
A Novel ALDH2 Activator AD-9308 Improves Diastolic and Systolic Myocardial Functions in Streptozotocin-Induced Diabetic Mice

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Echocardiography
2.3. Hematoxylin and Eosin (H&E) Staining
2.4. Serum 4-HNE Level Measurement
2.5. ImmunoblottingAnalysis
2.6. ALDH2 Enzymatic Activity Assays
2.7. Gene Expression Analysis
2.8. Immunohistochemistry (IHC) Staining
2.9. Cell Culture and Treatment
2.10. Cell Counting Kit (CCK)-8 Assay
2.11. Measurement of ATP Level
2.12. NF-kB Activity Assay
2.13. Subcellular Fractionation
2.14. Ca2+ Measurement
2.15. Mitochondrial Respiratory Chain Complex Activity Assay
2.16. Measurements of Mitochondrial Functions
2.17. Statistical Analysis
3. Results
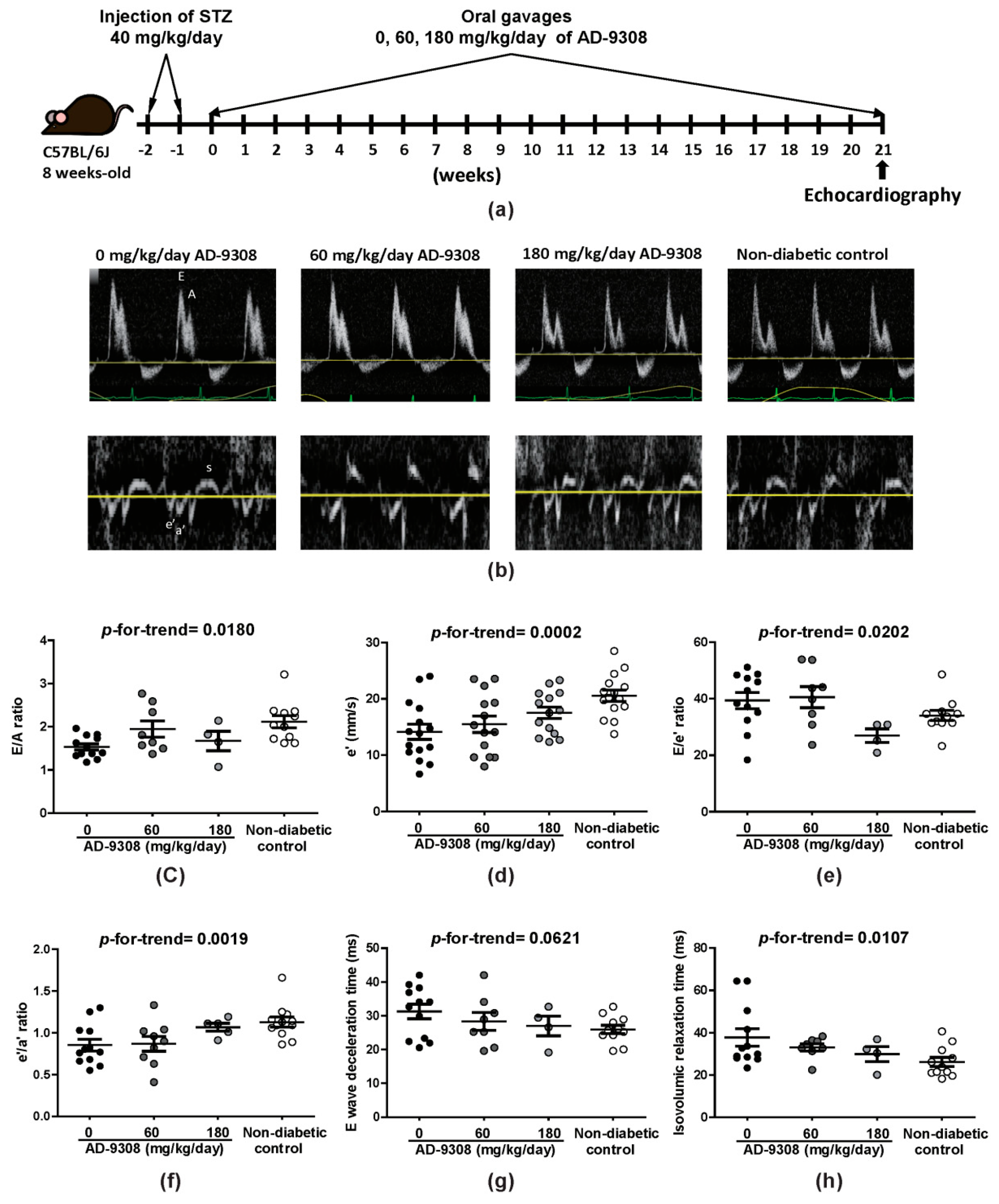
3.1. ALDH2 Activator, AD-9308, Ameliorated Diastolic Dysfunction in STZ-Induced Diabetic Mice
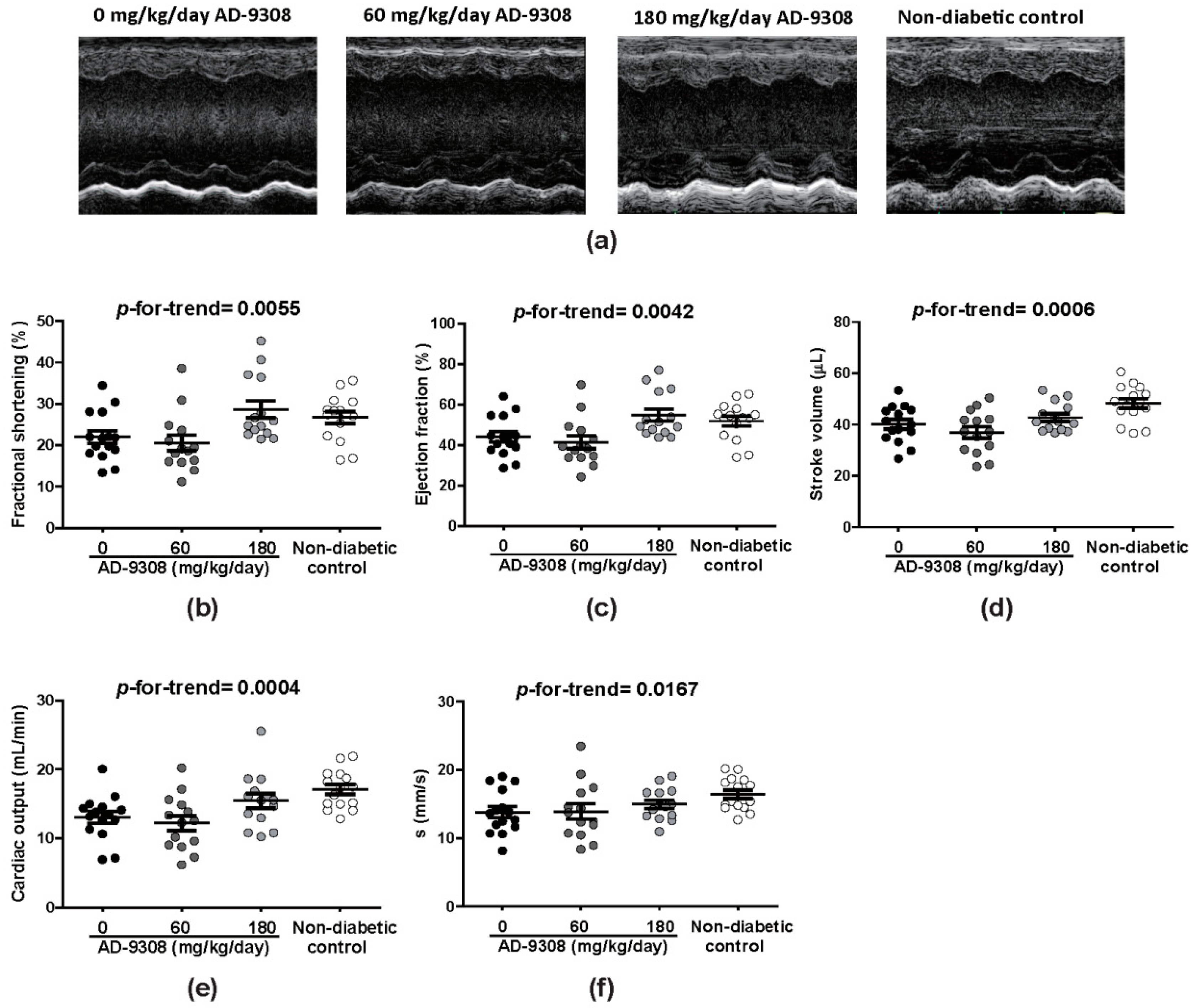
3.2. AD-9308 Ameliorated Systolic Dysfunction in STZ-Induced Diabetic Mice
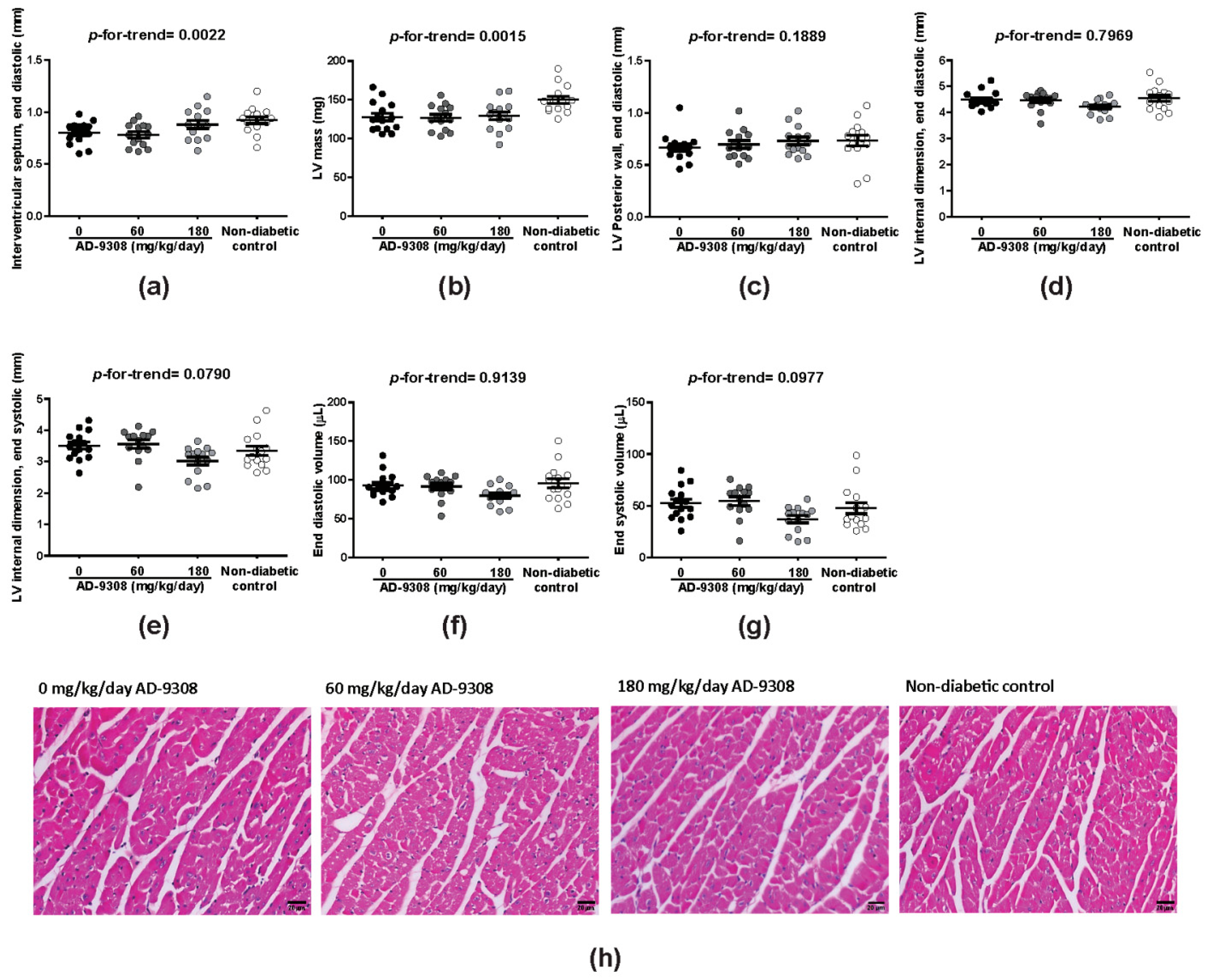
3.3. AD-9308 Partially Ameliorated LV Structural Changes in STZ-Induced Diabetic Mice
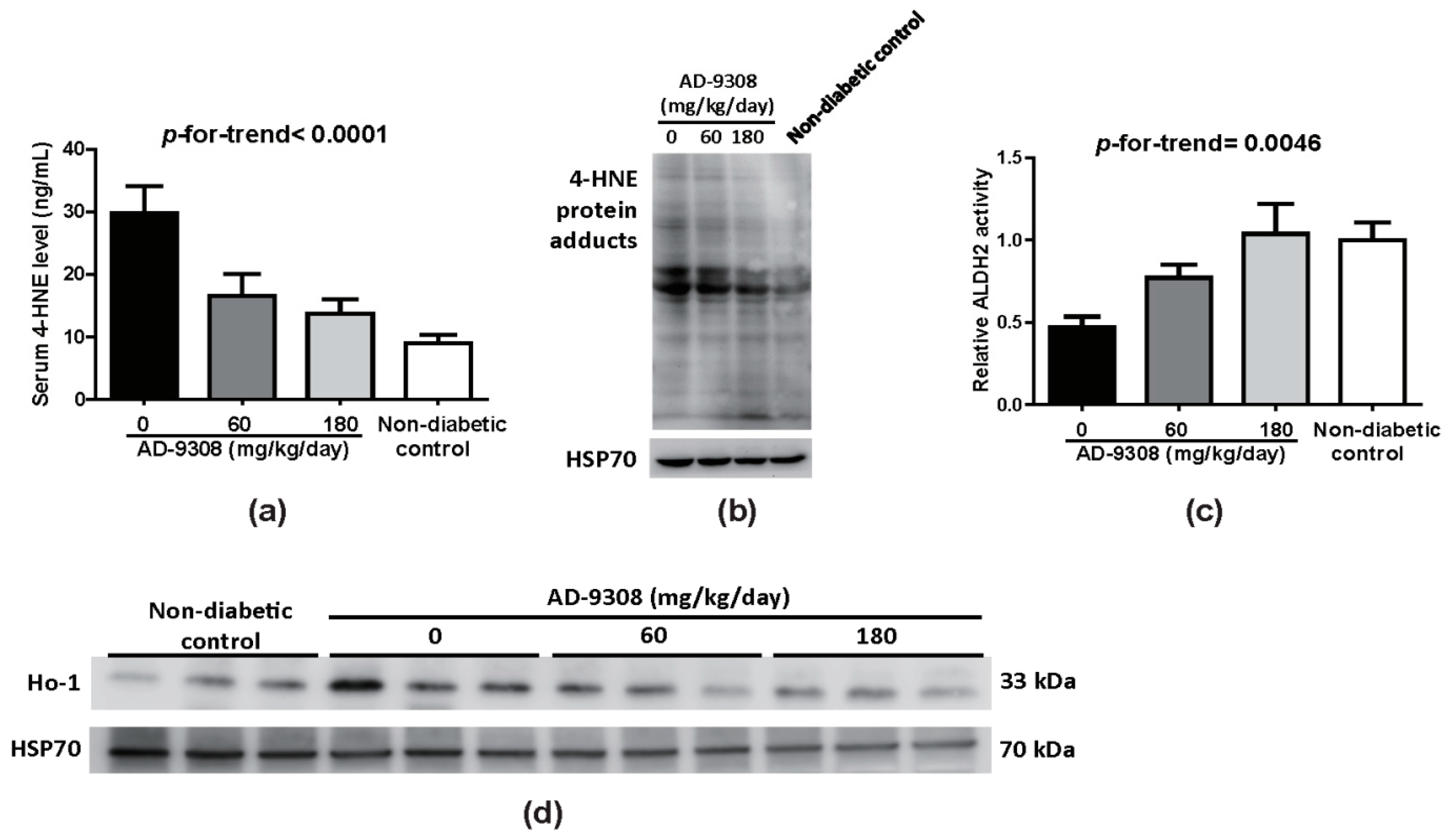
3.4. AD-9308 Treatment Decreased 4-HNE Level and Enhanced ALDH2 Activity
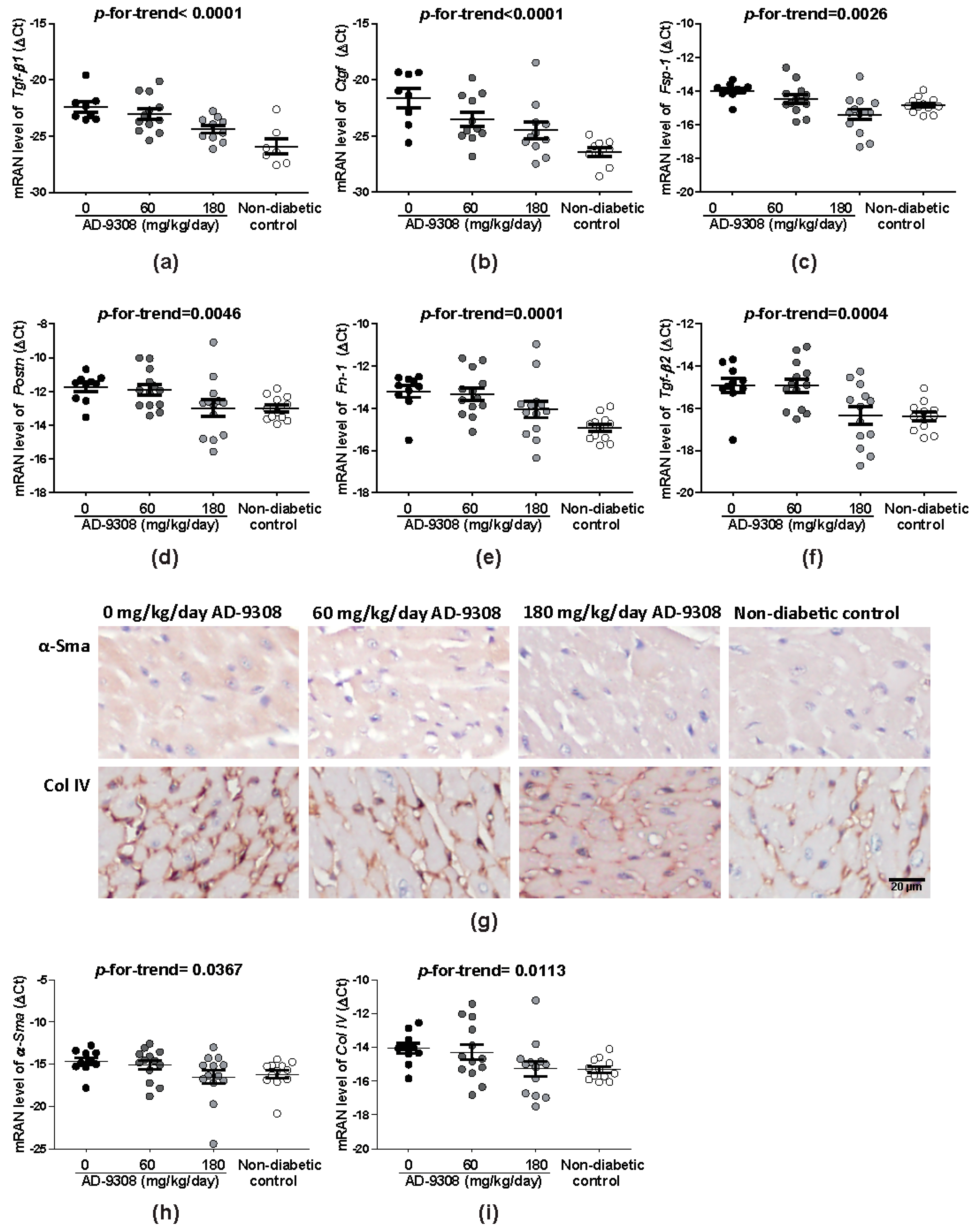
3.5. AD-9308 Treatment Reduced Fibrosis of Diabetic Hearts
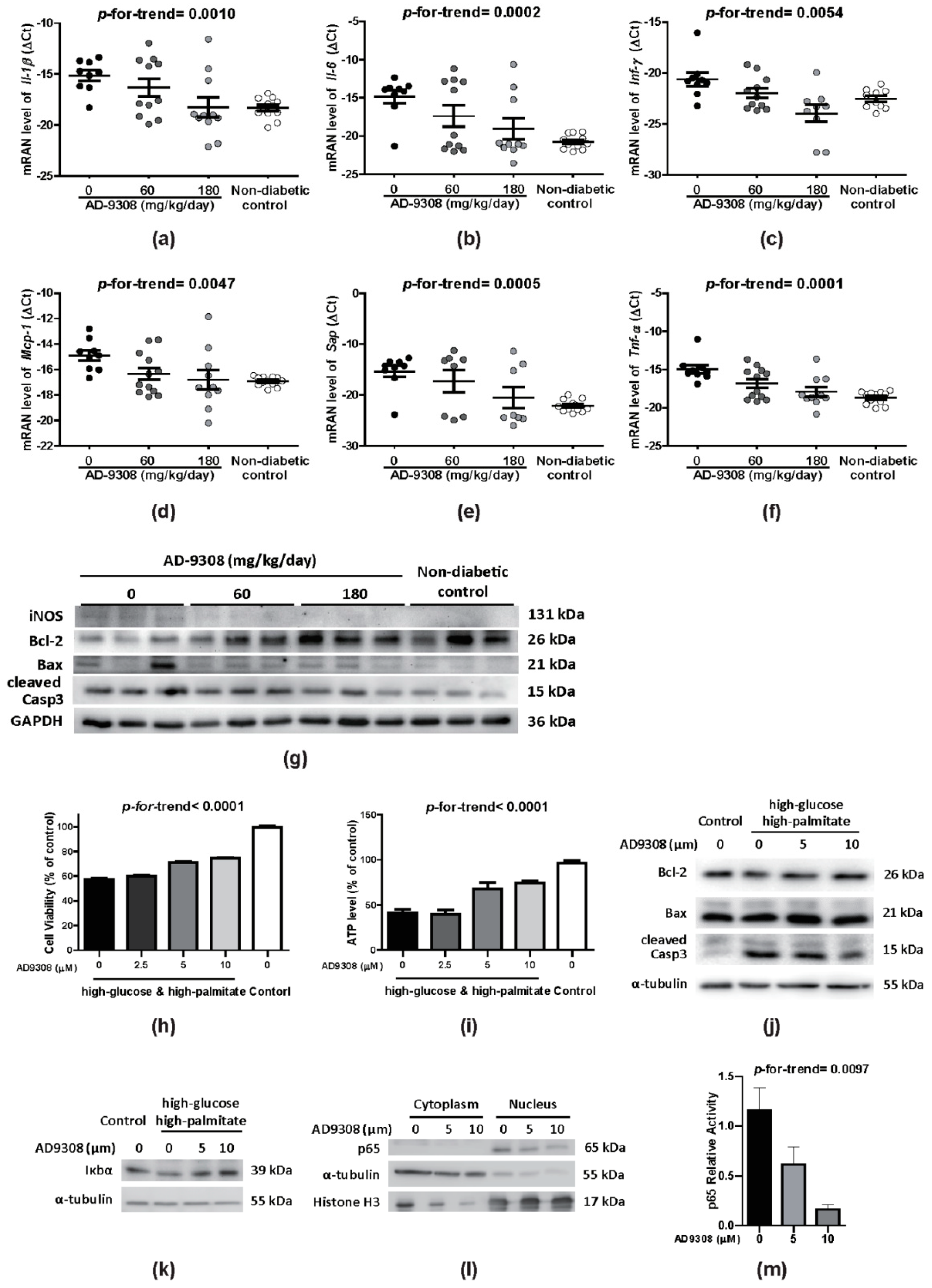
3.6. AD-9308 Treatment Attenuated Inflammation and Apoptosis in Diabetic Hearts
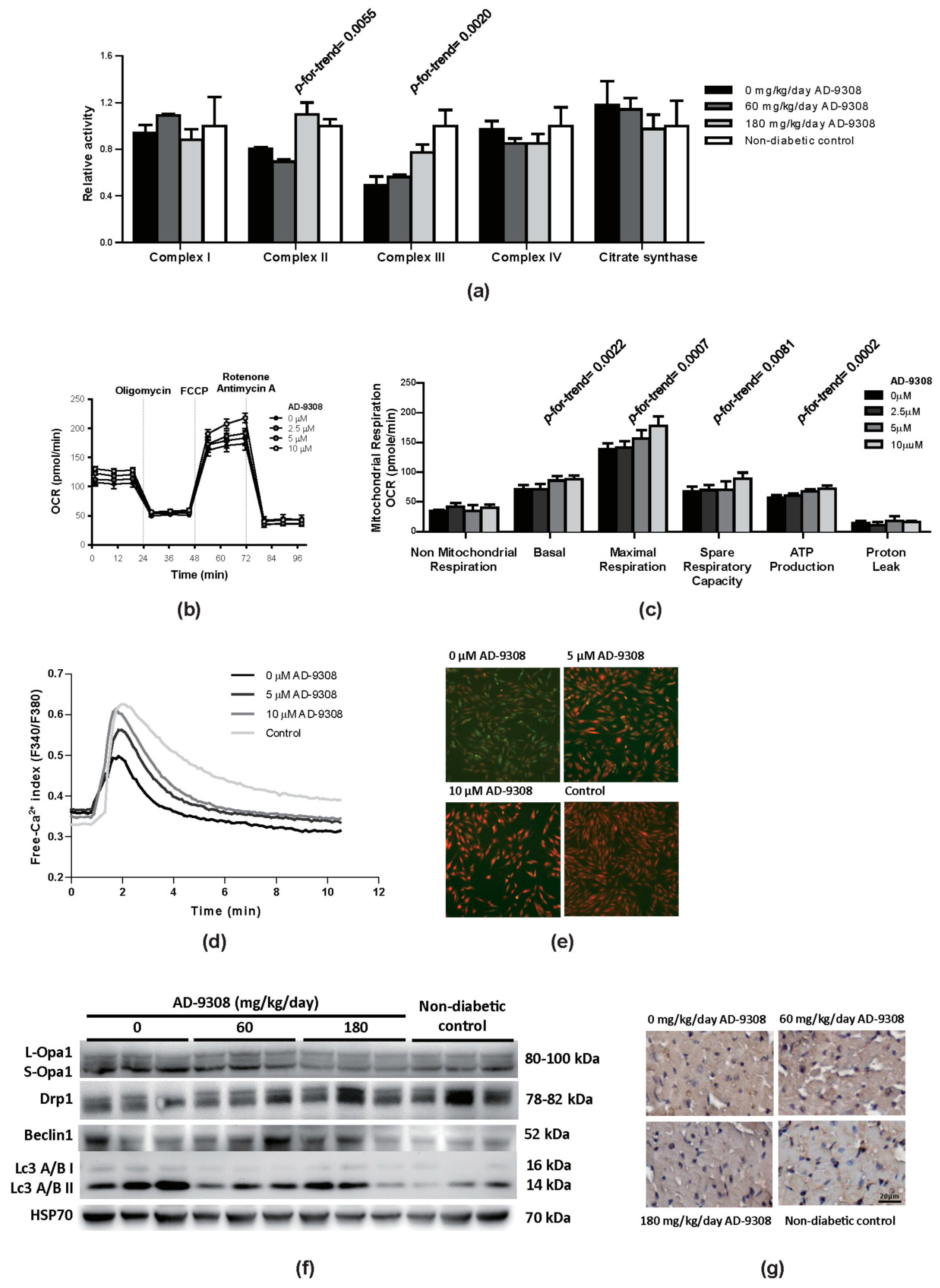
3.7. AD-9308 Treatment Improved Mitochondrial Functions, Suppressed Autophagy and Calcium Handling in Diabetic Mice
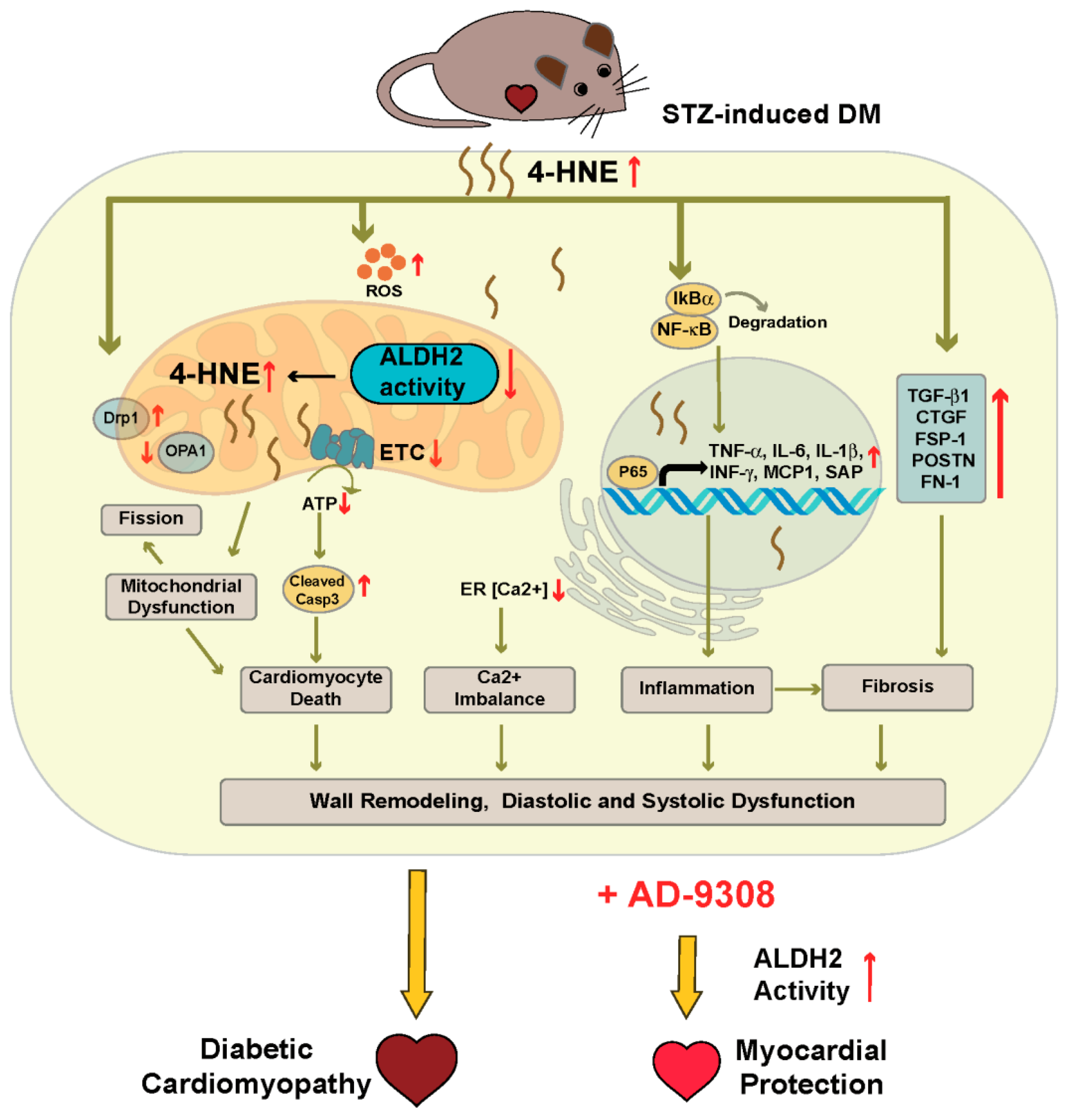
