Can the Hexagonal Ice-like Model Render the Spectroscopic Fingerprints of Structured Water? Feedback from Quantum-Chemical Computations


Abstract
:
1. Introduction
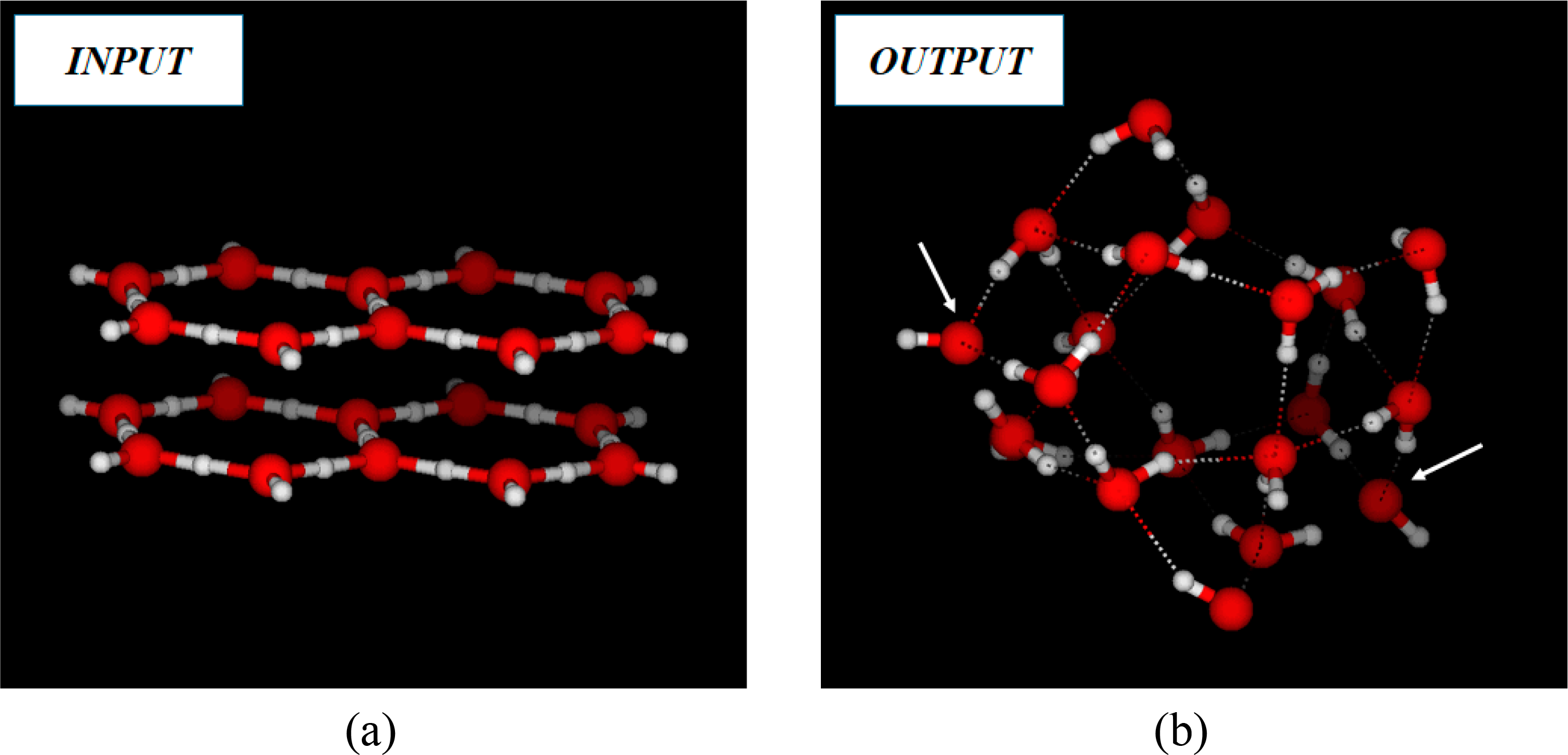
2. Quantum Chemistry and Computational Strategies
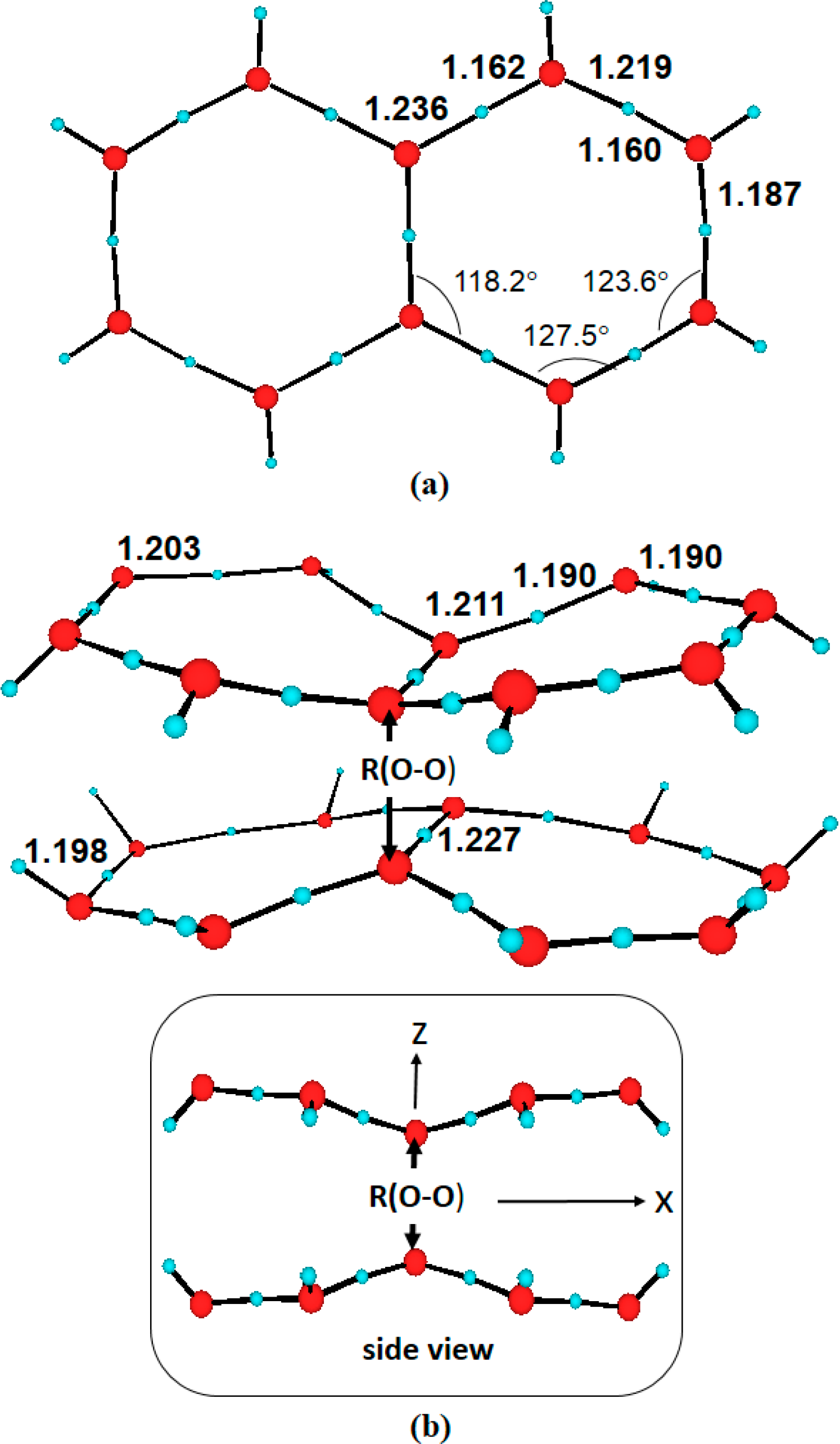
3. Results and Discussion
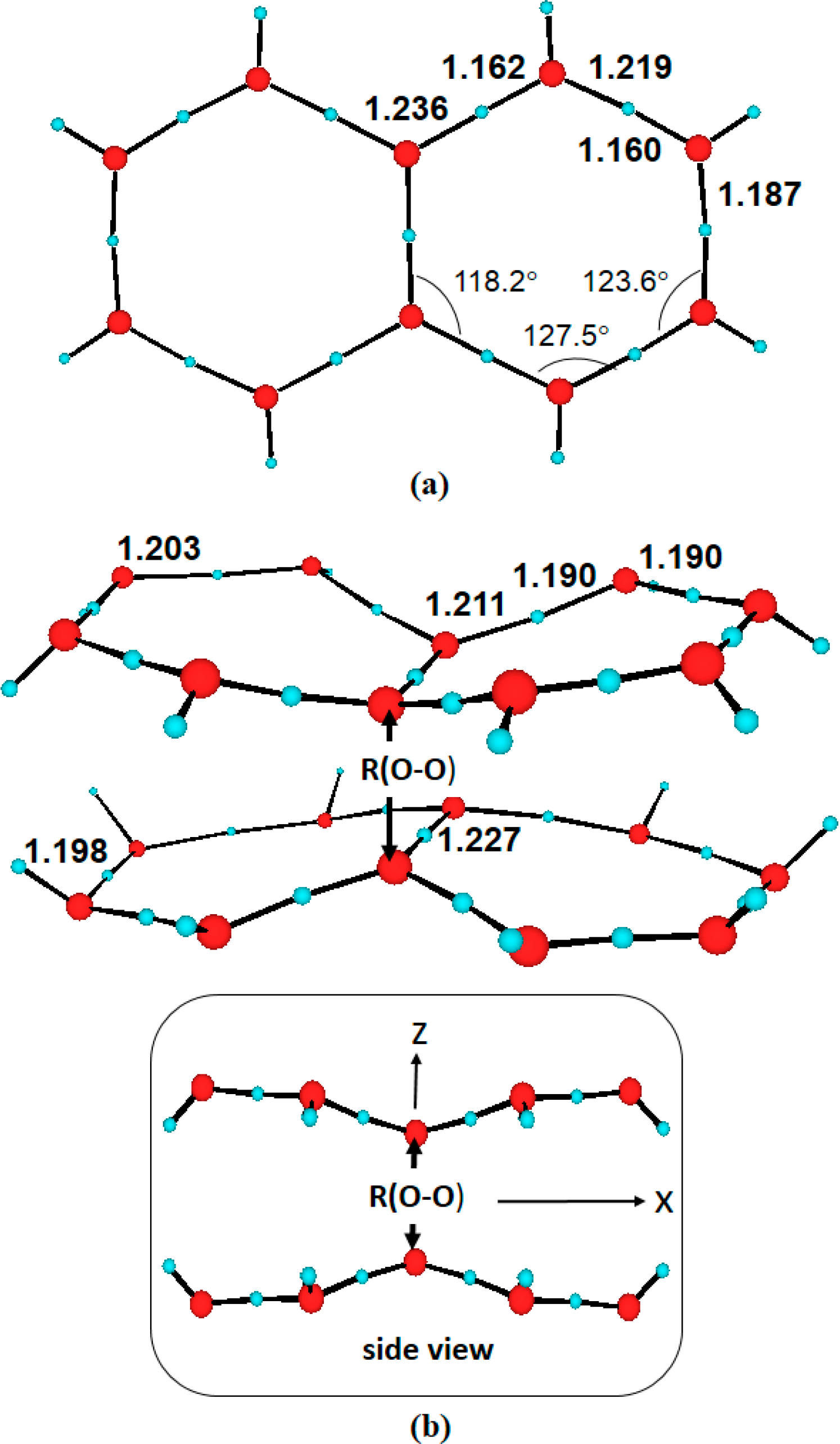
