Relative Entropy, Interaction Energy and the Nature of Dissipation


Abstract
: Many thermodynamic relations involve inequalities, with equality if a process does not involve dissipation. In this article we provide equalities in which the dissipative contribution is shown to involve the relative entropy (a.k.a. Kullback-Leibler divergence). The processes considered are general time evolutions both in classical and quantum mechanics, and the initial state is sometimes thermal, sometimes partially so. By calculating a transport coefficient we show that indeed—at least in this case—the source of dissipation in that coefficient is the relative entropy.1. Introduction
The distinction between heat and work, between the uncontrollable flow of energy of molecular processes and the controllable flow of energy usable by an agent, underlies all of thermodynamics, and is implicitly incorporated in the equation dE = đW + đQ. This distinction is defined by human subjectivity and by the human technological ability to extract work from the flow of energy of microscopic processes.
On the other hand, assuming that the evolution is given by the fundamental laws of dynamics, classical or quantum, one must represent the evolution of a system at a fundamental microscopic level by the action of a unitary operator on a quantum state (density operator) in quantum situations or by the action of a symplectic operator on a classical state (probability distribution) in classical situations. The state of the system evolves according to the Heisenberg equation of motion or according to the Liouville equation respectively. An immediate consequence is that the entropy of the exact microscopic state of a system that is isolated or is coupled only to an external source of work stays constant during the evolution. In particular, the microscopic state cannot tend to an equilibrium state. Thus, the information content of the exact microscopic state stays constant during the evolution. The problem is that in practice it is impossible both to define the microscopic state and to follow its exact evolution. The definition of the state and the representation of the exact evolution using unitary or symplectic dynamics are thus untenable idealizations. Nevertheless, and this constitutes a paradox, these idealizations cannot be ignored or dismissed, because it is precisely the difference between the exact evolution and its standard approximations which explains and can be used to predict dissipative effects, both of energy and information.
In thermodynamics, in kinetic theories or in stochastic dynamics, the exact microscopic state of a system is replaced by an approximate or “coarse-grained” state and the corresponding exact evolution is replaced by an evolution of the corresponding coarse-grained state (or, in standard thermodynamics by a quasi-static or formal evolution). There are two main reasons for using these approximate states and evolutions:
- (1)
As discussed, it is impossible—even in principle—to specify the exact state of a large system and follow its evolution. An attempt at extremely high precision would modify the system, even in a classical context (related to Maxwell’s demon). And it is even worse for quantum systems. Moreover, this would be useless.
- (2)
Only slow variables (on the time scales of microscopic processes) can be measured with confidence and stability. As a result, an observer can only describe the system as a state of minimal information (or maximal entropy) compatible with the observed slow variables [1,2].
The coarse-grained state is thus a statistical data structure which summarizes at a given moment the knowledge of the observer. The evolution of this coarse-grained state merely reflects the evolution of the knowledge of the observer about the system. The observer cannot follow the microscopic processes, but only the slow variables which can be measured and used, and as a consequence there is a loss of information about the details of the microscopic processes; in the traditional language of thermodynamics, entropy increases or is produced. The observer, reflecting a particular state of knowledge (or more precisely, a lack of knowledge), describes the state of the system as a state of minimal information (or maximal entropy) compatible with observation. Thus, entropy is not a kind of substance flowing from one part of a system to another part or mysteriously produced internally by the physical system, as is often suggested by many texts of thermodynamics or statistical physics: it is only the observer’s partial inability to relate the exact microscopic theory to a reduced macroscopic description in order to use the system as a source of useful work or information. This is what is measured as an increase of entropy or by entropy production. The macroscopic state is the result of a statistical inference (specifically, maximum entropy) for the given, observed, macroscopic variables (which are the slow variables of the system [1,2]. This point of view on the nature of entropy was emphasized by Jaynes, who observed [3,4], “The expression ‘irreversible process’ represents a semantic confusion…”
The difference between the exact evolution of the microscopic ideal state and the evolution of its coarse-grained approximation is what is called “dissipation,” both of information content and of energy or other “useful” variables. Standard thermodynamics uses the maximal coarse graining of equilibrium, and the idealized evolution is not modeled explicitly, so dissipation can be taken into account only by inequalities. For more detailed coarse-graining (as in hydrodynamics, Boltzmann’s equation, kinetic theories or stochastic thermodynamics) one can obtain an estimate for the dissipative effects, for example, by the calculation of transport coefficients.
In this article, our main purpose is to prove that the relative entropy term between the initial and final states measures dissipation. In our approach, “dissipation” is defined as the difference between the maximal work that the physicist thinks could be extracted from a system when using the thermodynamic or quasi-static theory to make predictions, and the work that is actually extracted because the system is evolving according to the exact dynamics, classical or quantum, independently of what the physicist thinks (see also our use of relative entropy in [5] and [6], where the context was more limited [7]). Moreover, in the present context we find that the relative entropy terms are proportional to the square of the interaction energy. In all standard theories, dissipative effects are measured by the transport coefficients of energy or momentum or concentration of chemical species. Thus, we need prove that the relative entropy allows the calculation of transport coefficients. Indeed, we show below that the relative entropy terms provide the calculation of the thermal conductivity between two general quantum systems, initially at thermal equilibrium at different temperatures. This is a kind of Fourier law, except that we do not suppose a linear regime, so that the temperature dependence is more complicated than simple linearity. Moreover, our exact calculation of the transport coefficient shows that it is indeed proportional to the square of the interaction energy, which confirms that for vanishingly small interaction energy no transfer occurs in finite time. In other words, no power or finite rate of information flow can be extracted from a system if one does not have at the same time dissipative effects.
In the following material, we first consider a system comprised of two components, A and B. We make no specific hypotheses on the size of the systems, and we do not introduce thermal reservoirs. Thus, the identities we derive are in effect exact tautologies. In Sections 3 to 5, we present several identities. We here mention two examples: (1) a derivation of the Brillouin-Landauer estimate of the energy necessary to change the information content of a system; (2) an estimate of the work that can be extracted from a two-part system in interaction with an external source of work in terms of non-equilibrium free energies and relative entropy of the state before and after the evolution. Similar identities were also obtained recently by Esposito et al. [8], Reeb and Wolf [9], and Takara et al. [10] Continuing, we study the effect of an external agent on an (otherwise) isolated system; again we obtain an identity relating the work to the difference of internal (not the free) energies along with the usual dissipative terms. Then, we derive the relation between the relative entropy and the heat conductivity in a quantum system. Finally, we define a general notion of coarse-graining or reduced description, which includes the usual notions. In some of our examples one or both systems are initially at thermal equilibrium, but only the initial temperatures appear explicitly in the definition of the non-equilibrium free energies. The latter are no longer state functions because they depend explicitly on the initial temperature and not on the actual effective temperature. No coarse graining by an effective final or intermediate thermal state is used, and neither system is a reservoir.
2. Notations and Basic Identities
2.1. States and Entropy
Many results will be valid both in classical and quantum contexts. We denote by ρ either a probability distribution function over a classical phase space, or a density matrix in the quantum case. We denote by Tr either the integral on the phase space, or the trace operation. Thus ρ is a positive quantity and satisfies Tr ρ = 1. The entropy of ρ is
It is defined up to a multiplicative constant. (Classically ρ should be divided by a dimensional constant to render it dimensionless.)
The relative entropy (see [11]) is defined by
where ρ and ρ′ are states.
One has
and S(ρ|ρ′) does not depend on the units in phase space. Moreover S(ρ|ρ′) = 0 if and only if ρ = ρ′.
Writing S(ρ|ρ′) as − Tr ρ log ρ′ − (− Tr ρ log ρ), suggests the following interpretation: Suppose the true state is ρ, but the observer thinks that the state is ρ′. S(ρ|ρ′) is then the true average of the missing information minus the estimate of the missing information.
2.2. The Basic Identity
If we add and subtract S(ρ′) in the second member of Equation (2), we obtain the basic identity
Most of our results follow from this identity.
When ρ′ is a thermal state at (inverse) temperature β [12],
where
is the partition function. With ρ′ = ρβ, the identity (4) reduces to
Here H is a given function or operator.
Defining the free energy of state ρ by
we obtain
and F (ρβ, H) is the equilibrium free energy related to the partition function by
Equation (9) is important for applications, because its right hand side can be related to energy dissipation (up to the factor β), which gives a clear physical meaning to the relative entropy (see Section 3).
2.3. Evolution Operators and Entropy
We assume that the system (classical or quantum) evolves under the action of an arbitrary operator U (symplectic or unitary). If ρ is a state, we denote by ρ(U) the new state after the evolution U.
Entropy is conserved by the evolution
For example, in the quantum case, we have ρ(U) = Uρ U†, where U is the propagator: , U|t=0 = 1, with H a possibly time-dependent Hamiltonian.
If ϕ(ρ) is a functional of ρ which evolves with U, and ϕ(ρ(U)) is the functional after evolution of ρ, we denote the variation of ϕ(ρ) after the evolution U in the following way
Remark 1: Many of our results are valid for a general evolution U which is not symplectic or unitary, for example stochastic evolution.