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef]
- Rask-Madsen, C.; King, G.L. Vascular Complications of Diabetes: Mechanisms of Injury and Protective Factors. Cell Metab. 2013, 17, 20–33. [Google Scholar] [CrossRef]
- Kannel, W.B.; Hjortland, M.; Castelli, W.P. Role of diabetes in congestive heart failure: The Framingham study. Am. J. Cardiol. 1974, 34, 29–34. [Google Scholar] [CrossRef]
- Dauriz, M.; Mantovani, A.; Bonapace, S.; Verlato, G.; Zoppini, G.; Bonora, E.; Targher, G. Prognostic Impact of Diabetes on Long-term Survival Outcomes in Patients With Heart Failure: A Meta-analysis. Diabetes Care 2017, 40, 1597–1605. [Google Scholar] [CrossRef]
- Rubler, S.; Dlugash, J.; Yuceoglu, Y.Z.; Kumral, T.; Branwood, A.W.; Grishman, A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am. J. Cardiol. 1972, 30, 595–602. [Google Scholar] [CrossRef]
- Jia, G.; Hill, M.A.; Sowers, J.R. Diabetic Cardiomyopathy: An Update of Mechanisms Contributing to This Clinical Entity. Circ. Res. 2018, 122, 624–638. [Google Scholar] [CrossRef] [PubMed]
- Murtaza, G.; Virk, H.U.H.; Khalid, M.; Lavie, C.J.; Ventura, H.; Mukherjee, D.; Ramu, V.; Bhogal, S.; Kumar, G.; Shanmugasundaram, M.; et al. Diabetic cardiomyopathy—A comprehensive updated review. Prog. Cardiovasc. Dis. 2019, 62, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, N.; Katare, R. Molecular mechanism of diabetic cardiomyopathy and modulation of microRNA function by synthetic oligonucleotides. Cardiovasc. Diabetol. 2018, 17, 43. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Zhang, Z.; Zheng, C.; Wintergerst, K.A.; Keller, B.B.; Cai, L. Mechanisms of diabetic cardiomyopathy and potential therapeutic strategies: Preclinical and clinical evidence. Nat. Rev. Cardiol. 2020, 17, 585–607. [Google Scholar] [CrossRef] [PubMed]