3.1. Anionic Systems
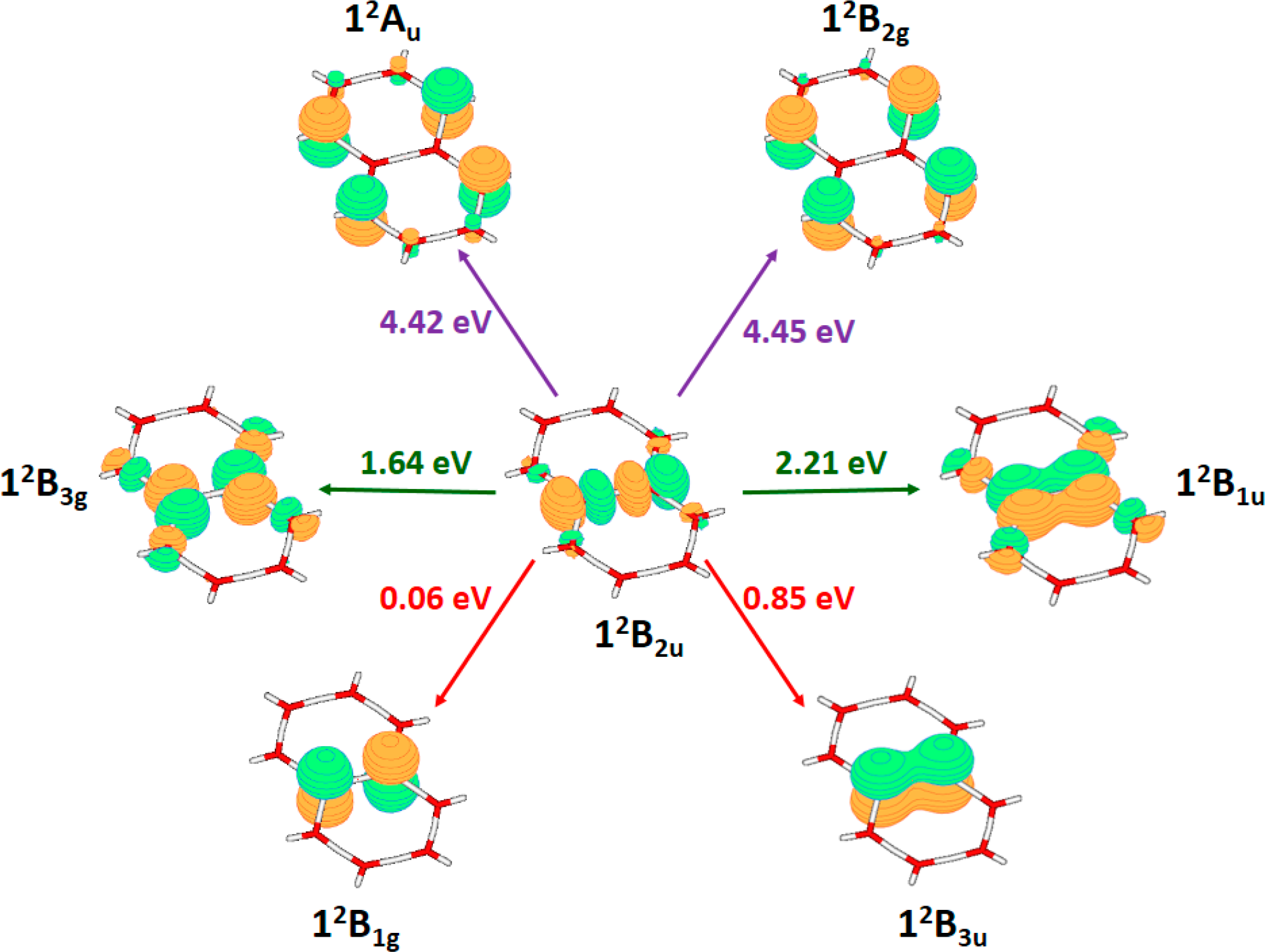
3.2. Neutral Systems
3.2.1. Absorption Spectroscopic Features
3.2.2. Fluorescence Spectroscopic Features
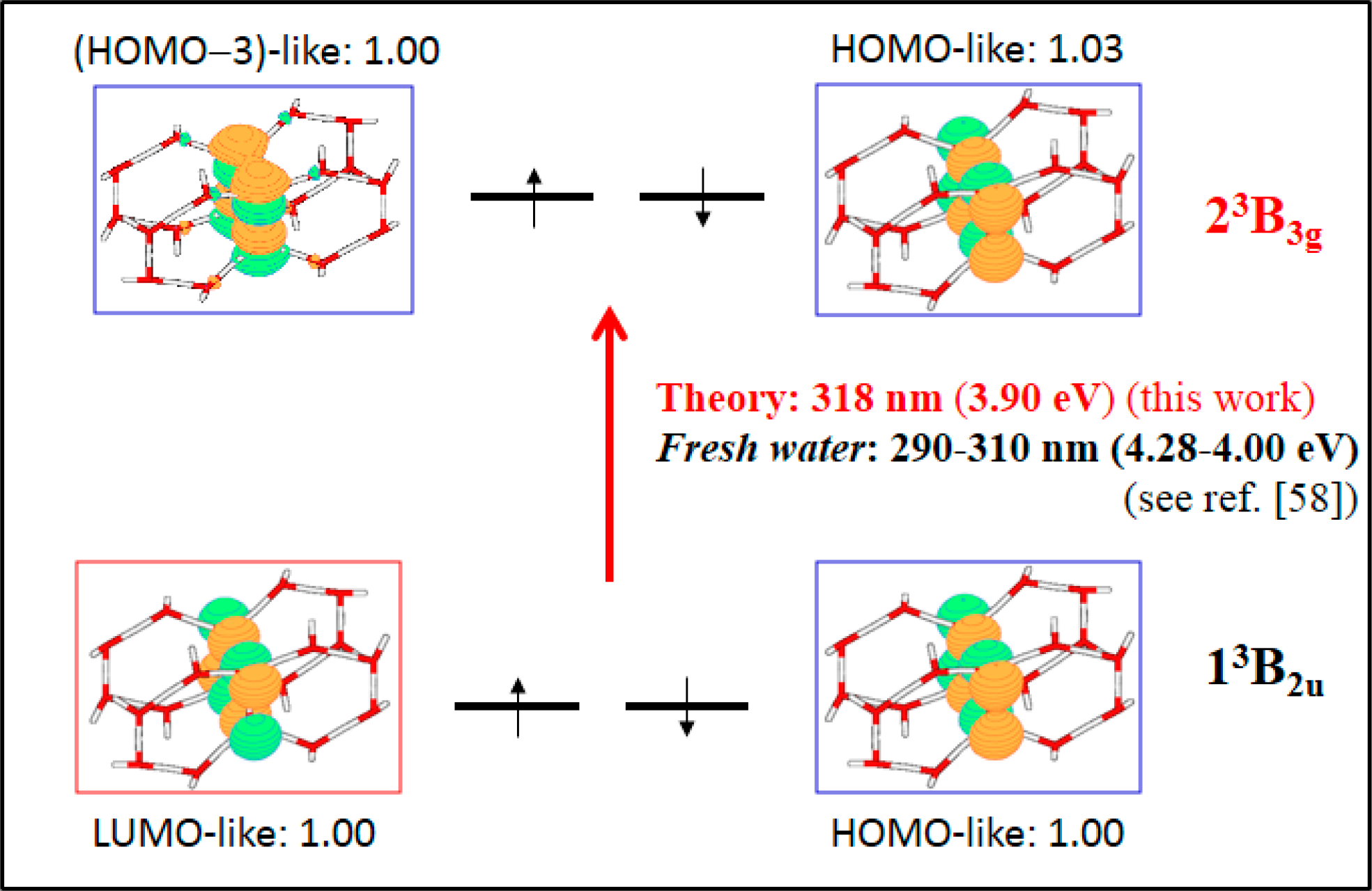
3.2.3. On the Relevance of the Triplet Manifold
- What is the “glue” that makes the singlet/triplet ground-state so stable?
