.C.; Condó, A.M.; Guillemet, A.F. Predictive Gibbs-energy approach to crystalline/amorphous relative stability of nanoparticles: Size-effect calculations and experimental test. J. Alloys Compd. 2016, 689, 161–168. [Google Scholar] [CrossRef]
- Leitner, J.; Sedmidubskỳ, D. Enhanced solubility of nanostructured paracetamol. Biomed. Phys. Eng. Express 2016, 2, 0555007. [Google Scholar] [CrossRef]
- Shardt, N.; Elliott, J.A.W. Thermodynamic Study of the Role of Interface Curvature on Multicomponent Vapor−Liquid Phase Equilibrium. J. Phys. Chem. A 2016, 120, 120–2194. [Google Scholar] [CrossRef] [PubMed]
- Lewis, G.N.; Randall, M. Thermodynamics; McGraw-Hill Book Co.: New York, NY, USA, 1923. [Google Scholar]
- Lupis, C.H.P. Chemical Thermodynamics of Materials; Elsevier: Amsterdam, The Netherland, 1983. [Google Scholar]
- Atkins, P.; de Paula, J. Physical Chemistry; Oxford University Press: Oxford, UK, 2002. [Google Scholar]
- Lukas, H.L.; Fries, S.G.; Sundman, B. Computational Thermodynamics: The Calphad Method; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Saunders, N.; Miodownik, A.P. CALPHAD (Calculation of Phase Diagrams): A Comprehensive Guide; Elsevier: Amsterdam, The Netherlands, 1998; p. 479. [Google Scholar]
- Stirling, J. Methodus Differentialis; Ap. Whiston & White: London, UK, 1764. (In Latin) [Google Scholar]
- Tweddle, I. James Stirling’s Methodus Differentialis: An Annotated Translation of Stirling’s Text; Springer: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Boltzmann, L. Über die beziehung dem zweiten Haubtsatze der mechanischen Wärmetheorie und der Wahrscheinlichkeitsrechnung respektive den Sätzen über das Wärmegleichgewicht. Wien. Ber. 1877, 76, 373–435. (In German) [Google Scholar]
- Sharp, K.; Matschinsky, F. Translation of Ludwig Boltzmann’s Paper “On the Relationship between the Second Fundamental Theorem of the Mechanical Theory of Heat and Probability Calculations Regarding the Conditions for Thermal Equilibrium” Sitzungberichte der Kaiserlichen Akademie der Wissenschaften. Mathematisch-Naturwissen Classe. Abt. II, LXXVI 1877, pp. 373–435 (Wien. Ber. 1877, 76:373–435). Reprinted in Wiss. Abhandlungen, Vol. II, reprint 42, pp. 164–223, Barth, Leipzig, 1909. Entropy 2015, 17, 1971–2009. [Google Scholar] [CrossRef]
- Leubner, C. Generalised Stirling approximations to N! Eur. J. Phys. 1985, 6, 299–301. [Google Scholar] [CrossRef]
- Nemes, G. New asymptotic expansion for the Gamma function. Arch. Math. 2010, 95, 161–169. [Google Scholar] [CrossRef]
- Mortici, C. Ramanujan formula for the generalized Stirling approximation. Appl. Math. Comput. 2010, 217, 699–704. [Google Scholar] [CrossRef]
- Lin, L. On Stirling’s formula remainder. Appl. Math. Comput. 2014, 247, 494–500. [Google Scholar] [CrossRef]
- Mortici, C. A new fast asymptotic series for the gamma function. Raman. J. 2015, 38, 549–559. [Google Scholar] [CrossRef]
- Lu, D.; Song, L.; Ma, C. Some new asymptotic approximations of the gamma function based on Nemes’ formula, Ramanujan’s formula and Burnside’s formula. Appl. Math. Comput. 2015, 253, 1–7. [Google Scholar] [CrossRef]
- Xu, A.; Hu, Y.; Tang, P. Asymptotic expansions for the gamma function. J. Number Theory 2016, 169, 134–143. [Google Scholar] [CrossRef]
- Chen, C.P. On the asymptotic expansions of the gamma function related to the Nemes, Gosper and Burnside formulas. Appl. Math. Comput. 2016, 276, 417–431. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Term Numbers Combined | Equation (10) | Equation (11) |
|---|---|---|
| 1 | 63.5 | 52.4 |
| 1 + 2 | −6.06 | 13.03 |
| 1 + 2 + 3 | 0.026 | −0.055 |
| 1 + 2 + 3 + 4 | −1.39 × 10−5 | −1.83 × 10−5 |
| 1 + 2 + 3 + 4 + 5 | 3.96 × 10−8 | −5.21 × 10−8 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dezso, A.; Kaptay, G. On the Configurational Entropy of Nanoscale Solutions for More Accurate Surface and Bulk Nano-Thermodynamic Calculations. Entropy 2017, 19, 248. https://doi.org/10.3390/e19060248
Dezso A, Kaptay G. On the Configurational Entropy of Nanoscale Solutions for More Accurate Surface and Bulk Nano-Thermodynamic Calculations. Entropy. 2017; 19(6):248. https://doi.org/10.3390/e19060248
Chicago/Turabian StyleDezso, Andras, and George Kaptay. 2017. "On the Configurational Entropy of Nanoscale Solutions for More Accurate Surface and Bulk Nano-Thermodynamic Calculations" Entropy 19, no. 6: 248. https://doi.org/10.3390/e19060248
APA StyleDezso, A., & Kaptay, G. (2017). On the Configurational Entropy of Nanoscale Solutions for More Accurate Surface and Bulk Nano-Thermodynamic Calculations. Entropy, 19(6), 248. https://doi.org/10.3390/e19060248