- Regan, T.J.; Lyons, M.M.; Ahmed, S.S.; Levinson, G.E.; Oldewurtel, H.A.; Ahmad, M.R.; Haider, B. Evidence for Cardiomyopathy in Familial Diabetes Mellitus. J. Clin. Investig. 1977, 60, 885–899. [Google Scholar] [CrossRef] [PubMed]
- Boonman-de Winter, L.; Rutten, F.; Cramer, M.; Landman, M.; Liem, A.; Rutten, G.; Hoes, A. High prevalence of previously unknown heart failure and left ventricular dysfunction in patients with type 2 diabetes. Diabetologia 2012, 55, 2154–2162. [Google Scholar] [CrossRef] [PubMed]
- Bugger, H.; Abel, E.D. Rodent models of diabetic cardiomyopathy. Dis. Model. Mech. 2009, 2, 454–466. [Google Scholar] [CrossRef] [PubMed]
- Rajesh, M.; Bátkai, S.; Kechrid, M.; Mukhopadhyay, P.; Lee, W.S.; Horváth, B.; Holovac, E.; Cinar, R.; Liaudet, L.; Mackie, K.; et al. Cannabinoid 1 Receptor Promotes Cardiac Dysfunction, Oxidative Stress, Inflammation, and Fibrosis in Diabetic Cardiomyopathy. Diabetes 2012, 61, 716–727. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Rajesh, M.; Batkai, S.; Mukhopadhyay, P.; Lee, W.S.; Horvath, B.; Cinar, R.; Liaudet, L.; Mackie, K.; Haskó, G. Cannabinoid 1 receptor promotes cardiac dysfunction, oxidative stress, inflammation, and fibrosis in diabetic cardiomyopathy. FASEB J. 2013, 27 (Suppl. 1), 1128.10. [Google Scholar] [CrossRef]
- Westermann, D.; Van Linthout, S.; Dhayat, S.; Dhayat, N.; Escher, F.; Spillmann, F.; Noutsias, M.; Riad, A.; Schultheiss, H.-P.; Bücker-Gärtner, C.; et al. Cardioprotective and Anti-Inflammatory Effects of Interleukin Converting Enzyme Inhibition in Experimental Diabetic Cardiomyopathy. Diabetes 2007, 56, 1834–1841. [Google Scholar] [CrossRef]