3. Two Systems in Interaction
A basic procedure in thermodynamics is to consider the evolution and properties of an otherwise isolated two-part system. Although it is often the case that the overall system conserves energy, for the subsystems more general behavior is often seen.
3.1. Hypotheses
We assume that the system is formed of two parts, A and B, in interaction. At time-0, the state is a product state
After the evolution U, the state is ρ(U) and we denote by and its marginals,
which are then states on A and B respectively. We also assume that there is a quantity H that is conserved by the evolution and H has the form
where HA and HB are quantities depending only on A and B respectively and VAB is an interaction term. Then, if we denote
our hypothesis is that
In particular this is the case if U is time-evolution with Hamiltonian H.
Remark 2: For this situation, certain results are also valid without the assumption that the evolution U preserves the energy H.
If ρ is a state corresponding to a system formed of two parts, A and B, and ρA and ρB are its marginals (as in Equation (14)), then the relative entropy S (ρ|ρA ⊗ ρB) is the same as the mutual information of the associated distributions. It can be interpreted as the amount of information in ρ that comes from the fact that A and B are in interaction (see [11]). This quantity will appear in many of our relations below (e.g., Equation (28)) as part of the dissipation.
3.2. Relation between a State and Its Marginals
Assuming Equation (13) (that the initial state is a product state), one has the identity
Indeed, using the conservation of the entropy of ρ during the evolution U,

This is because one evidently has . Note that Equation (21) requires that U preserve the entropy. One has also the well-known inequality
which is a particular case of
for any state ρ.
Note that the stronger result, Equation (21), is obtained by retaining the relative entropy term in this equation. The same remark will apply in most of the following results.
3.3. The Case Where A is Initially in a Thermal State
At time 0 we take ρA,0 to be thermal with temperature βA,
where ZA(βA) = ZA(βA, HA) is the partition function, (6). From Equation (7) with and , we deduce (note that this requires that HA be independent of time)
and as a consequence
The last two equations do not require that U be a unitary evolution conserving the entropy, nor that it conserve the energy.
Remark 3: This inequality can be found in [13] as an unnumbered equation. Its consequences were not deduced in that reference.
Remark4: Note that it is the initial temperature that appears in Equations (25) and (26). Moreover, is not in general an equilibrium state.
Suppose that B starts in an arbitrary initial state ρB,0, while A begins in the thermal state . Combining Equations (21) and (25), we obtain
The last equation requires that U preserve entropy, since that feature is used in the derivation of Equation (21). It also remains valid if the Hamiltonian of B, HB, depends on an external parameter varying with time, so that B receives work from an external agent. This is because the entropy-preserving property only depends on U being unitary (or symplectic). On the other hand, HA should be time independent (see the parenthetical remark before Equation (25)). Then if U conserves energy
These relations imply the following inequalities:
- (1)
If U preserves entropy, even if HB depends on an external parameter varying with time
- (2)
If U conserves entropy and the total energy, one has
with the following interpretations. Suppose U conserves the entropy; then we couple a system B (initially in an arbitrary state ρB,0) to system A (initially in thermal equilibrium) and that we want to lower the entropy of B so that δ(U(t)) S(ρB) ≤ 0. Then, the energy of A must increase by at least
even if B receives work from an external source (so that HB depends on an external parameter). Moreover, if the total energy is conserved, the sum of the energy of B and the coupling energy must decrease by at least:
Thus lowering the entropy of a system B, coupled to a system initially at equilibrium, costs transfers of energy from B to A or to the interaction energy; thus the sum of B’s energy and the interaction energy must decrease, but B’s energy alone need not decrease. This is a result analogous to those of Brillouin [14] and Landauer [15] (reprinted in Leff and Rex [16]), even if system B receives work from an external source. But note again that only the temperature βA appears. This is the initial temperature at the beginning of the evolution U, so that system A is not necessarily a thermal bath, because its temperature may vary during the evolution U.
3.4. The Case of Equality in Equation (31)
It is important to study the case where the previous inequalities are changed into equalities, because this occurs if and only if strong conditions are verified: expressing these conditions is one of the advantages of our approach.
Equation (31) was derived under the hypothesis δ(U) S(ρB) < 0. Then by Equation (27) one has . This implies and . Because , so by Equation (31), δ(U) S(ρB) = 0. But we could also derive δ(U) S(ρB) = 0 from Equation (21), because δ(U(t)) S(ρA) = 0 and .
3.5. Both Systems A and B are at Equilibrium
Assume that A and B are initially at thermal equilibrium at different temperatures. Then, one has
- (1)
For a general evolution
- (1)
If U conserves entropy
- (1)
If U conserves energy
Then, we conclude
- (A)
If U conserves entropy: Combining Equations (33), (34), and (35) yields
It is easy to check directly that
Thus
![Entropy 16 03173f2]()
This last identity implies the Clausius-like inequality
- (B)
For a general evolution U: Combining Equations (33) and (34)
and thus
This may be viewed as an inequality for free energies of A and B at the respective temperatures TA and TB. Note again that during the time evolution neither A nor B need remain in thermal states.
3.6. Case of Equality in (39) (2nd) and (41)
- (A)
U conserves entropy. Equality in Equation (39) implies immediately that and , in which case the energy of A and the energy of B have not changed and δ(U(t)) S(ρA) = δ(U(t)) S(ρB) = 0. From the first equality in Equation (38) one has
and one deduces that the state ρ has not changed.
- (B)
General evolution U. If one has equality in Equation (41), it follows from Equation (40) the same results as above: the state ρ has not changed.
3.7. Interaction Energy and Relative Entropy
It is often assumed that the interaction energy between the parts of the complete system can be neglected but, obviously, if this were exactly true the subsystems would evolve independently. Of course, an interaction can be small but nevertheless have significant impact when it persists for long times. However, there are cases where even for short times the interaction cannot be neglected. Assume then that U conserves entropy and energy. Divide Equations (33) and (34) by βB and add; then use the conservation of energy Equation (36) to eliminate δ(U) EB(ρ) and deduce after some calculations
so that
In case of equality in Equation (44), one deduces that so that the state has not changed and δ(U) EA(ρA) = δ(U) EV (ρ) = 0. Moreover if δ(U) EA(ρA) is positive and TA is larger than TB, the interaction energy V is necessarily not zero and δ(U) EV (ρ) is negative.
Finally, if one could neglect the interaction energy, Equation (44) implies that energy flows from the hot to the cold system.
3.8. The Case βA = βB
Again assume that U conserves both entropy and energy. From Equation (43) and the conservation of energy, one deduces
so that δ(U) EV (ρ) ≤ 0. Thus when A and B are initially at thermal equilibrium at the same temperature, the sum of the energies of A and B can only increase at the expense of the interaction energy [17].
4. Two Systems in Interaction With a Work Source
The problem of converting heat into work, first treated by Carnot, was at the origin of classical thermodynamics. Here, to address this issue, we explicitly introduce a work source interacting with two systems A and B, before focusing in Section 5 on the interaction of one system with a work source.
4.1. Hypotheses
We consider two systems A and B in interaction, with system A coupled to a work source. We represent the action of the work source by parameters, collectively denoted by λ, so that HA = HA(λ). Thus we assume that HB and V are independent of λ. The action of the work source is given by an evolution of the parameters λ(t) imposed by an external agent. The total system A+B has a unitary or symplectic evolution U(t) depending explicitly on time-t. Clearly, U(t) conserves entropy but does not conserve energy, and instead one has the identity
with the following notation
Here, λ0 is the initial value of the parameter λ and λ(U) is its final value at the end of the evolution U, this being an abbreviation for U(t), t being the final time. Note that Equation (49) extends the definition given near Equation (12). Such an extension is needed because we now allow changes in the Hamiltonian, represented by the additional variable λ. Equation (46) defines the work δ(U)W, which is taken to be positive if the source receives work from the system A + B. (Note that this is opposite to the usual convention which was implicit in the opening paragraph of this paper.)
We assume that initially A and B are in independent thermal states, but A depends on the work source parameter λ. The complete initial state at time 0 is thus
with
and
Here, FA(βA, λ0) denotes the equilibrium free energy for A. For a general state ρ of a system with energy H we define the non equilibrium free energy of the state ρ at temperature T to be
In particular, for subsystem A one can define the non equilibrium free energy of the state at temperature βA to be
In both of the above formulas temperature is not necessarily related to the state ρ.
4.2. Identities for the Work
We next establish the following two relations
![Entropy 16 03173f3]()
and
![Entropy 16 03173f4]()
with the non equilibrium free energy of calculated at the initial temperature TA, namely Equation (55), . We will comment on these relations in Par. 4.3 Remark 5: Here the free energy of Equation (55) is not a state function, because it is calculated at the initial temperature of A. Note our notation: When we write FA(βA, λ0) this is the equilibrium free energy of the thermal state of A at temperature βA and external parameter λ0. When we write we mean the non-equilibrium free energy, as defined above.
Proof of Equation (56): One again starts from the fundamental identities Equations (7) and (25)
and
Note that Equation (59) is just Equation (7) with the substitutions and . Therefore it contains the initial HA(λ0) (referring to ), not the final one. Equation (60) is likewise a rewriting of Equation (35).