- What determines the population of the singlet or triplet ground state?
- What about the most intense transition in the triplet manifold?
4. Conclusions
- For negatively charged multilayers, i.e., anionic systems, the charge is not expected to be delocalized over the entire honeycomb layer. The system becomes polarized into hydrated hydroxyl anions and cationic moieties. Accordingly, the negative potential observed inside the EZ can be related to the presence of hydroxyl anions. The lowest-lying excited states found for the charged monolayer considered are about ∼7.4 eV (∼168 nm), well above the structured-water spectroscopic fingerprints determined experimentally ∼4.59 (∼270 nm). Therefore, we confidently conclude that heavily negatively charged hexagonally arrangements are not responsible for the observed spectroscopy.
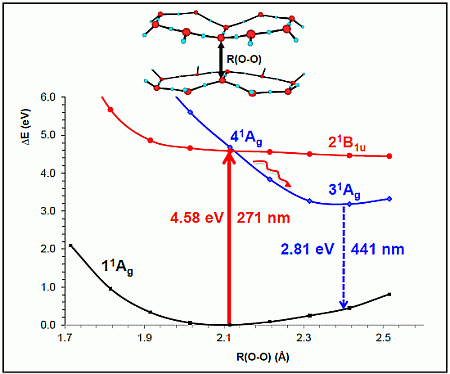
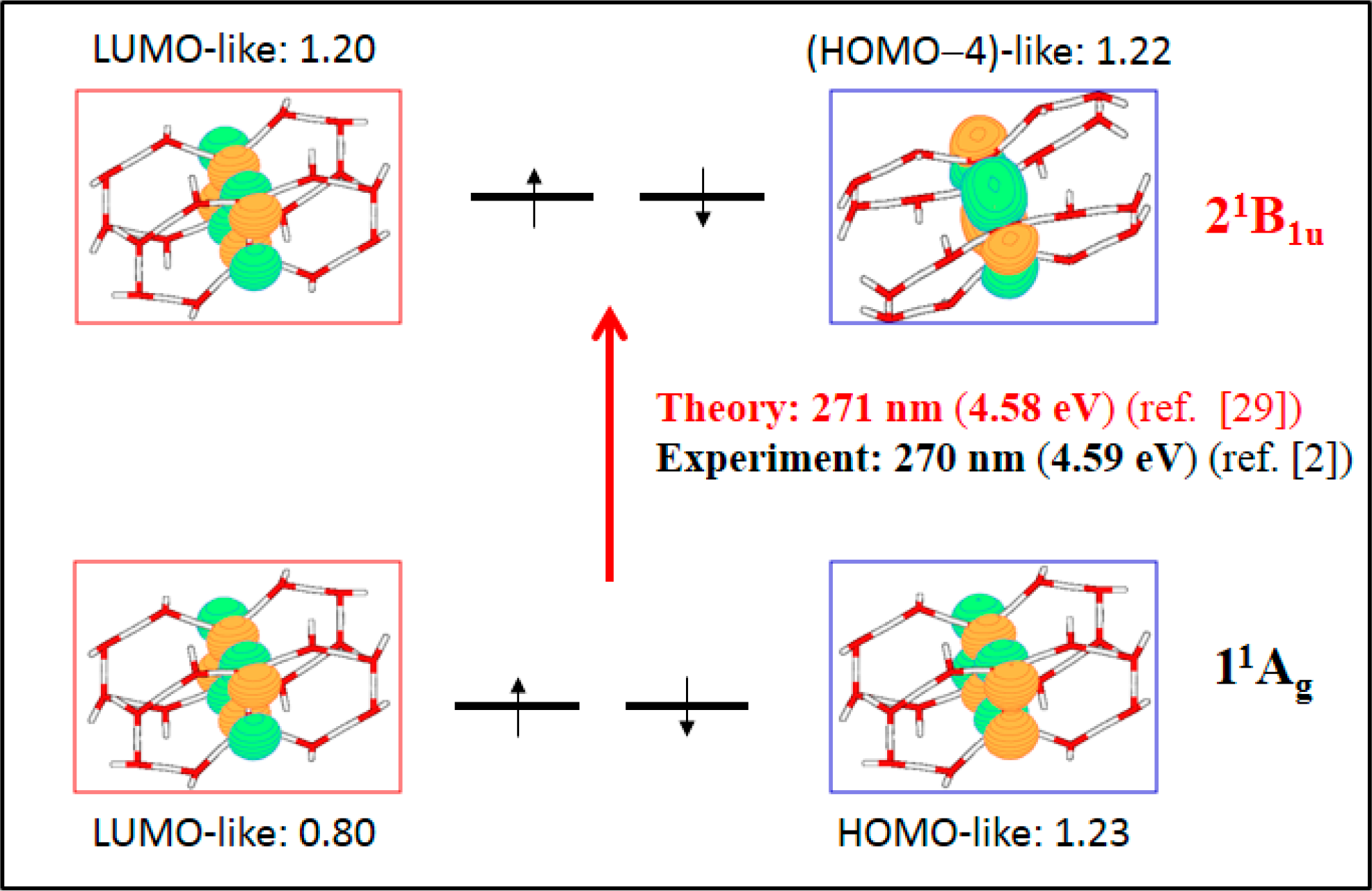
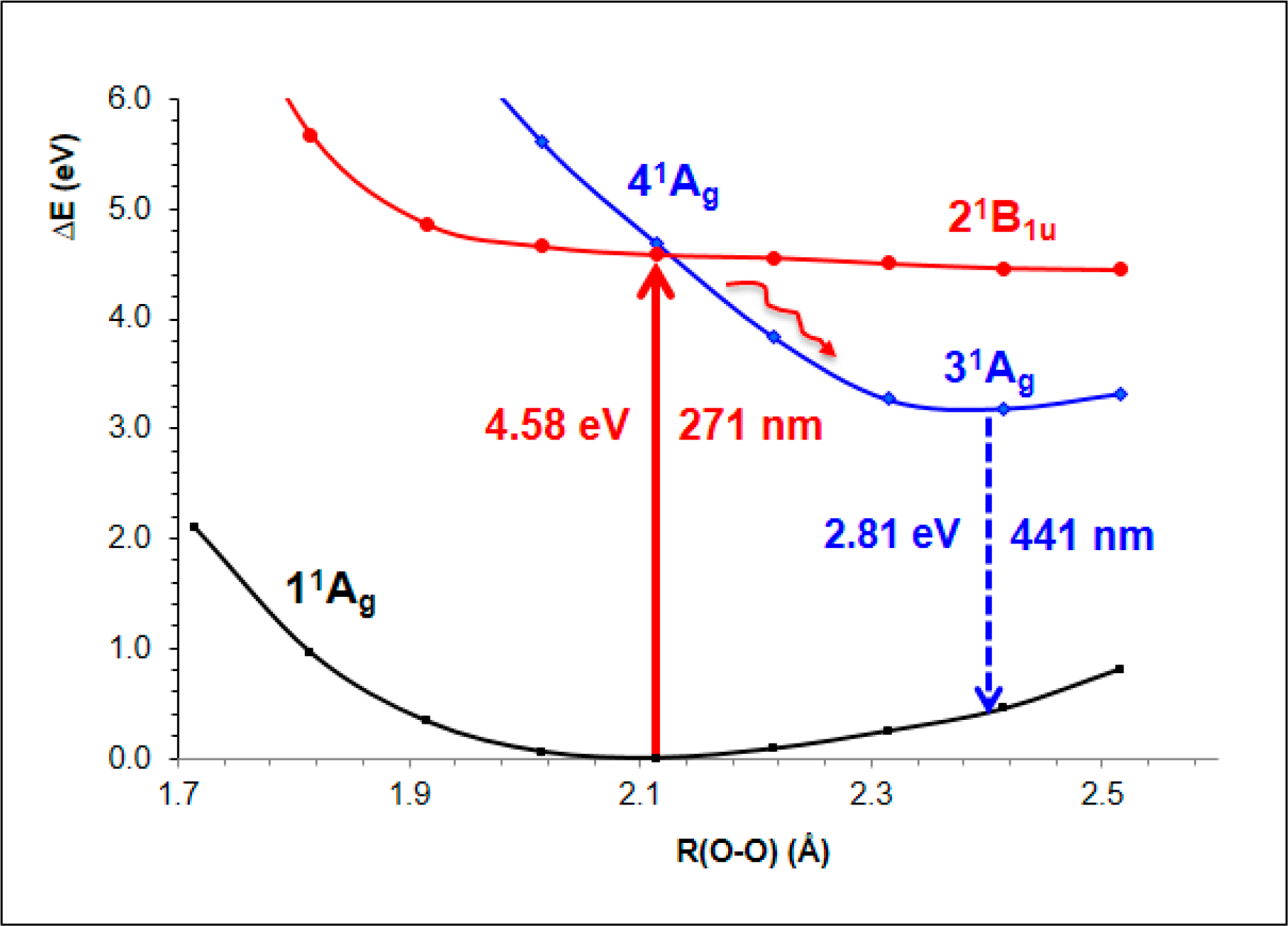
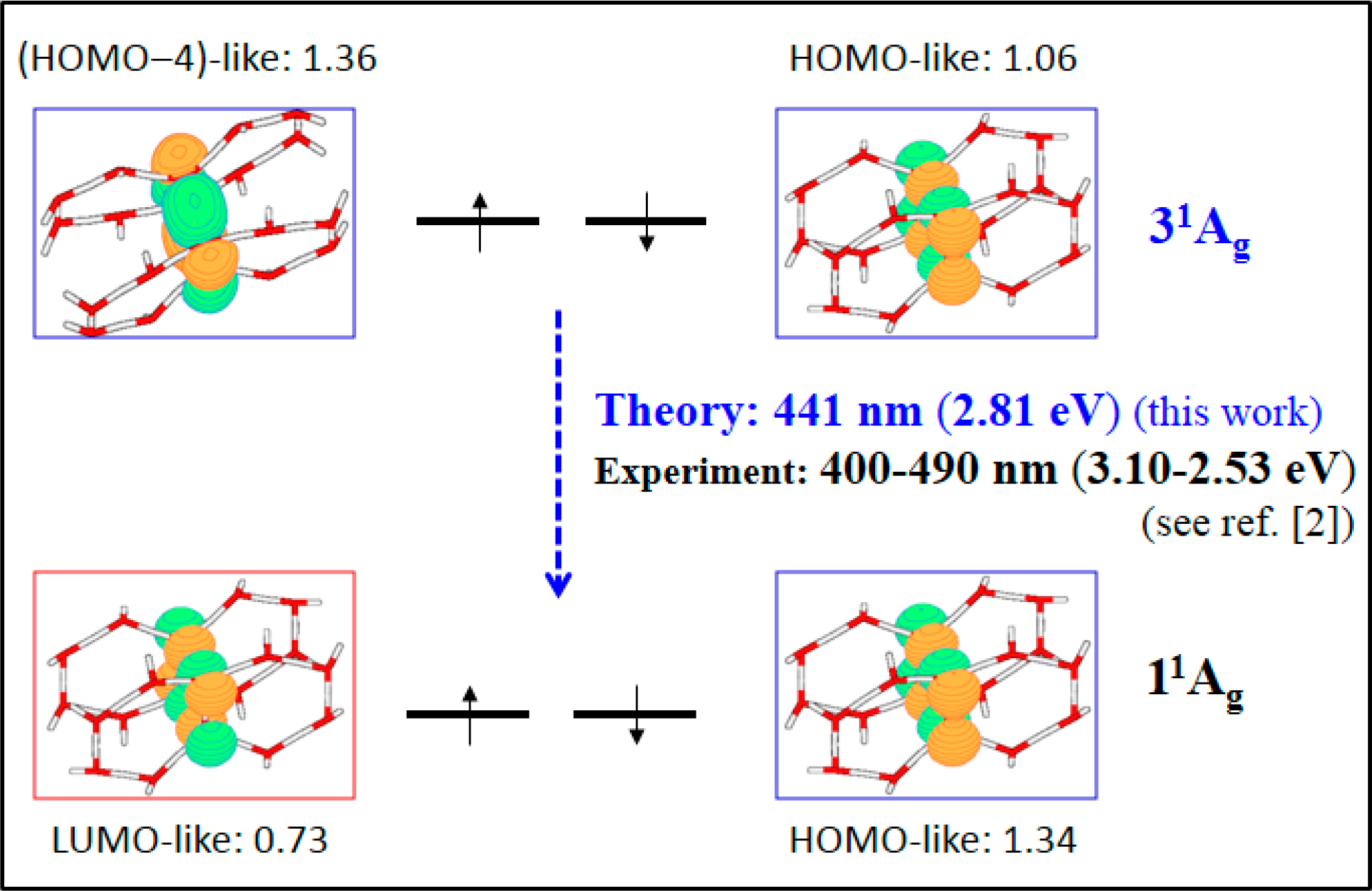
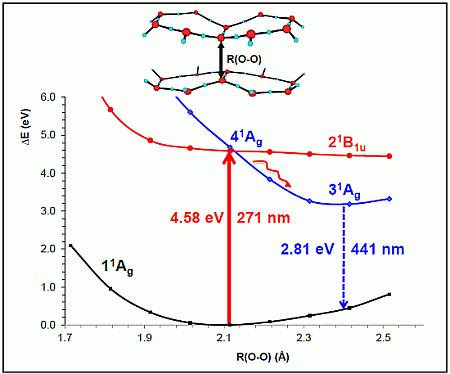
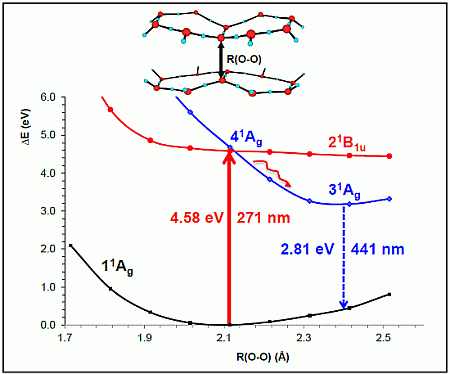
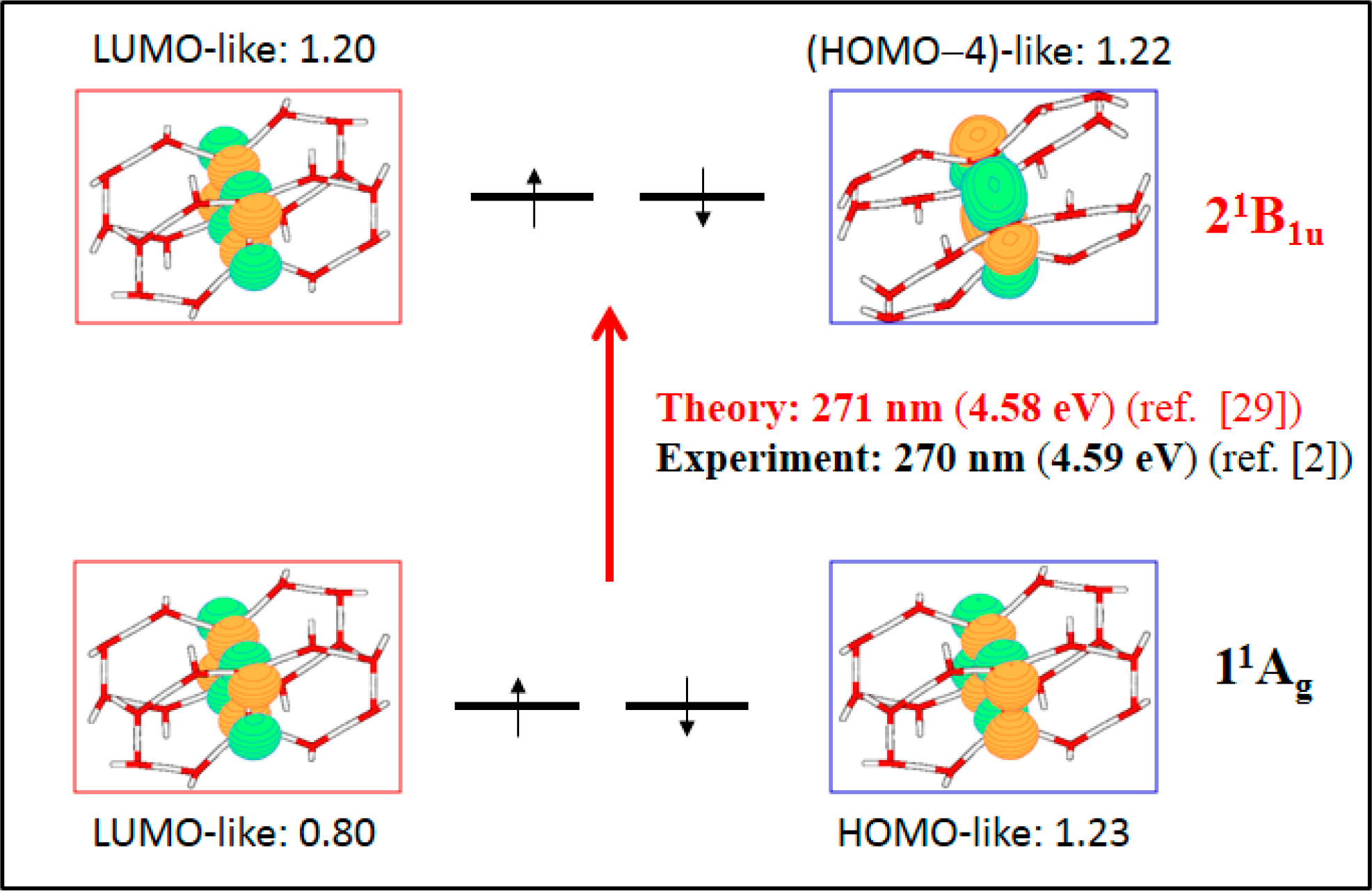
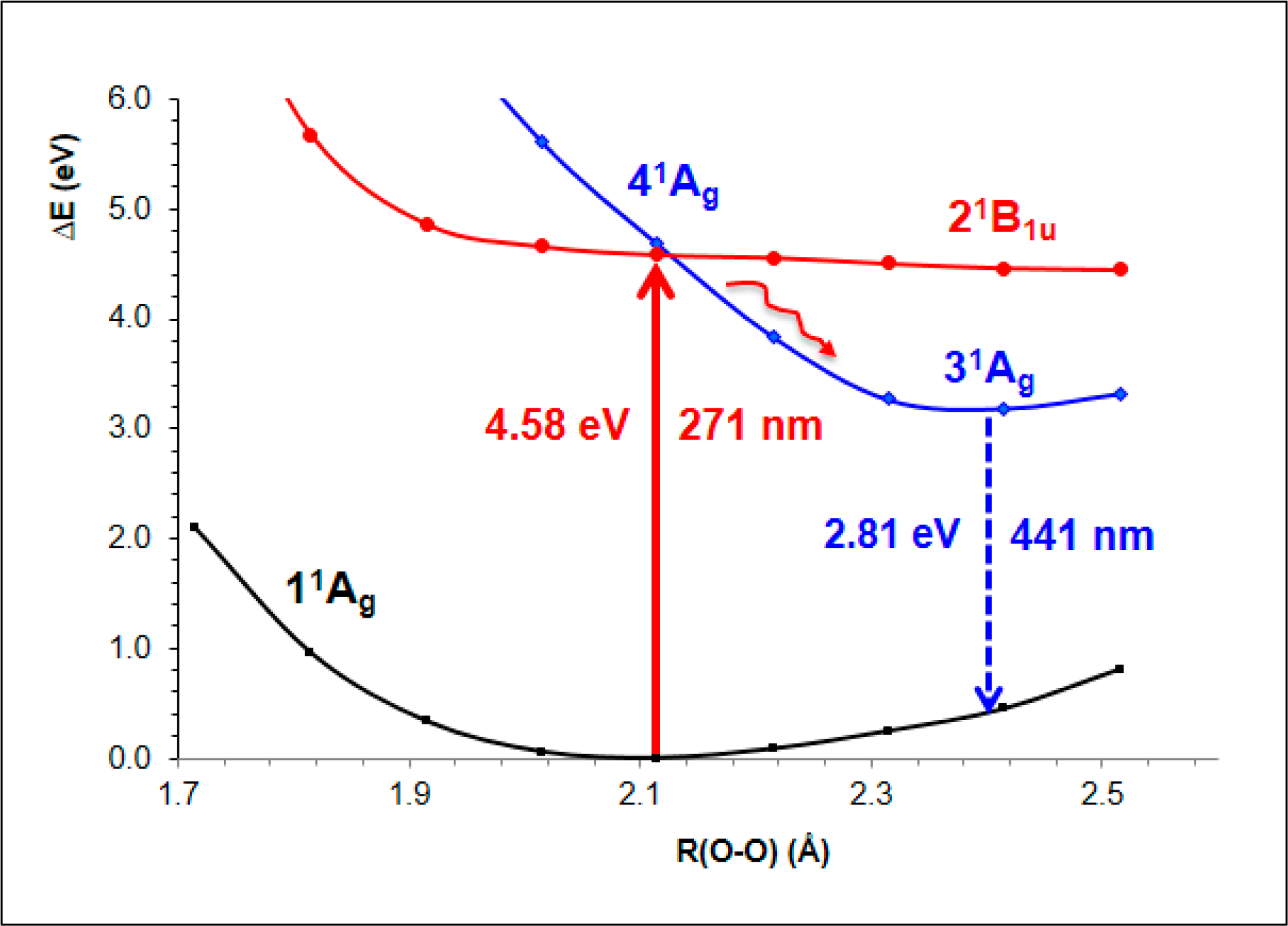