- Westermann, D.; Rutschow, S.; Van Linthout, S.; Linderer, A.; Bücker-Gärtner, C.; Sobirey, M.; Riad, A.; Pauschinger, M.; Schultheiss, H.-P.; Tschöpe, C. Inhibition of p38 mitogen-activated protein kinase attenuates left ventricular dysfunction by mediating pro-inflammatory cardiac cytokine levels in a mouse model of diabetes mellitus. Diabetologia 2006, 49, 2507–2513. [Google Scholar] [CrossRef]
- Chiu, J.; Farhangkhoee, H.; Xu, B.Y.; Chen, S.; George, B.; Chakrabarti, S. PARP mediates structural alterations in diabetic cardiomyopathy. J. Mol. Cell. Cardiol. 2008, 45, 385–393. [Google Scholar] [CrossRef]
- Ye, G.; Metreveli, N.S.; Donthi, R.V.; Xia, S.; Xu, M.; Carlson, E.C.; Epstein, P.N. Catalase protects cardiomyocyte function in models of type 1 and type 2 diabetes. Diabetes 2004, 53, 1336–1343. [Google Scholar] [CrossRef]
- Kralik, P.M.; Ye, G.; Metreveli, N.S.; Shen, X.; Epstein, P.N. Cardiomyocyte Dysfunction in Models of Type 1 and Type 2 Diabetes. Cardiovasc. Toxicol. 2005, 5, 285–292. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Tahiliani, A.G.; Vadlamudi, R.; Katz, S.; McNeill, J. Cardiac sarcoplasmic reticulum function in insulin- or carnitine-treated diabetic rats. Am. J. Physiol. Heart Circ. Physiol. 1983, 245, H969–H976. [Google Scholar] [CrossRef]
- Bugger, H.; Abel, E.D. Molecular mechanisms for myocardial mitochondrial dysfunction in the metabolic syndrome. Clin. Sci. 2008, 114, 195–210. [Google Scholar] [CrossRef]