Now add Equations (58) and (59) and subtract Equation (60), using the fact that
(this is the same as our unnumbered equation between Equations (37) and (38)) we obtain
Conservation of energy Equation (46) gives
![Entropy 16 03173f5]()
We eliminate the second trace in the right hand side of Equation (63) using Equation (62), multiply by TA to obtain Equation (56).
Proof of Equation (57): In Equation (56), we replace the relative entropy term, using
![Entropy 16 03173f6]()
and use the definition of of Equation (55)
4.3. Inequalities for the Work
From the identities of Equations (56) and (57), we deduce immediately corresponding inequalities
and
The interpretation of inequality (67) is straightforward. If one can neglect the interaction energy, and if TA = TB, one gets an analogue of the familiar thermodynamic inequality giving an upper bound between the work received by the work source and the variation of the free energy of A,
Note that this relation is not restricted to cycles, nor to exchanges with thermal baths (which would stay in their initial thermal states).
Remark 6: Equation (57) contains much more information than inequalities Equations (67) and (68), since it expresses the difference between the maximum work that can be delivered by system A and the work effectively extracted from A, which is the energy dissipated in the process. It is expressed in terms of relative entropies, and it will be shown in Section 6 that it can be explicitly estimated, which yields a calculation of transport coefficients from first principles.
4.4. The Case of Equalities in Equations (66) and (67)
If one has equality in Equation (66), the relative entropy of Equation (56) must be equal to 0,
so and the final state has come back to its initial value. If we have equality in Equation (67), both relative entropies of Equation (57) are equal to 0. In this case has come back to its initial value and δ(U)EB(ρ) = 0. Then, one has
4.5. Case Where A is not Initially in Thermal Equilibrium
We shall now assume that the initial state is
ρA,0 being a general state.
The following identity also holds:
This equation, true for any initial state ρA, can be found in [8,18]. Note the temperature of B appearing in the non equilibrium free energy of A
If no work is performed, Equation (72) reduces to Equation (28) upon exchanging the labels A and B. Proof: Using S(ρ(U)) = S(ρ0) and the definition of the thermal state, one has
Then,
and Equation (74) becomes
Using the conservation of energy, Equation (46), one obtains
Here
is the variation of the non equilibrium free energy of A calculated at the initial temperature TB of B and
In particular
which gives a general upper bound for the work production from heat exchanges between an arbitrary system A and a system B initially at equilibrium (not necessarily a heat bath). In this relation, equality is realized if and only if the two relative entropy terms of Equation (77) are zero, which means that
Remark 7: By convention, a thermal bath is in a thermal state which is assumed to remain constant during the evolution. Our system B is not a thermal bath in this sense; its state varies during the evolution.
5. A System Coupled Only to an External Work Source
While Carnot and many others primarily considered model machines exchanging heat with several reservoirs, new thermodynamic relations have recently been announced [19] concerning exchanges of a single system with a work source. We now focus on this case.
5.1. Hypotheses
We consider a system coupled only to an external work source, so that the Hamiltonian of the system is H(λ).
At time t = 0, the state of the system is supposed to be a thermal state . The external observer imposes an evolution λ(t) of the parameter λ from λ0 to λ(U), inducing a unitary or symplectic evolution U of the whole system. The work that the external observer must perform to realize this evolution is obviously the variation of the energy of the system. With the convention of Section 4.1, we denote by δ(U(t))W the work counted positive if the external source receives it from the system. We are now in a particular case of Section 4.1 when the system is A, there is no system B and no V. Thus from Equation (46)
5.2. Identities for the Work
From Equations (56) and (57) we obtain immediately
and
with F(U) the non equilibrium free energy at temperature β0.
We now prove the following identity
This is a particular case of the result of [19].
Proof of Equation (86): We start from Equation (84) written as
Now
But
so that comparing Equations (88) and (89), one has
and from Equation (87) we then deduce Equation (86).
Remark 8: Since the transition under discussion is adiabatic, free energy is less suitable for inequalities of the form (86) than is internal energy. See Section 5.6.
5.3. Inequalities for the Work
5.3.1. From Equation (83)
From Equation (83) we deduce
with equality if and only if ,i.e., the final state is the initial state.
5.3.2. From Equation (86)
From Equation (86) we deduce
with equality if and only if
That is, ρ(U) is the thermal state at the initial temperature and final value λ(U) of λ. Note that a necessary condition for this is that the entropy of the final thermal state is the same as the entropy of the initial state.
5.4. Relation to the Identity of Jarzynski
Let z denote a point in the phase space of the system. In this section we assume that the dynamics is classical.
We denote by z(s|z0) the classical trajectory of the phase space point at time s starting from z0 at time s = 0, for the classical evolution U. The external observer imposes the variation λ(s) of λ from λ0 to λ(U) = λ(t). The identity of Jarzynski is [20]:
![Entropy 16 03173f7]()
Because the exponential function is strictly convex, Jensen’s inequality implies that
![Entropy 16 03173f8]()
so that using Equation (94) and taking the logarithm, one obtains
which is the inequality (92).
But if the inequality (96) is an equality, we deduce as in Equation (93), but we also deduce that the inequality of Jensen (95) is an equality. Because the exponential function is strictly convex, this implies that the differences
where C is a constant independent of z0 (but obviously dependent on λ0, λ(U) and t); in other words, the “microscopic work” is independent of the microscopic trajectory. Although this equality would seem impossible, it turns out that identity (97) can be realized for certain systems and evolutions of λ (see appendix A).
5.5. Effective Temperatures
Let H(λ) be a Hamiltonian depending on λ and ρ a state (classical or quantum) with energy E(ρ) = Tr (ρH(λ)). We can define two temperatures for ρ.
- (i)
The temperature βe(ρ, λ) is the temperature such that
with . It is known that ∂E(β, λ)/∂β < 0, so that Equation (98) defines βe unambiguously. The basic identity (4) shows that
so that
which is the well known fact that maximizes the entropy among all states ρ having a fixed energy. The quantity βe(ρ, λ) can be called the effective temperature.
- (ii)
There is a second temperature βa(ρ, λ) such that
In this definition, S(β, λ) is the entropy of a thermal state with temperature β and external parameter λ. We call this the adiabatic temperature, and by the same arguments as given above it is well-defined. From Equation (100) and Equation (101), one has
Because
we deduce from Equation (102) that
and
Because S is a strictly increasing function of E (for λ fixed), one sees that in Equation (102) or (104), one has equality if and only if βa = βe. Moreover, one has the identity
which can immediately be verified.
5.6. A More Precise Expression for the Work
In thermodynamics, for an adiabatic evolution, the work is related to the internal energy by dE = −dW, rather than to the free energy. Similarly, the work is related to the adiabatic temperature rather than to the effective energy temperature.
Given the state ρ(U) (corresponding to the evolution U, the parameter varying from λ0 to λ(U)) we can define the adiabatic temperature such that
We prove the following identity
Proof of Equation (108): One has by definition (82)
Then
![Entropy 16 03173f9]()
because by the definition (107). From this result and Equation (109) we deduce Equation (108).
As a consequence of Equation (108), we deduce the inequality
In standard thermodynamics, for system thermally isolated and coupled to a work source, one has dE = −dW, because δ(U(t)) S = 0 for an adiabatic (thermally isolated) process and we recover equality in Equation (111). In this situation, the inequality (92) comparing the work to the difference of free energies is not relevant, because the temperature does not remain constant.
Note that the work upper bound (111), given in terms of energy and the adiabatic temperature, is sharper than the bound given by (92), which is in terms of free energy. This is proved in the next subsection.
5.7. Upper Bounds on the Work Delivered by a System. Comparison of Equations (92) and (111)
We next show that using internal energy for the work inequality gives a sharper result than using the free energy. Specifically,
Proof of Equation (112): We need only prove that
Using the definition of the equilibrium free energy and Equation (107) we have
Note that in Equation (114) all terms involving λ are evaluated at λ(U). Therefore
But
Using Equation (116) in Equation (115), we obtain
But < 0, so that ∆ ≥ 0. Note that this does not depend on which of β0 and is larger.
5.8. The Case of Equalities in Equations (111) and (92)
5.8.1. Equality in Equation (111)
In this case, one has in Equation (108) so
In particular, ρ(U) is a thermal state so that
However, if one has equality in Equation (111), this does not improve the upper bound of Equation (92) for the free energy,
In other words, the optimal bound for δ(U(t)) W is given by the internal energy and not the free energy (so that the internal energy in general yields a better bound than that given in [20]).
5.8.2. Equality in Equation (92)
From Equation (93) we deduce that
so that ρ(U) is a thermal state and thus
This implies that we also have equality in Equation (111)
5.9. The Case λ(U) = λ0
If one assumes that the final value λ(U) of λ is equal to its initial value, we see immediately that .Indeed
so that the temperatures are equal . In this case, one has from Equation (108)
with equality if and only if
so that the state has returned to its initial value.
Remark 9: If the external observer imposes a variation λ(t) of the control parameter with λ(0) = λ0, λ(t1) = λ1, λ(tf) = λ0, inequality (125) says that at the end of the cycle, the observer has always lost work. In particular, the work that the external observer has put in the system in the time interval [0, t1] cannot be entirely recovered in the time interval [t1, tf] whatever one does, except if the final state ρ(U) is the initial state.