- The spectroscopic features recorded for structured water are fully consistent with those computed for the neutral molecular models used to describe the essentials of the multilayer honeycomb arrangements. The neutral π-stacked dimer [H38O20] has been revealed to be particularly suited for this purpose, both for the absorption and fluorescence events. The ground state of this unique system is equally well described by the 11Ag and 13B2u states. Namely, the lowest singlet and the lowest triplet states show degeneracy. The relative large attractive interaction (more than 2 eV) between the two monomers [H19O10] exerted by the (singlet/triplet) ground state of the dimer [H38O20] can be related to the exclusion of many substances from the zone adjacent to various hydrophilic surfaces determined experimentally. The neutral π-stacked dimer [H38O20] can be regarded as the main responsible for the recorded absorption with a computed band maximum at 271 nm (4.58 eV). On the other hand, the most intense vertical triplet→triplet transition is predicted to be at 318 nm (3.90 eV), which has been tentatively related to the absorption maximum observed in the range 290–310 nm (4.28–4.00 eV) for pristine water. The computed fluorescence features with band maximum at 441 nm (2.81 eV) is consistent with that registered experimentally in the range 400–490 nm (3.10–2.53 eV). In the singlet manifold, the absorbing bright excited state (21B1u) and the fluorescent emitting state (31Ag) have distinct nature, which nicely rationalizes the large Stokes shift concomitant to the spectroscopy of structured water. The population transfer from the bright to the dark state occurs in the vicinity of the ground-state equilibrium structure (R(O-O) = 2.115 Å). The release of radiation as fluorescence can be accomplished by a slight separation of the two monomers in the π-stacked dimer [H38O20] (R(O-O) = 2.390 Å).
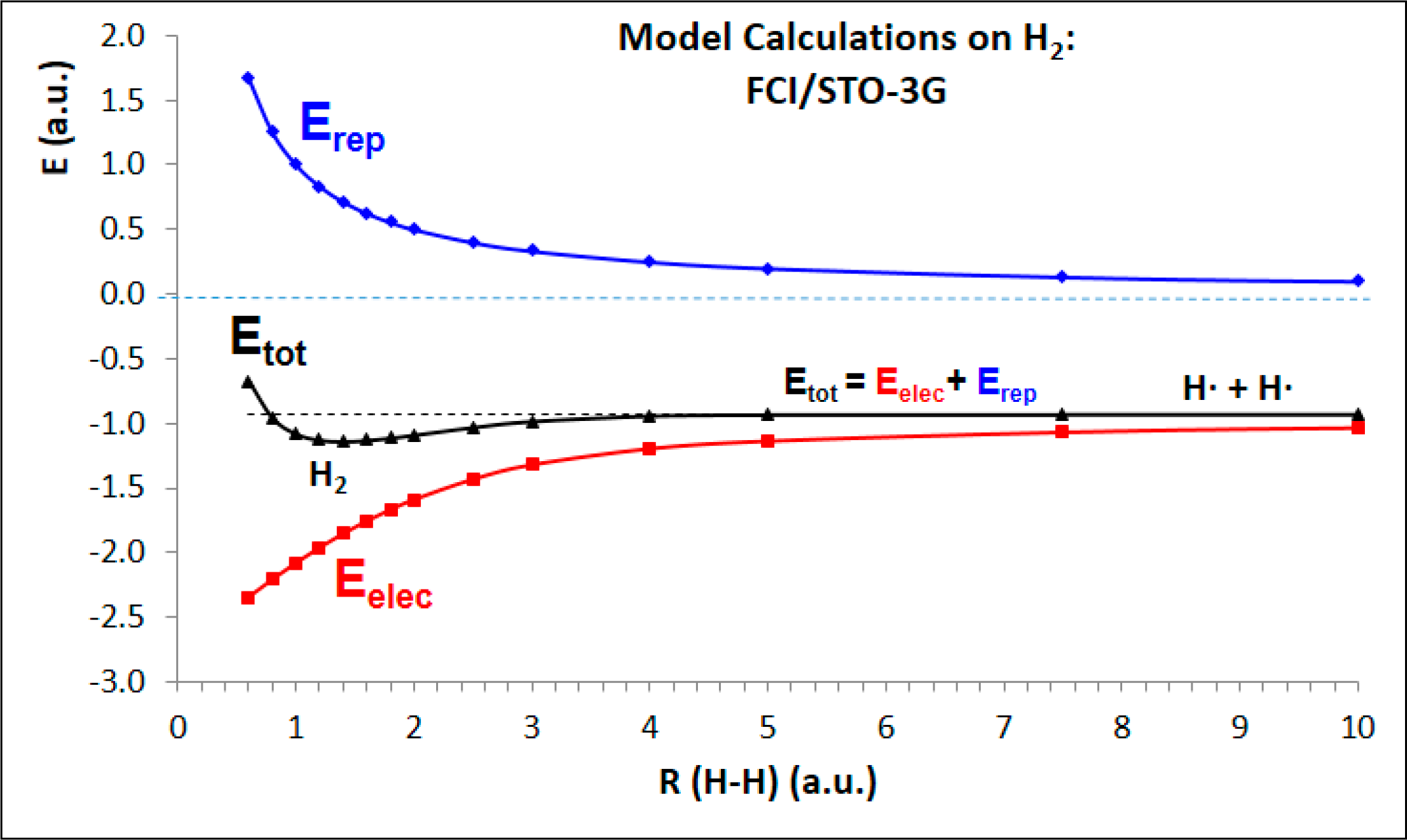
- The underlying reason behind the successful description of the molecular systems considered is grounded on the ability of the CASPT2 method to treat in a balance manner the electron correlation effects of the electronic states in the different hypersurfaces explored. Despite the latter statement is supported up to date by more than two thousand calculations of excited states dealing with many different types of compounds, the fact is particularly outstanding in this case involving crystalline-like systems formed by hexagonally arranged water moieties, where a basic concept such as atomic valence becomes uncertain and quantum-mechanical effects able to describe the attractive potential properly have to be accounted for in a large extent.