- Negre-Salvayre, A.; Coatrieux, C.; Ingueneau, C.; Salvayre, R. Advanced lipid peroxidation end products in oxidative damage to proteins: Potential role in diseases and therapeutic prospects for the inhibitors. Br. J. Pharmacol. 2008, 153, 6–20. [Google Scholar] [CrossRef] [PubMed]
- Mali, V.R.; Palaniyandi, S.S. Regulation and therapeutic strategies of 4-hydroxy-2-nonenal metabolism in heart disease. Free Radic. Res. 2014, 48, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Dham, D.; Roy, B.; Gowda, A.; Pan, G.; Sridhar, A.; Zeng, X.; Thandavarayan, R.A.; Palaniyandi, S.S. 4-Hydroxy-2-nonenal, a lipid peroxidation product, as a biomarker in diabetes and its complications: Challenges and opportunities. Free Radic. Res. 2020, 18, 1–15. [Google Scholar] [CrossRef]
- Zhao, Y.; Song, W.; Wang, Z.; Wang, Z.; Jin, X.; Xu, J.; Bai, L.; Li, Y.; Cui, J.; Cai, L. Resveratrol attenuates testicular apoptosis in type 1 diabetic mice: Role of Akt-mediated Nrf2 activation and p62-dependent Keap1 degradation. Redox Biol. 2018, 14, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Toyokuni, S.; Yamada, S.; Kashima, M.; Ihara, Y.; Yamada, Y.; Tanaka, T.; Hiai, H.; Seino, Y.; Uchida, K. Serum 4-Hydroxy-2-Nonenal-Modified Albumin Is Elevated in Patients with Type 2 Diabetes Mellitus. Antioxid. Redox Signal. 2000, 2, 681–685. [Google Scholar] [CrossRef] [PubMed]
- Mali, V.R.; Ning, R.; Chen, J.; Yang, X.-P.; Xu, J.; Palaniyandi, S.S. Impairment of aldehyde dehydrogenase-2 by 4-hydroxy-2-nonenal adduct formation and cardiomyocyte hypertrophy in mice fed a high-fat diet and injected with low-dose streptozotocin. Exp. Biol. Med. 2014, 239, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Budas, G.R.; Churchill, E.N.; Disatnik, M.H.; Hurley, T.D.; Mochly-Rosen, D. Activation of Aldehyde Dehydrogenase-2 Reduces Ischemic Damage to the Heart. Science 2008, 321, 1493–1495. [Google Scholar] [CrossRef]