Remark 10: When λ(U) = λ0, one can also recover Equation (125) from the identity (86). This identity reduces to
6. Relative Entropy, Energy Dissipation and Fourier’s Law
In this Section we derive dissipation in the quantum context and show it to be intimately related to the relative entropy.
6.1. The Born Approximation
A quantum system has a Hamiltonian
Let be the eigenstates and eigenvalues of H0. In the Born approximation, the state becomes at time t a state with
The quantities satisfy
where
We assume here Vn,n = 0 for all n. One readily deduces that in the Born approximation
and by unitarity , so to second order in
Let ρ0 be an initial state diagonal in the basis
Then, at time t, the state becomes
![Entropy 16 03173f10]()
If L is a Hermitian operator diagonal in the basis with eigenvalues λn, using Equation (135) one obtains in the Born approximation
6.2. Two Interacting Systems
We consider two quantum systems A, B with Hamiltonians HA, HB respectively, interacting. Denote by V = VA,B the interaction energy and
We call the eigenstates and eigenvalues of HA (resp. HB), and we apply the Born approximation to H, with H0 = HA + HB. The non perturbed Hamiltonian H0 has eigenstates with eigenvalues .
We assume that at time t = 0, the state of the system A + B is ρ0 = ρA ⊗ ρB with
![Entropy 16 03173f11]()
so that they are diagonal in the eigenbasis of HA and HB and therefore commute with HA +HB. At time t, the initial state ρ0 = ρA ⊗ ρB evolves to ρ(t). Then
But S(ρ(t)) = S(ρ0) by unitarity of the evolution, so that
This is of the form of Equation (136) with
L has eigenvectors with eigenvalues log pA,k + log pB,l; ρA ⊗ ρB has the same eigenvectors with eigenvalues pA,k + pB,l. Applying Equation (136), one obtains in the Born approximation
![Entropy 16 03173f12]()
Notice that the quantity in the right hand side is automatically non-negative. Here, we have
![Entropy 16 03173f13]()
with .
We also deduce from this result that in this approximation S(ρ(t)|ρA ⊗ ρB) = 0 if and only if V = 0 (recall that the diagonal elements of V are 0).
6.3. The Case Where Both Initial States are Thermal
Assume that at time and are the thermal states of A and B respectively. From Equation (37) one has
Moreover, from conservation of energy
so that eliminating δ(U(t)) EB(ρB), one obtains
We now estimate both terms on the right hand side of Equation (146).
6.3.1. Estimate of the Relative Entropy
From Equation (142) we deduce
![Entropy 16 03173f14]()
Moreover, when t → ∞, as in the usual Born approximation, Equation (143) shows that
Thus if fA and fB denote the density of states for A and B, we obtain from Equation (147)
![Entropy 16 03173f15]()
6.3.2. Estimate of the Interaction Energy
Because δ(U(t)) V (ρ) = −δ(U(t)) EA(ρ) − δ(U(t)) EB(ρ), one has
This is of the form of Equation (136) with L = −HA − HB, and so
![Entropy 16 03173f16]()
Up to a sign, this expression is formally identical to the expression Equation (142), except that the difference of energies (EA,k + EB,l − EA,n − EB,m) replaces the quantity βA(EA,k − EA,n) + βB(EB,l − EB,m). As a consequence EA,k + EB,l − EA,n − EB,m partially cancels the denominator of and one sees that is negligible when t → ∞.
Then from Equations (146) and (149), one sees that
In Equation (152) K is the positive constant
![Entropy 16 03173f17]()
with
![Entropy 16 03173f18]()
It is obvious that φ ≥ 0. Note that K does not vanish for βA close to βB.
The expression (152) is a form of Fourier’s law for heat transport from B to A, (βA − βB)K being the rate of dissipation. In this case, one sees that the significance of the relative entropy is that of a transport coefficient, here the transport of energy from one part of a system to another part.
7. Coarse Grained States
Coarse-graining is omnipresent in in macroscopic and mesoscopic physics, since microscopic variables are often not what is observed. In general coarse-graining represents a loss of information, hence an increase in entropy. In this section we consider a variety of coarse-graining procedures, and consistent with our work in this article, relative entropy plays a significant role both in the definition of coarse-graining and in the measures of entropy increase.
7.1. Definition
Let ρ and ρ′ be two states of the same system (classical or quantum). We say that ρ′ is obtained from ρ by a coarse graining operation if
The idea is that the information associated with ρ′ (namely log ρ′) is the same whether one averages with ρ′ or with the more detailed distribution ρ. Using the basic identity, Equation (4), we can say that ρ′ is obtained from ρ by a coarse graining operation, if and only if
In particular, S(ρ′) ≥ S(ρ), so that the entropy increases by coarse-graining. (See the comment after Equation (3).)
A coarse-graining mapping is a mapping Γ which associates to any state ρ (or to some states of a given class), a coarse grained state ρ′ = Γ(ρ).
7.2. Examples of Coarse-graining Mappings
Example 1: Maximum entropy.
Let A1,…, An be observables of the system, so they are either functions in the phase space or hermitian operators on the Hilbert space of the system. We consider the class of states ρ such that
One can then consider the state ρ′ such that ρ′ has maximal entropy given the relation
It is immediately seen that
where C is a normalization constant and αi are the “conjugate parameters”, (provided ρ′ is normalizable). The mapping Γ: ρ → ρ′ is indeed a coarse grain mapping in the sense of the previous definition, because by Equation (159)
In particular, one has Equation (156).
The case of the thermal state is the best known, where one takes A = H, the Hamiltonian of the system.
Example 2: Naive coarse graining; the observables as characteristic functions.
- (i)
Classical case: Let Z be the phase space of the system and {Zi} a finite partition of Z ( and Zi∩Zj = Ø for i ≠ j). We choose (i.e. the characteristic function of Zi). This is a particular case of example 1 and if ρ is a state
Using the condition (156), namely,
one can deduce from Equations (161) and (162)
This equation implies that ρ′ is normalized ∫ ρ′dz = 1. We recover the usual coarse graining.
- (ii)
Quantum case: Let be the Hilbert space of the system and Pi a resolution of the identity by orthogonal projectors
Then the analogue of Equation (163) is
Example 3: Coarse graining by marginals.
- (i)
Classical case: We assume that the system consists of several parts, and that its phase space is a Cartesian product, , corresponding to various subsystems with phase space Zi. If ρ is a state on Z, we denote by ρi its marginal probability distribution on Zi, so
Let Γ be the mapping that associates the product of its marginals to ρ(z)
Then the condition (155) is satisfied. It is easy to see that is the state ρ′ that maximizes the entropy among all the states ρ″ such that for any i.
- (i)
Quantum case. The Hilbert space of the system is
(168)where the are the Hilbert spaces of the subsystems. If ρ is a state, then its marginal state on is the partial trace on the Hilbert space , which is the tensor product of the Hilbert spaces for j different from i
and the mapping Γ,
is a coarse grained mapping. Γ(ρ) is again the state ρ′ which maximizes the entropy among all states ρ″ such that for all i.
Example 4: Decomposition of Z.
If , but the Zi do not form a partition of Z (they can have intersections of non-zero measure), one can still apply Example 1 to and obtain
But now Equation (163) is no longer valid because, for given z, there will be in general several i with z ∈ Zi.
7.3. Coarse Graining and Relative Entropy
- (i)
The case of the naive coarse-graining is distinguished among all types of coarse-graining by the following property. Let a partition of the phase space and p, q two probability distributions on Z. Let and be the coarse grained states of p and q associated to this partition. Then one has
Proof: call and . We have, using the definition of and
Now
![Entropy 16 03173f19]()
But . We use the fact that the function x log x is convex, so that for each i
![Entropy 16 03173f20]()
Therefore from Equation (175)
- (ii)
For the coarse-graining associated to subsystems one has and if p, q are states on Z, the coarse grained states are and we deduce immediately that
Consider the case i = 1, and call z = (z1, z′) with z′ = (z2,…, zn) and call Z′ = Z2 × ⋯ × Zn. Then
Back to TopTop![Entropy 16 03173f21]()
Now . As in Equation (175), we use the convexity of x log x and deduce that
![Entropy 16 03173f22]()
From Equation (177) we deduce that for the coarse graining mapping associated to the division of in n subsystems, one has
Remark 11: The upper bound of Equation (180) cannot be improved. Indeed consider the case where: p(z1,…, zn) = p1(z1)δ(z1 − z2) … δ(zn−1 − zn) q(z1,…, zn) = q1(z1)δ(z1 − z2) … δ(zn−1 − zn). Then pi = p1 and qi = q1, but S(p|q) = S(p1|q1) and .
- (iii)
Thermal coarse graining.
Let Z be a phase space, and p and q two probability distributions on Z, H(z) a function of z ∈ Z.
Let and be the thermal coarse grained probability distributions of p and q, respectively, with respect to H. So
where β(p) is the effective temperature of p, i.e., .
Assuming that p − q is small, an obvious bound, after straightforward calculations (expanding to second order in p − q), is
This bound is surely not optimal, because if p and q are already thermal states, . Note though that even without the hypotheses on p and q, .