- The research work presented emphasizes the quantization of matter at the molecular level, yielding the resonance conditions for the interaction with the electromagnetic radiation. Both aspects are complementary to describe the event actually taking place. Therefore, the results provided fully support the scenario envisioned in the realm of biology by Albert Szent-Györgyi. In the conclusion section of the book entitled “Bioenergetics” [59], published in 1957, he stated: “Lucretian biochemistry involves the assumption that no interaction can take place between molecules without their touching one another. Support is given in this book to the idea that manifold interactions can take place without such bodily contact, either through energy bands or through the electromagnetic field, which thus appears with water and its structures as the matrix of biological reactions.” Quantum Chemistry shows indeed that water may form structures that absorb and transmit energy precisely in the same energy range as that arising from aromatic amino acids and DNA/RNA nucleobases.
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Ball, P. Water as an active constituent in cell biology. Chem. Rev 2008, 108, 74–108. [Google Scholar]
- Chai, B.H.; Zheng, J.M.; Zhao, Q.; Pollack, G.H. Spectroscopic studies of solutes in aqueous solution. J. Phys. Chem. A 2008, 112, 2242–2247. [Google Scholar]
- Cheng, J.X.; Pautot, S.; Weitz, D.A.; Xie, X.S. Ordering of water molecules between phospholipid bilayers visualized by coherent anti-Stokes Raman scattering microscopy. Proc. Natl. Acad. Sci. USA 2003, 100, 9826–9830. [Google Scholar]
- Feng, I.; Kuo, W.; Mundy, C.J. An ab initio molecular dynamics study of the aqueous liquid-vapor interface. Science 2004, 303, 658–660. [Google Scholar]
- Brown, G.E. How minerals react with water. Science 2001, 294, 67–69. [Google Scholar]
- Ostroverkhov, V.; Waychunas, G.A.; Shen, Y.R. New information on water interfacial structure revealed by phase-sensitive surface spectroscopy. Phys. Rev. Lett 2005, 94, 046102. [Google Scholar]
- Jena, K.C.; Covert, P.A.; Hore, D.K. The Effect of Salt on the Water Structure at a Charged Solid Surface: Differentiating Second- and Third-order Nonlinear Contributions. J. Phys. Chem. Lett 2011, 2, 1056–1061. [Google Scholar]
- Yoo, H.; Paranji, R.; Pollack, G.H. Impact of Hydrophilic Surfaces on Interfacial Water Dynamics Probed with NMR Spectroscopy. J. Phys. Chem. Lett 2011, 2, 532–536. [Google Scholar]
- Chen, X.; Yang, T.; Kataoka, S.; Cremer, P.S. Specific ion effects on interfacial water structure near macromolecules. J. Am. Chem. Soc 2007, 129, 12272–12279. [Google Scholar]
- Tychinsky, V. High Electric Susceptibility is the Signature of Structured Water in Water-Containing Objects. Water 2011, 3, 95–99. [Google Scholar]
- Bunkin, N.F.; Ignatiev, P.S.; Kozlov, V.A.; Shkirin, A.V.; Zakharov, S.D.; Zinchenko, A.A. Study of the Phase States of Water Close to Nafion Interface. Water 2013, 4, 129–154. [Google Scholar]
- Pollack, G.H. The Fourth Phase of Water: Beyond Solid, Liquid, and Vapor; Ebner and Sons: Seattle, WA, USA, 2013. [Google Scholar]
- Zheng, J.M.; Chin, W.C.; Khijniak, E.; Pollack, G.H. Surfaces and interfacial water: Evidence that hydrophilic surfaces have long-range impact. Adv. Colloid Interface Sci 2006, 127, 19–27. [Google Scholar]
- Ho, M.-W. Living Rainbow H2O; World Scientific: Singapore, Singapore, 2012. [Google Scholar]
- Bunkin, N.F.; Gorelik, V.S.; Kozlov, V.A.; Shkirin, A.V.; Suyazov, N.V. Colloidal Crystal Formation at the “Nafion-Water” Interface. J. Phys. Chem. B 2014, 118, 3372–3377. [Google Scholar]
- Chaplin, M. Do We Underestimate the Importance of Water in Cell Biology? Nat. Rev. Mol. Cell Biol 2006, 7, 861–866. [Google Scholar]
- Yoo, H.; Nagornyak, E.; Das, R.; Wexler, A.D.; Pollack, G.H. Contraction-Induced Changes in Hydrogen Bonding of Muscle Hydration Water. J. Phys. Chem. Lett 2014, 5, 947–952. [Google Scholar]
- Schee, C.R.V.D.; Ooms, K.J. Investigating Water Interactions with Collagen Using 2H Multiple Quantum Filtered NMR Spectroscopy To Provide Insights into the Source of Double Quantum Filtered Signal in Tissue. J. Phys. Chem. B 2014, 118, 3491–3497. [Google Scholar]
- So, E.; Stahlberg, R.; Pollack, G.H. Exclusion zone as an intermediate between ice and water. In Water and Society: Ecology and the Environment; Jorgensen, S.E., Brebbia, A., Popov, V., Eds.; WIT: Southampton, UK, 2012; Volume 153, pp. 1–9. [Google Scholar]
- Mota, R.; Parafita, R.; Giuliani, A.; Hubin-Franskin, M.J.; Lourenco, J.M.C.; Garcia, G.; Hoffmann, S.V.; Mason, N.J.; Ribeiro, P.A.; Raposo, M.; Limao-Vieira, P. Water VUV electronic state spectroscopy by synchrotron radiation. Chem. Phys. Lett 2005, 416, 152–159. [Google Scholar]
- Wernet, P.; Nordlund, D.; Bergmann, U.; Cavalleri, M.; Odelius, M.; Ogasawara, H.; Naslund, L.A.; Hirsch, T.K.; Ojamae, L.; Glatzel, P.; Pettersson, L.G.M.; Nilsson, A. The structure of the first coordination shell in liquid water. Science 2004, 304, 995–999. [Google Scholar]