- Zhao, M.X.; Zhou, B.; Ling, L.; Xiong, X.Q.; Zhang, F.; Chen, Q.; Li, Y.H.; Kang, Y.M.; Zhu, G.Q. Salusin-beta contributes to oxidative stress and inflammation in diabetic cardiomyopathy. Cell Death Dis. 2017, 8, e2690. [Google Scholar] [CrossRef]
- Liu, M.; Verma, N.; Peng, X.; Srodulski, S.; Morris, A.; Chow, M.; Hersh, L.B.; Chen, J.; Zhu, M.H.; Netea, M.G.; et al. Hyperamylinemia Increases IL-1beta Synthesis in the Heart via Peroxidative Sarcolemmal Injury. Diabetes 2016, 65, 2772–2783. [Google Scholar] [CrossRef]
- Pan, G.; Deshpande, M.; Pang, H.; Palaniyandi, S.S. Precision medicine approach: Empagliflozin for diabetic cardiomyopathy in mice with aldehyde dehydrogenase (ALDH) 2*2 mutation, a specific genetic mutation in millions of East Asians. Eur. J. Pharmacol. 2018, 839, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Chandra, A.; Wang, L.F.; Seifert, W.E., Jr.; DaGue, B.B.; Ansari, N.H.; Srivastava, S.K.; Bhatnagar, A. Metabolism of the lipid peroxidation product, 4-hydroxy-trans-2-nonenal, in isolated perfused rat heart. J. Biol. Chem. 1998, 273, 10893–10900. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, H.; Hao, P.; Xue, L.; Wei, S.; Zhang, Y.; Chen, Y. Inhibition of Aldehyde Dehydrogenase 2 by Oxidative Stress Is Associated with Cardiac Dysfunction in Diabetic Rats. Mol. Med. 2011, 17, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Yu, Y.T.; Jiang, C. Mitochondrial Aldehyde Dehydrogenase-2 Binding Compounds and Methods of Use Thereof. U.S. Patent 9,879,036 B2, 30 January 2018. [Google Scholar]