8. Conclusions
The results in this article are used to obtain upper bounds for entropy production or energy variation in various situations of thermodynamic interest, with many such results either new or sharper than similar known bounds. Furthermore, the energy dissipated in these processes is expressed in terms of relative entropies, which not only gives a general microscopic interpretation of dissipation, but also, in relevant examples, leads to an explicit, first principles, evaluation of dissipation terms, analogous to the Fourier law.
Although relative entropy has made appearances in many contexts, especially with respect to information theory, our results on a generalized Fourier heat law relates it in a direct way to the notion of dissipation as understood in physics.
Acknowledgments
We (B.G. and L.S.) are grateful to the visitor program of the Max Planck Institute for the Physics of Complex Systems, Dresden, for its hospitality during the time that much of this work was performed. We also thank one of the referees for an extremely careful reading of the manuscript, leading we believe to considerable improvement in presentation and content.
Author Contributions
The authors contributed equally to the presented mathematical framework and the writing of the paper.
Conflicts of Interest
The authors declare no conflict of interest.
Appendixe
A. An Example of Trajectory-Independent Microscopic Work
We exhibit a Hamiltonian H(z, λ) and an evolution λ(t) of the external parameter such that
with C independent of z0.
Take the harmonic oscillator
Call
![Entropy 16 03173f23]()
the solution with λ = 0.
For λ(s) a function of time s, the solutions of the Hamiltonian equations starting from (x0, p0) at s = 0 are
with and Assume that λ0 = 0. Define and . Then
![Entropy 16 03173f24]()
We can impose a condition on t such that this quantity does not depend on x0 and p0, namely
![Entropy 16 03173f25]()
Then using these two equalities, one has
Thus if λ(t) ≠ 0, we can arrange that the microscopic work is independent of the initial condition and is non zero.
B. An Exactly Solvable Model
The system A + B is formed of two two-levels atoms. The Hamiltonians of A and B are
with eigenstates |0A〉, |+A〉, |0B〉, |+B〉, so that the total Hamiltonian is in the basis |0A, 0B〉, |+A, 0B〉, |0A, +B〉, |+A, +B〉:
where w is the interaction energy.
Calling E0 = EA + EB, the eigenvalues of H are
as well as EA and EB. The eigenstates of EA and EB are |+A, 0B〉, |0A, +B〉, and the eigenstates of λ± are
so that
Here is the normalization factor.
The initial state is :
![Entropy 16 03173f26]()
Using these formulas one can compute
and verify that
Then
and
![Entropy 16 03173f27]()
![Entropy 16 03173f28]()
Using Equation (146), one obtains
Here these quantities are periodic functions of period . Near resonance, where λ+ ≃ λ−, w ≃ 0, E0 = EA +EB ≃ 0 and we recover that δ(U(t)) EA(ρ) ≃ K(βA − βB)t from Equation (A22).
C. Example: Forced Harmonic Oscillator
We take the Hamiltonian
with the condition λ(0) = 0. The classical action is
![Entropy 16 03173f29]()
where C(t) does not depend on x or x′. The quantum propagator is
where “≃” indicates that we have not written the normalization factor. This factor does not depend on x or x′ and is at the moment unimportant. The thermal state for λ = 0 is
The time-evolved state at time-t is
The energy at time-t, using λ(t) = 0, is
with . Define
The calculation of the double Gaussian integral in Equation (A27) gives
where N(t) is the normalization factor
The action of the Hamiltonian on the propagated state is
We define the variable X as
Then the energy of the propagated state at time t is
![Entropy 16 03173f30]()
and E(0) is the value of E(t) at t = 0, so that
Finally using the values of A and A′ in terms of I1 and I2, we obtain
This is independent of β and is positive. As a corollary, this result is valid if one propagates any eigenstate of the Hamiltonian H0. One can also derive the classical energy
where ρβ(λ = 0) is the classical thermal state. One uses the equations of motion
and then
If λ(0) = 0 but λ(t) ≠ 0, one gets
![Entropy 16 03173f31]()
This can be negative, for example if λ(t) = t:
References and Notes
- Landau, L.D.; Lifshitz, E.M. Statistical Physics; Pergamon Press: Oxford, UK, 1980. [Google Scholar]
- Schulman, L.S.; Gaveau, B. Coarse grains: The emergence of space and order. Found. Phys 2001, 31, 713–731. [Google Scholar]
- Jaynes, E.T. Information theory and statistical mechanics. Phys. Rev 1957, 106, 620–630. [Google Scholar]