- Carrasco, J.; Santra, B.; Klimes, J.; Michaelides, A. To Wet or Not to Wet? Dispersion Forces Tip the Balance for Water Ice on Metals. Phys. Rev. Lett 2011, 106, 026101. [Google Scholar]
- Carrasco, J.; Hodgson, A.; Michaelides, A. A molecular perspective of water at metal interfaces. Nat. Mater 2012, 11, 667–674. [Google Scholar]
- McGeoch, J.E.M.; McGeoch, M.W. Entrapment of water by subunit c of ATP synthase. J. R. Soc. Interface 2008, 5, 311–318. [Google Scholar]
- Cicero, G.; Grossman, J.C.; Schwegler, E.; Gygi, F.; Galli, G. Water Confined in Nanotubes and between Graphene Sheets: A First Principle Study. J. Am. Chem. Soc 2008, 130, 1871–1878. [Google Scholar]
- Kimmel, G.A.; Matthiesen, J.; Baer, M.; Mundy, C.J.; Petrik, N.G.; Smith, R.S.; Dohnálek, Z.; Kay, B.D. No Confinement Needed: Observation of a Metastable Hydrophobic Wetting Two-Layer Ice on Graphene. J. Am. Chem. Soc 2009, 131, 12838–12844. [Google Scholar]
- Michaelides, A.; Morgenstern, K. Ice nanoclusters at hydrophobic metal surfaces. Nat. Mater 2007, 6, 597–601. [Google Scholar]
- Segarra-Martí, J.; Coto, P.B.; Rubio, M.; Roca-Sanjuán, D.; Merchán, M. Towards the understanding at the molecular level of the structured-water absorption and fluorescence spectra: A fingerprint of π-stacked water. Mol. Phys 2013, 111, 1308–1315. [Google Scholar]
- Segarra-Martí, J.; Roca-Sanjuán, D.; Merchán, M. On the hexagonal ice-like model of structured water: Theoretical analysis of the low-lying excited states. Comput. Theor. Chem 2014. [Google Scholar] [CrossRef]
- Szabo, A.; Ostlund, N.S. Modern Quantum Chemistry: Introduction to Advanced Electronic Structure Theory; Dover Publications: Mineola, NY, USA, 1996. [Google Scholar]
- Andersson, K.; Malmqvist, P.-Å.; Roos, B.O. 2nd-Order Perturbation-Theory with a Complete Active Space Self-Consistent Field Reference Function. J. Chem. Phys 1992, 96, 1218–1226. [Google Scholar]
- Merchán, M.; Serrano-Andrés, L. Ab Initio Methods for Excited States. In Computational Photochemistry; Olivucci, M., Ed.; Elsevier: Amsterdam, The Netherlands, 2005; Volume 16, pp. 35–91. [Google Scholar]
- Roca-Sanjuán, D.; Aquilante, F.; Lindh, R. Multiconfiguration second-order perturbation theory approach to strong electron correlation in chemistry and photochemistry. Comput. Mol. Sci 2012, 2, 585–603. [Google Scholar]
- Roos, B.O.; Andersson, K.; Fulscher, M.P.; Malmqvist, P.A.; Serrano-Andrés, L.; Pierloot, K.; Merchán, M. Multiconfigurational perturbation theory: Applications in electronic spectroscopy. Adv. Chem. Phys 1996, 93, 219–331. [Google Scholar]
- Serrano-Andrés, L.; Merchán, M. Quantum chemistry of the excited state: 2005 overview. J. Mol. Struct 2005, 729, 99–108. [Google Scholar]
- Merchán, M.; Serrano-Andrés, L.; Fülscher, M.P.; Roos, B.O. Multiconfigurational Perturbation Theory Applied to Excited States of Organic Molecules. In Recent Advances in Multireference Theory; Hirao, K., Ed.; World Scientific Publishing: Singapore, Singapore, 1999; Volume 4, pp. 161–195. [Google Scholar]
- Serrano-Andrés, L.; Merchán, M. Spectroscopy: Applications. In Encyclopedia of Computational Chemistry; Schleyer, P.V.R., Ed.; Wiley: Chichester, UK, 2004. [Google Scholar]
- Aquilante, F.; De Vico, L.; Ferré, N.; Ghigo, G.; Malmqvist, P.A.; Neogrady, P.; Pedersen, T.B.; Pitonak, M.; Reiher, M.; Roos, B.O.; Serrano-Andrés, L.; Urban, M.; Veryazov, V.; Lindh, R. Software News and Update MOLCAS 7: The Next Generation. J. Comput. Chem 2010, 31, 224–247. [Google Scholar]
- Aquilante, F.; Pedersen, T.B.; Veryazov, V.; Lindh, R. MOLCAS—a software for multiconfigurational quantum chemistry calculations. Comput. Mol. Sci 2013, 3, 143–149. [Google Scholar]
- Forsberg, N.; Malmqvist, P.-Å. Multiconfiguration perturbation theory with imaginary level shift. Chem. Phys. Lett 1997, 274, 196–204. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H.P.; Izmaylov, A.F.; Bloino, J.; Zheng, G.; Sonnenberg, J.L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgometry, J.A., Jr; Peralta, J.E; Ogliaro, F.; Bearpark, M.; Heyd, J.J.; Brothers, E.; Kudin, K.N.; Staroverov, V.N.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J.C.; Iyengar, S.S.; Tomasi, J.; Coss, M.; Rega, N.; Millam, J.M.; Klene, M.; Knox, J.E.; Cross, J.B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R.E.; Yazyev, O.; Austin, A.J.; Cammi, R.; Pomelli, C.; Ochterski, J.W.; Martin, R.L.; Morokuma, K.; Zakrzewski, V.G.; Voth, G.A.; Salvador, P.; Dannenberg, J.J.; Dapprich, S.; Daniels, A.D.; Farkas, O.; Foresman, J.B.; Ortiz, J.V.; Cioslowski, J.; Fox, D.J. GAUSSIAN 09 Revision B.01; Gaussian, Inc: Wallingford, CT, USA, 2010. [Google Scholar]
- Serrano-Andrés, L.; Merchán, M.; Borin, A.C. A Three-State Model for the Photophysics of Guanine. J. Am. Chem. Soc 2008, 130, 2473–2484. [Google Scholar]