- Spinazzi, M.; Casarin, A.; Pertegato, V.; Salviati, L.; Angelini, C. Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat. Protoc. 2012, 7, 1235–1246. [Google Scholar] [CrossRef]
- Mali, V.R.; Pan, G.D.; Deshpande, M.; Thandavarayan, R.A.; Xu, J.; Yang, X.-P.; Palaniyandi, S.S. Cardiac Mitochondrial Respiratory Dysfunction and Tissue Damage in Chronic Hyperglycemia Correlate with Reduced Aldehyde Dehydrogenase-2 Activity. PLoS ONE 2016, 11, e0163158. [Google Scholar] [CrossRef]
- Zhang, Y.; Babcock, S.A.; Hu, N.; Maris, J.R.; Wang, H.; Ren, J. Mitochondrial aldehyde dehydrogenase (ALDH2) protects against streptozotocin-induced diabetic cardiomyopathy: Role of GSK3beta and mitochondrial function. BMC Med. 2012, 10, 40. [Google Scholar] [CrossRef] [PubMed]
- Penpargkul, S.; Fein, F.; Sonnenblick, E.H.; Scheuer, J. Depressed cardiac sarcoplasmic reticular function from diabetic rats. J. Mol. Cell Cardiol. 1981, 13, 303–309. [Google Scholar] [CrossRef]
- Pierce, G.N.; Russell, J.C. Regulation of intracellular Ca2+ in the heart during diabetes. Cardiovasc. Res. 1997, 34, 41–47. [Google Scholar] [CrossRef]
- Hayashi, T.; Mori, T.; Yamashita, C.; Miyamura, M. Regulation of Oxidative Stress and Cardioprotection in Diabetes Mellitus. Curr. Cardiol. Rev. 2008, 4, 251–258. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ding, M.; Feng, N.; Tang, D.; Feng, J.; Li, Z.; Jia, M.; Liu, Z.; Gu, X.; Wang, Y.; Fu, F.; et al. Melatonin prevents Drp1-mediated mitochondrial fission in diabetic hearts through SIRT1-PGC1α pathway. J. Pineal Res. 2018, 65, e12491. [Google Scholar] [CrossRef]