- Jaynes, E.T. Information theory and statistical mechanics II. Phys. Rev 1957, 108, 171–190. [Google Scholar]
- Gaveau, B.; Schulman, L.S. Master equation based formulation of non-equilibrium statistical mechanics. J. Math. Phys 1996, 37, 3897–3932. [Google Scholar]
- Gaveau, B.; Schulman, L.S. A general framework for non-equilibrium phenomena: The master equation and its formal consequences. Phys. Lett. A 1997, 229, 347–353. [Google Scholar]
- In [5] and [6] we found the relative entropy to be related to dissipation, but we there dealt with stochastic dynamics and our conclusions were limited to short times.
- Esposito, M.; Lindenberg, K.; van den Broeck, C. Entropy production as correlation between system and reservoir. New J. Phys 2010, 12, 013013. [Google Scholar]
- Reeb, D.; Wolf, M.M. (Im-)Proving Landauer’s Principle 2013, arXiv, 1306.4352v2.
- Takara, K.; Hasegawa, H.H.; Driebe, D.J. Generalization of the second law for a transition between nonequilibrium states. Phys. Lett. A 2010, 375, 88–92. [Google Scholar]
- Cover, T.M.; Thomas, J.A. Elements of Information Theory; Wiley: New York, NY, USA, 1991. [Google Scholar]
- Henceforth we suppress the qualifier “inverse” in referring to various β’s as temperatures.
- Partovi, M.H. Quantum thermodynamics. Phys. Lett. A 1989, 137, 440–444. [Google Scholar]
- Brillouin, L. Science and Information Theory, 2nd ed.; Academic Press: New York, NY, USA, 1962. [Google Scholar]
- Landauer, R. Irreversibility and heat generation in the computing process. IBM J. Res. Dev 1961, 5, 183–191. [Google Scholar]
- Leff, H.S.; Rex, A.F. Maxwell’s Demon: Entropy, Information, Computing; Princeton University Press: Princeton, NJ, USA, 1990. [Google Scholar]
- Schulman, L.S.; Gaveau, B. Ratcheting Up Energy by Means of Measurement. Phys. Rev. Lett 2006, 97, 240405. [Google Scholar]
- Esposito, M.; van den Broeck, C. Second law and Landauer principle far from equilibrium. Europhys. Lett 2011, 95, 40004. [Google Scholar]
- Kawai, R.; Parrondo, J.M.R.; Van den Broeck, C. Dissipation: The Phase-Space Perspective. Phys. Rev. Lett 2007, 98, 080602. [Google Scholar]
- Jarzynski, C. Nonequilibrium Equality for Free Energy Differences. Phys. Rev. Lett 1997, 78, 2690–2693. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS StyleGaveau, B.; Granger, L.; Moreau, M.; Schulman, L.S. Relative Entropy, Interaction Energy and the Nature of Dissipation. Entropy 2014, 16, 3173-3206. https://doi.org/10.3390/e16063173