- Serrano-Andrés, L.; Merchán, M.; Borin, A.C. Adenine and 2-Aminopurine: Paradigms of Modern Theoretical Photochemstry. Proc. Natl. Acad. Sci. USA 2006, 103, 8691–8696. [Google Scholar]
- Lippincott, E.R.; Stromberg, R.R.; Grant, W.H.; Cessac, G.L. Polywater. Science 1969, 164, 1482–1487. [Google Scholar]
- Chai, B.H.; Yoo, H.; Pollack, G.H. Effect of Radiant Energy on Near-Surface Water. J. Phys. Chem. B 2009, 113, 13953–13958. [Google Scholar]
- Rubio, M.; Serrano-Andrés, L.; Merchán, M. Excited states of the water molecule: Analysis of the valence and Rydberg character. J. Chem. Phys 2008, 128, 104305. [Google Scholar]
- Segarra-Martí, J.; Roca-Sanjuán, D.; Merchán, M.; Lindh, R. On the photophysics and photochemistry of the water dimer. J. Chem. Phys 2012, 137, 244309. [Google Scholar]
- Olaso-González, G.; Roca-Sanjuán, D.; Serrano-Andrés, L.; Merchán, M. Toward the understanding of DNA fluorescence: The singlet excimer of cytosine. J. Chem. Phys 2006, 125. [Google Scholar] [CrossRef]
- Roca-Sanjuán, D.; Olaso-González, G.; González-Ramírez, I.; Serrano-Andrés, L.; Merchán, M. Molecular basis of DNA photodimerization: Intrinsic production of cyclobutane cytosine dimers. J. Am. Chem. Soc 2008, 130, 10768–10779. [Google Scholar]
- González-Ramírez, I.; Climent, T.; Serrano-Pérez, J.J.; González-Luque, R.; Merchán, M.; Serrano-Andrés, L. The role of pyrimidine nucleobases excimers in DNA photoreactivity. Pure Appl. Chem 2009, 81, 1695–1705. [Google Scholar]
- Olaso-González, G.; Merchán, M.; Serrano-Andrés, L. The Role of Adenine Excimers in the Photophysics of Oligonucleotides. J. Am. Chem. Soc 2009, 131, 4368–4377. [Google Scholar]
- Climent, T.; González-Ramírez, I.; González-Luque, R.; Merchán, M.; Serrano-Andrés, L. Cyclobutane Pyrimidine Photodimerization of DNA/RNA Nucleobases in the Triplet State. J. Phys. Chem. Lett 2010, 1, 2072–2076. [Google Scholar]
- González-Ramírez, I.; Roca-Sanjuán, D.; Climent, T.; Serrano-Pérez, J.J.; Merchán, M.; Serrano-Andrés, L. On the photoproduction of DNA/RNA cyclobutane pyrimidine dimers. Theor. Chem. Acc 2011, 128, 705–711. [Google Scholar]
- Giussani, A.; Segarra-Martí, J.; Roca-Sanjuán, D.; Merchán, M. Excitation of Nucleobases from a Computational Perspective I: Reaction Paths. Top. Curr. Chem 2013. [Google Scholar] [CrossRef]
- Lange, K.K.; Tellgren, E.I.; Hoffmann, M.R.; Helgaker, T. A Paramagnetic Bonding Mechanism for Diatomics in Strong Magnetic FIelds. Science 2012, 337, 327–331. [Google Scholar]
- Pokorny, J.; Pokorny, J. Biophysical Pathology in Cancer Transformation. J. Clin. Exp. Oncol 2013, S1, 1–9. [Google Scholar]
- Roy, R.; Tiller, W.A.; Hoover, M.R. The Structure of Liquid Water: Novel Insights from Materials Research. Indian J. Res. Homeopath 2009, 3, 1–24. [Google Scholar]
- DeMeo, J. Water as a Resonant Medium for Unusual External Environmental Factors. Water 2011, 3, 1–47. [Google Scholar]
- Szent-Györgyi, A. Bioenergetics; Academic Press: New York, NY, USA, 1957. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
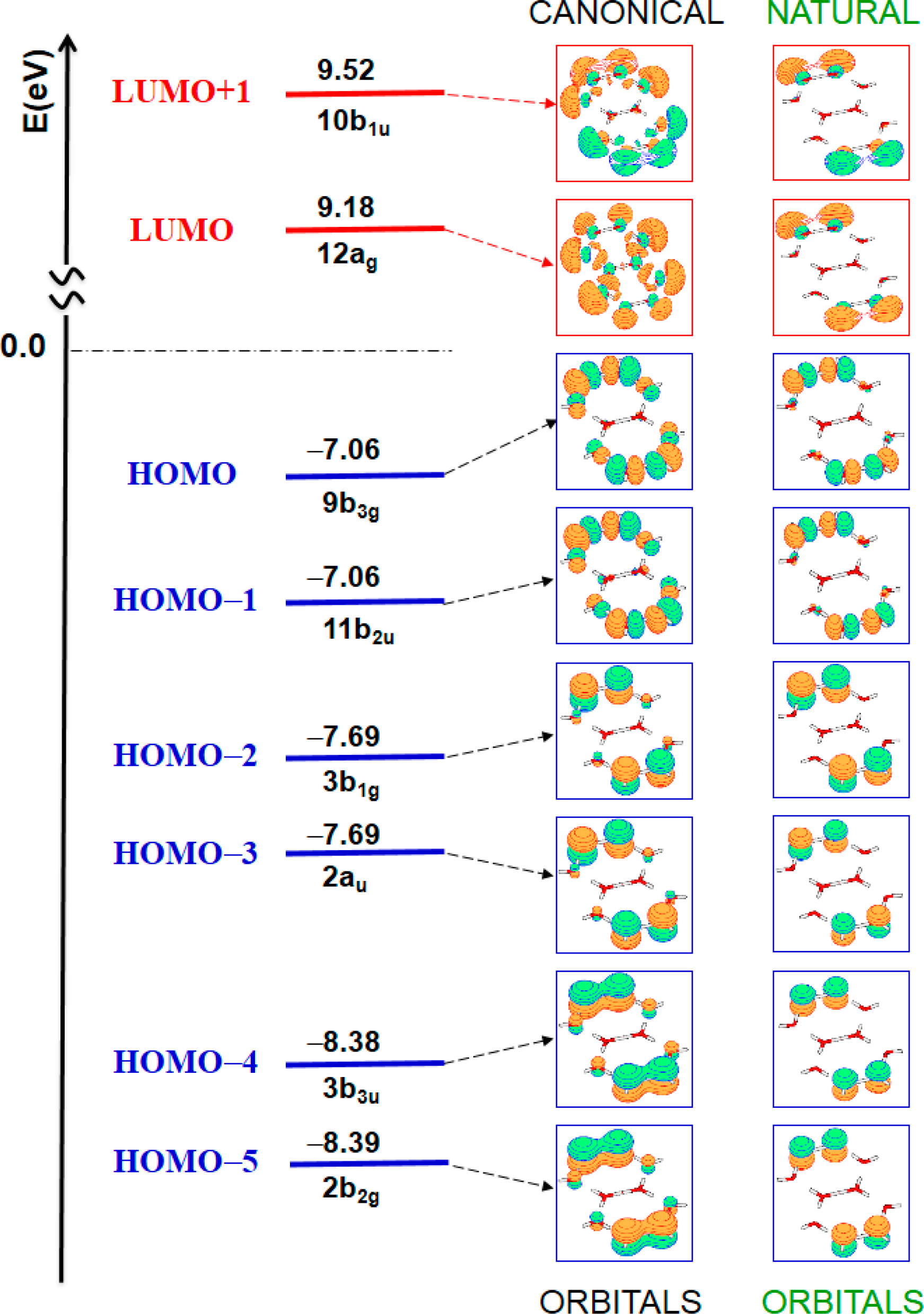
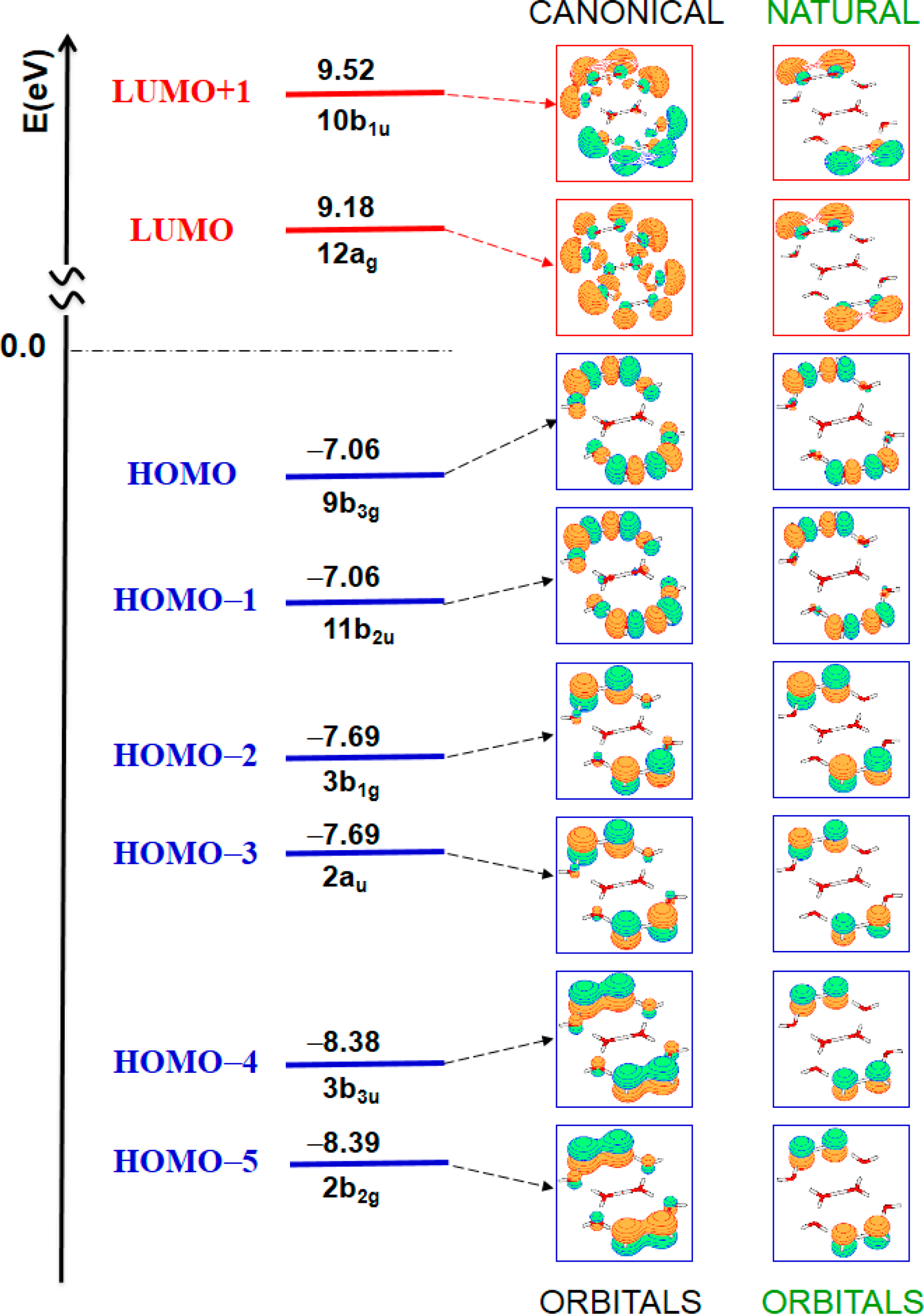
| State | ΔE [eV] | ΔE [nm] | Oscillator strength | Assignment (Weight) a | |
|---|---|---|---|---|---|
| 11Ag (ground state) | closed-shell HF | (98%) | |||
| 11B1g | 7.42 | 167 | forbidden | HOMO–2 → LUMO | (46%) |
| HOMO–3 → LUMO+1 | (45%) | ||||
| 11Au | 7.42 | 167 | forbidden | HOMO–3 → LUMO | (46%) |
| HOMO–2 → LUMO+1 | (45%) | ||||
| 11B3g | 7.49 | 166 | forbidden | HOMO → LUMO | (51%) |
| HOMO–1 → LUMO+1 | (48%) | ||||
| 11B2u | 7.51 | 165 | 0.2431 | HOMO–1 → LUMO | (49%) |
| HOMO → LUMO+1 | (49%) | ||||
| 11B3u | 8.34 | 149 | 0.0382 | HOMO–4 → LUMO | (44%) |
| HOMO–5 → LUMO+1 | (42%) | ||||
| 11B2g | 8.34 | 149 | forbidden | HOMO–5 → LUMO | (43%) |
| HOMO–4 → LUMO+1 | (43%) | ||||
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Segarra-Martí, J.; Roca-Sanjuán, D.; Merchán, M. Can the Hexagonal Ice-like Model Render the Spectroscopic Fingerprints of Structured Water? Feedback from Quantum-Chemical Computations. Entropy 2014, 16, 4101-4120. https://doi.org/10.3390/e16074101
Segarra-Martí J, Roca-Sanjuán D, Merchán M. Can the Hexagonal Ice-like Model Render the Spectroscopic Fingerprints of Structured Water? Feedback from Quantum-Chemical Computations. Entropy. 2014; 16(7):4101-4120. https://doi.org/10.3390/e16074101
Chicago/Turabian StyleSegarra-Martí, Javier, Daniel Roca-Sanjuán, and Manuela Merchán. 2014. "Can the Hexagonal Ice-like Model Render the Spectroscopic Fingerprints of Structured Water? Feedback from Quantum-Chemical Computations" Entropy 16, no. 7: 4101-4120. https://doi.org/10.3390/e16074101
APA StyleSegarra-Martí, J., Roca-Sanjuán, D., & Merchán, M. (2014). Can the Hexagonal Ice-like Model Render the Spectroscopic Fingerprints of Structured Water? Feedback from Quantum-Chemical Computations. Entropy, 16(7), 4101-4120. https://doi.org/10.3390/e16074101