AMA Style
Gaveau B, Granger L, Moreau M, Schulman LS. Relative Entropy, Interaction Energy and the Nature of Dissipation. Entropy. 2014; 16(6):3173-3206. https://doi.org/10.3390/e16063173
Chicago/Turabian Style
Gaveau, Bernard, Léo Granger, Michel Moreau, and Lawrence S. Schulman. 2014. "Relative Entropy, Interaction Energy and the Nature of Dissipation" Entropy 16, no. 6: 3173-3206. https://doi.org/10.3390/e16063173
APA Style
Gaveau, B., Granger, L., Moreau, M., & Schulman, L. S. (2014). Relative Entropy, Interaction Energy and the Nature of Dissipation. Entropy, 16(6), 3173-3206. https://doi.org/10.3390/e16063173
Article Metrics
Entropy, EISSN 1099-4300, Published by MDPIFurther Information
Article Processing Charges Pay an Invoice Open Access Policy Contact MDPI Jobs at MDPIGuidelines
For Authors For Reviewers For Editors For Librarians For Publishers For Societies For Conference OrganizersMDPI Initiatives
Sciforum MDPI Books Preprints.org Scilit SciProfiles Encyclopedia JAMS Proceedings Series
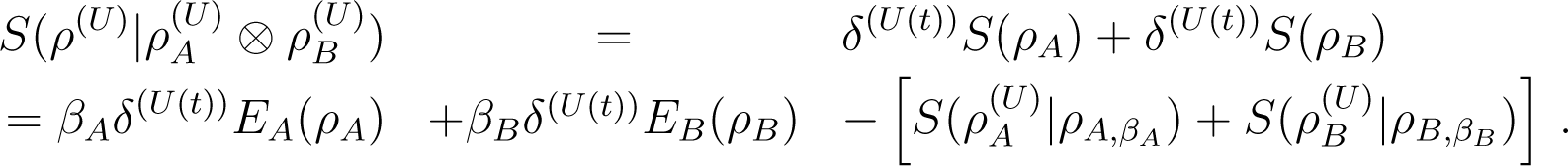
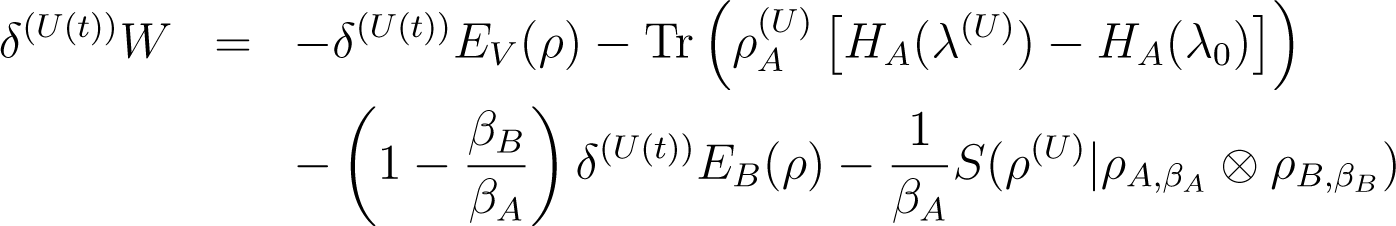
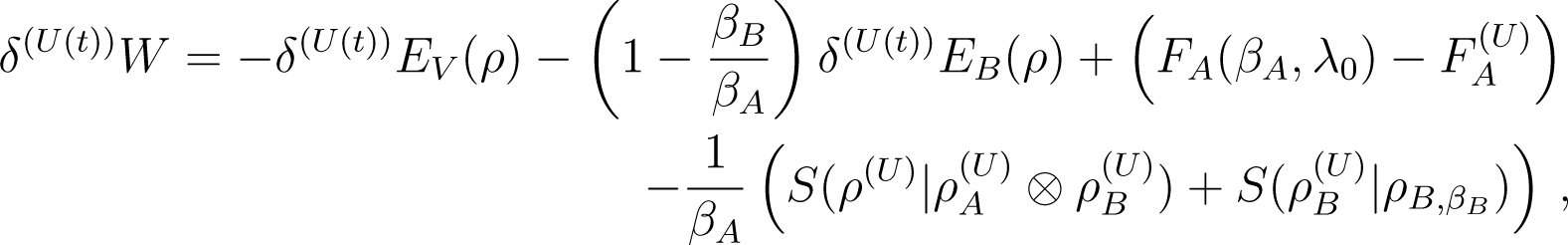
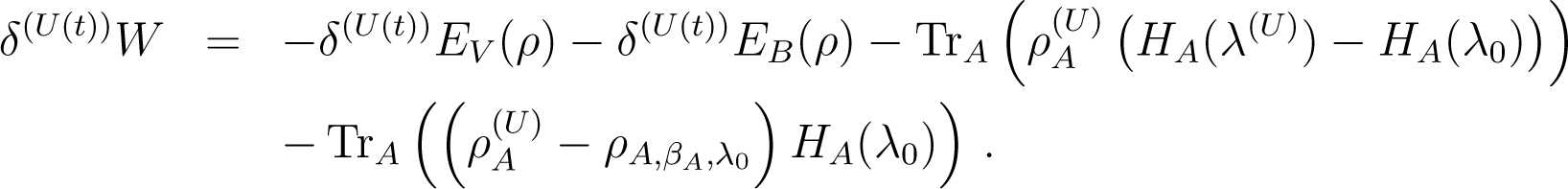
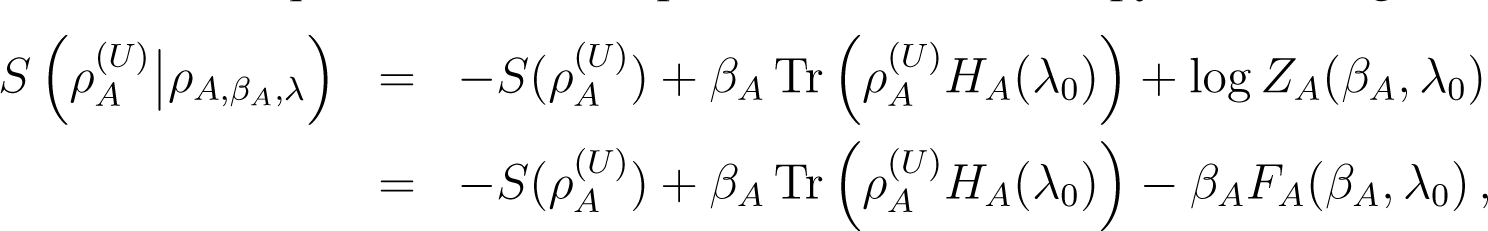
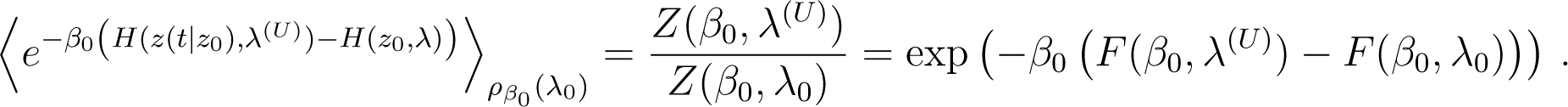
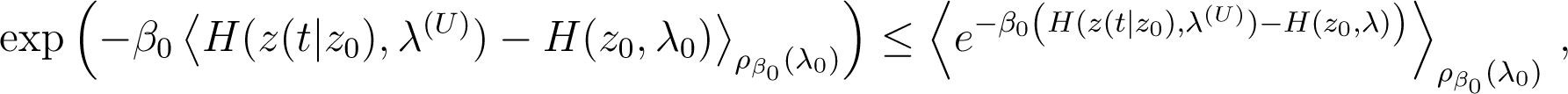
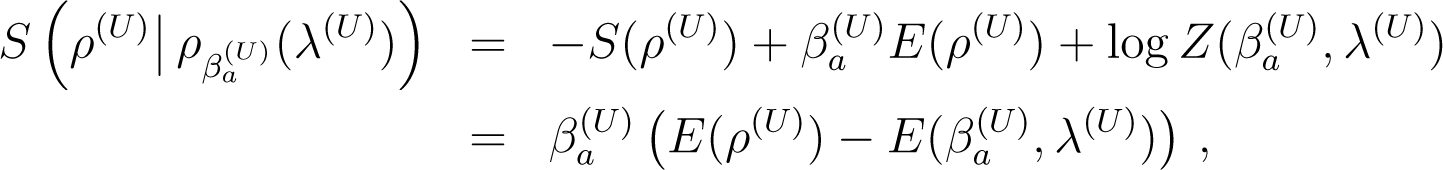
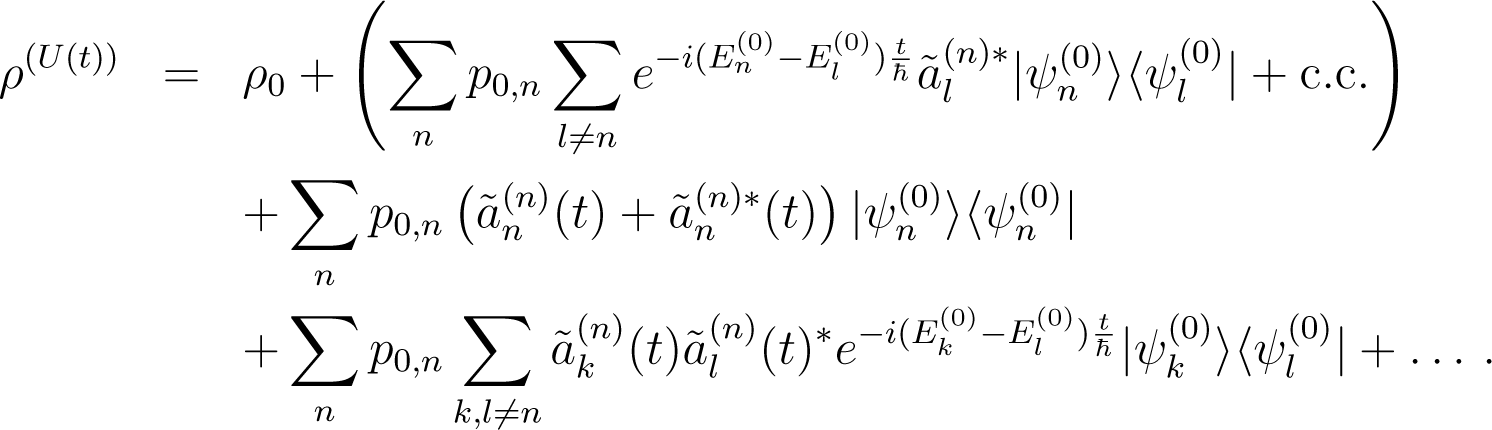



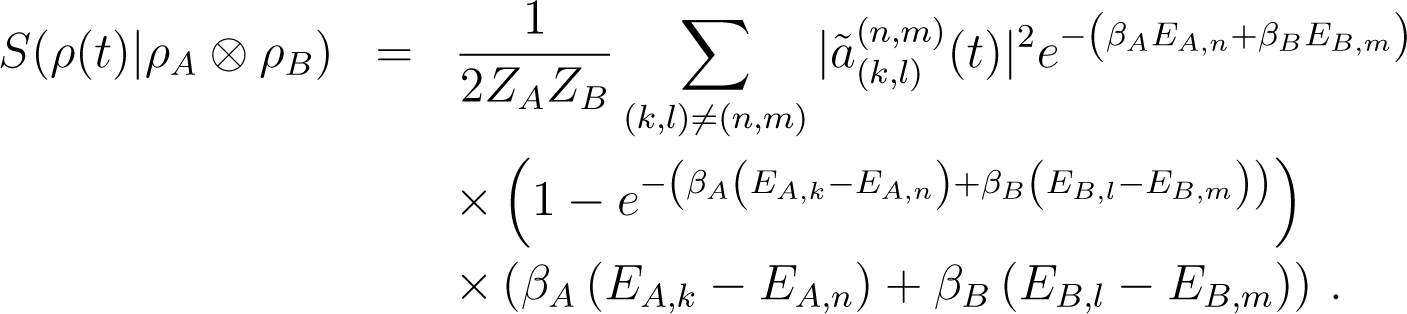
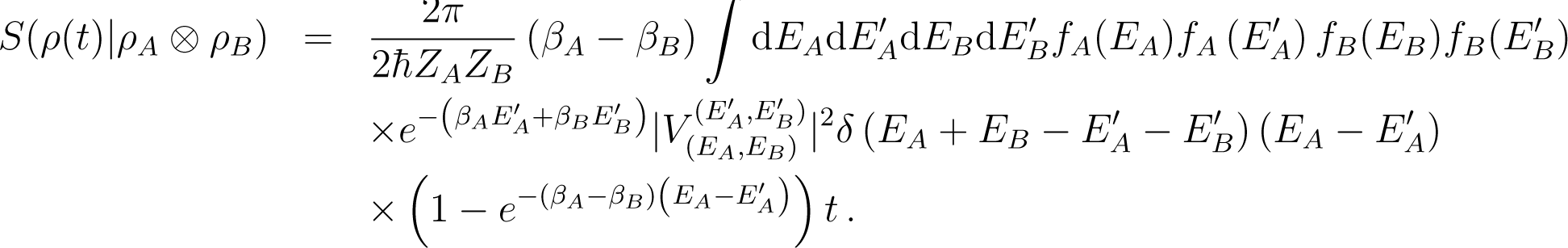
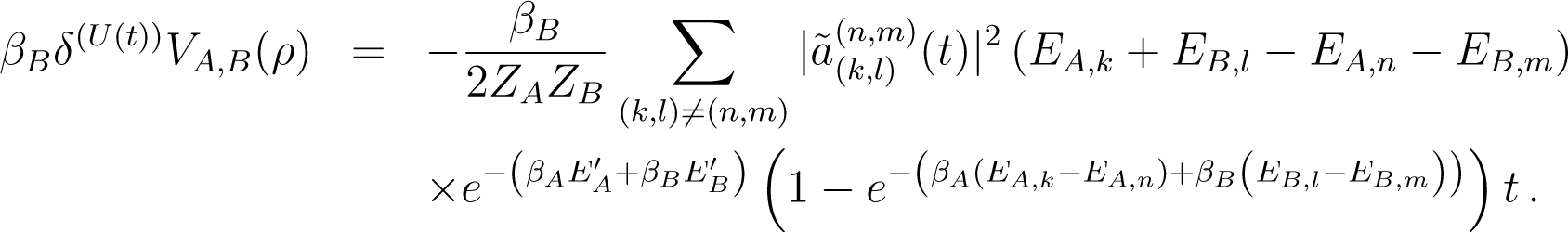
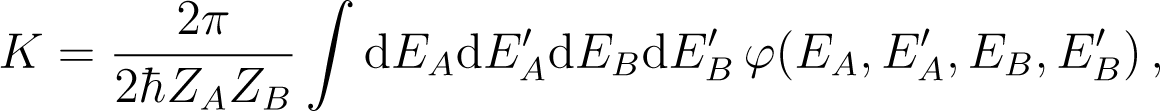
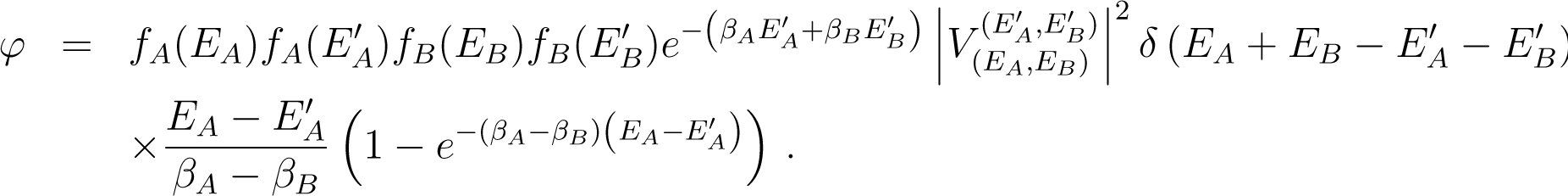
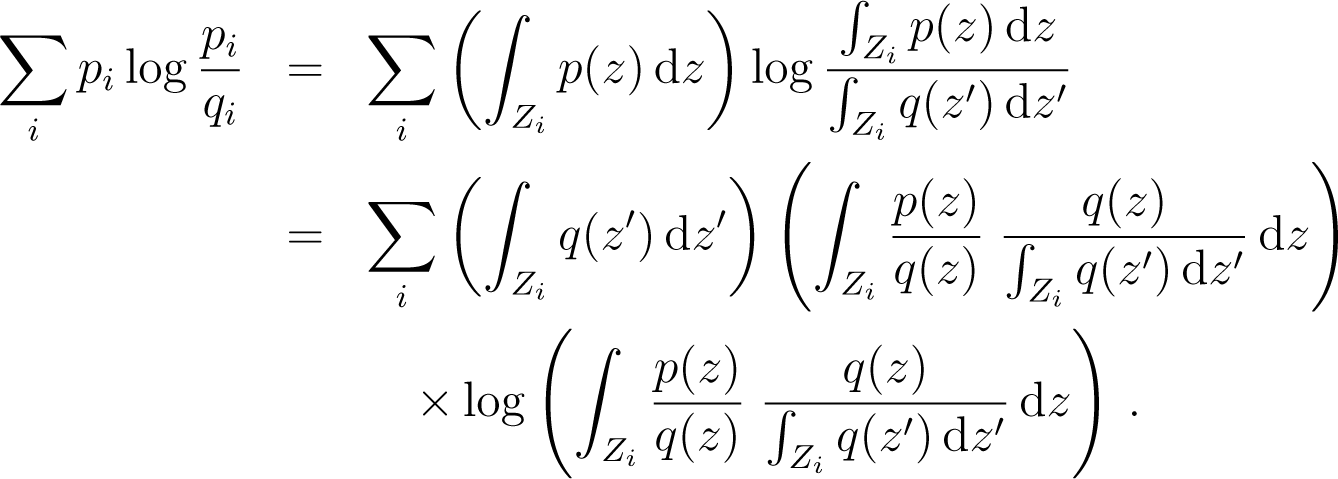
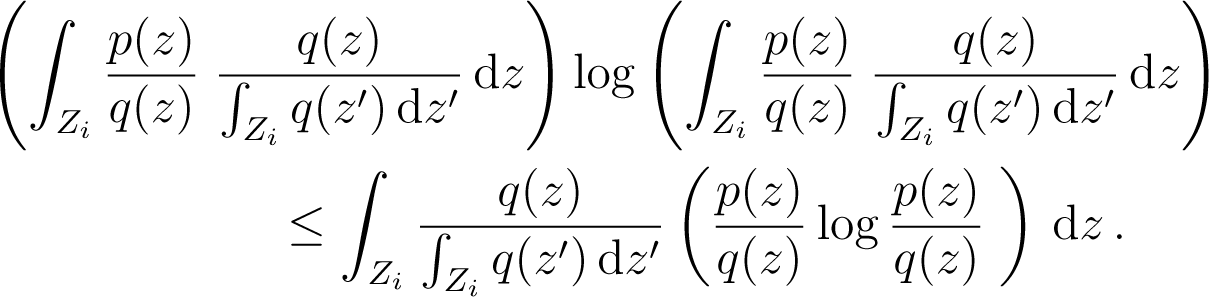
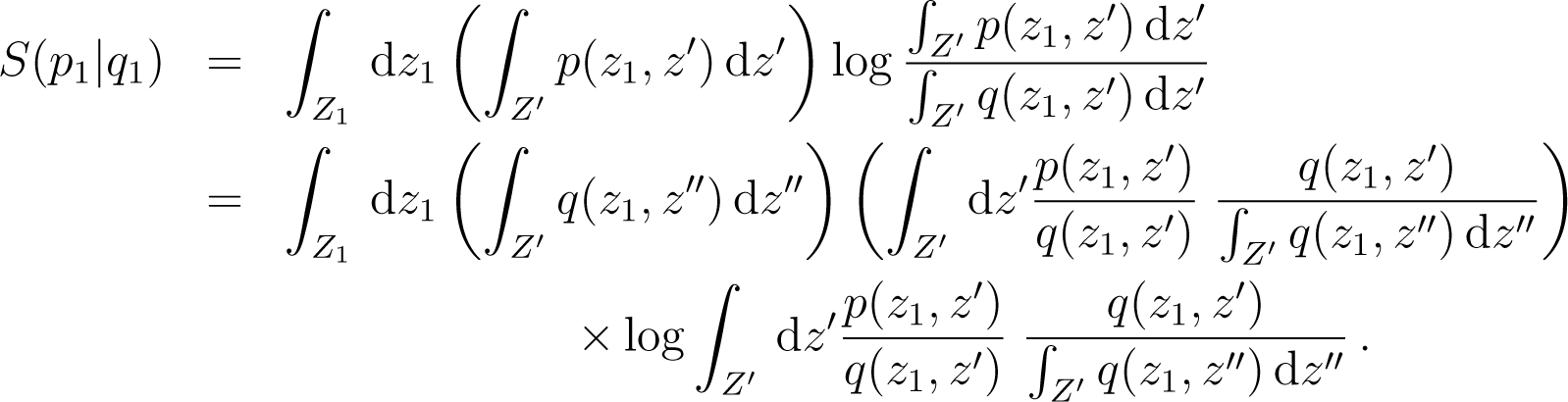
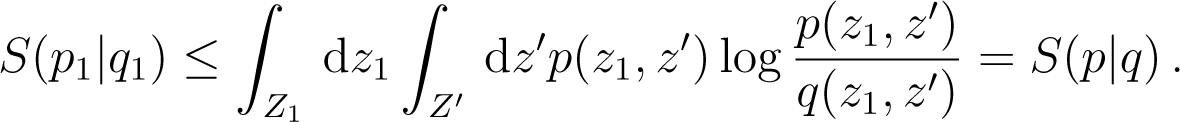


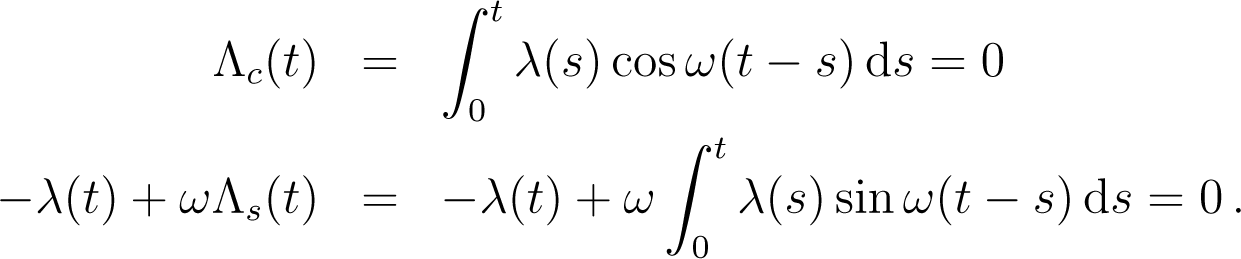

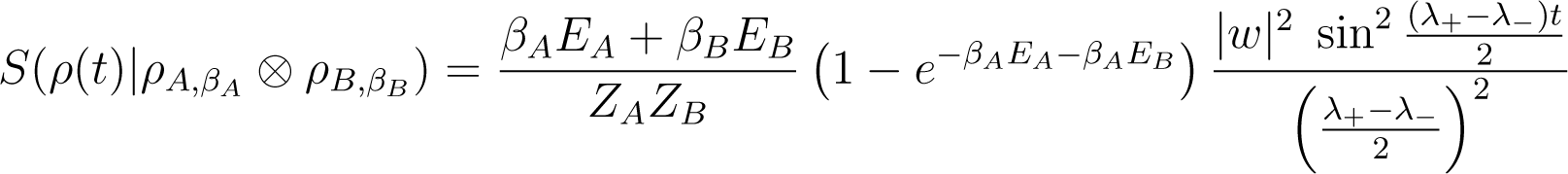
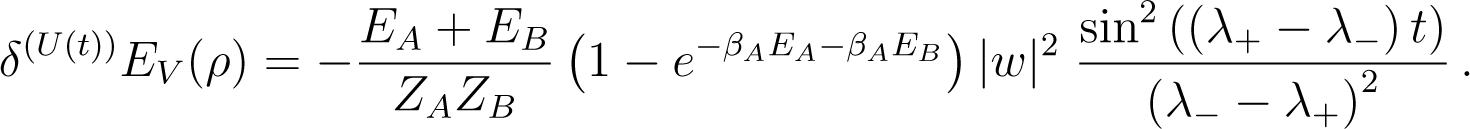
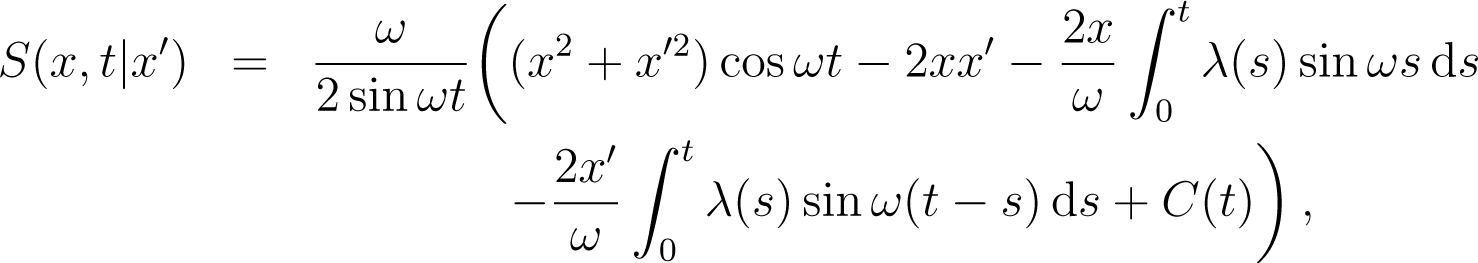


{kind=link}
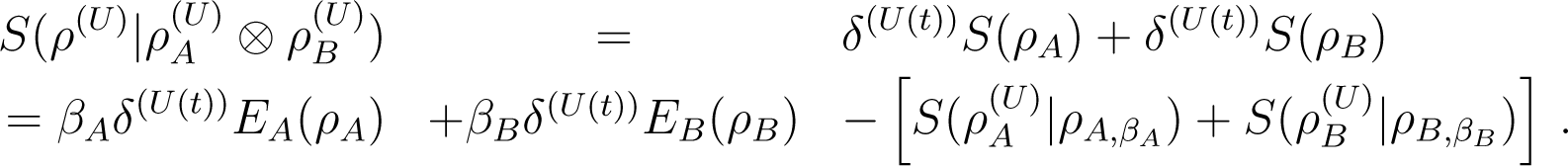{kind=link}
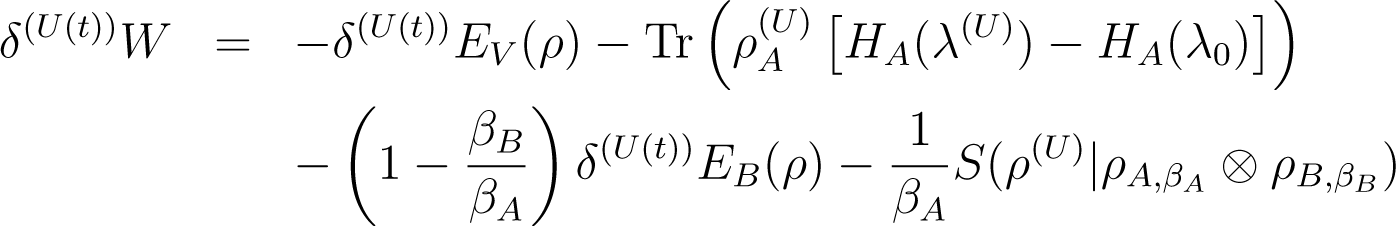{kind=link}
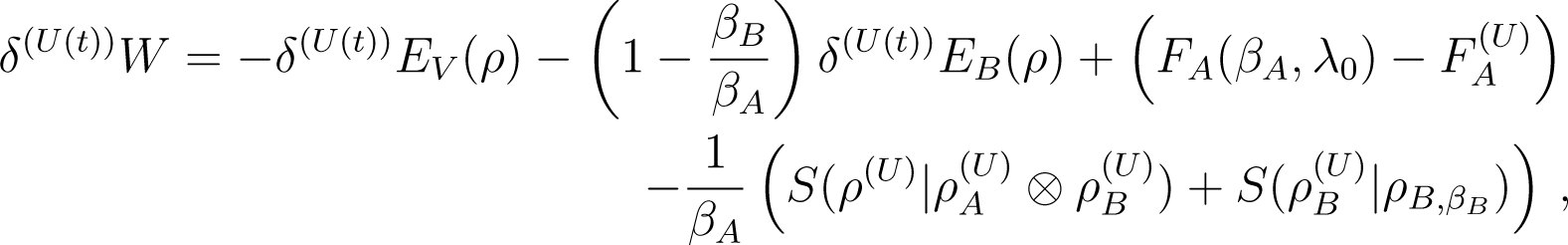{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}