- Archer, S.L. Mitochondrial Dynamics—Mitochondrial Fission and Fusion in Human Diseases. N. Engl. J. Med. 2013, 369, 2236–2251. [Google Scholar] [CrossRef] [PubMed]
- Chiong, M.; Cartes-Saavedra, B.; Norambuena-Soto, I.; Mondaca-Ruff, D.; Morales, P.E.; García-Miguel, M.; Mellado, R. Mitochondrial metabolism and the control of vascular smooth muscle cell proliferation. Front. Cell Dev. Biol. 2014, 2, 72. [Google Scholar] [CrossRef]
- Ong, S.B.; Hall, A.R.; Hausenloy, D.J. Mitochondrial Dynamics in Cardiovascular Health and Disease. Antioxid. Redox Signal. 2013, 19, 400–414. [Google Scholar] [CrossRef]
- Wai, T.; García-Prieto, J.; Baker, M.J.; Merkwirth, C.; Benit, P.; Rustin, P.; Rupérez, F.J.; Barbas, C.; Ibañez, B.; Langer, T. Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science 2015, 350, aad0116. [Google Scholar] [CrossRef] [PubMed]
- Martinet, W.; Knaapen, M.W.; Kockx, M.M.; De Meyer, G.R. Autophagy in cardiovascular disease. Trends Mol. Med. 2007, 13, 482–491. [Google Scholar] [CrossRef]
- Xu, X.; Kobayashi, S.; Chen, K.; Timm, D.; Volden, P.; Huang, Y.; Gulick, J.; Yue, Z.; Robbins, J.; Epstein, P.N.; et al. Diminished Autophagy Limits Cardiac Injury in Mouse Models of Type 1 Diabetes. J. Biol. Chem. 2013, 288, 18077–18092. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Yu, W.; Sun, D.; Wang, J.; Li, C.; Zhang, R.; Babcock, S.A.; Li, Y.; Liu, M.; Ma, M.; et al. A novel protective mechanism for mitochondrial aldehyde dehydrogenase (ALDH2) in type i diabetes-induced cardiac dysfunction: Role of AMPK-regulated autophagy. Biochim. Biophys. Acta 2015, 1852, 319–331. [Google Scholar] [CrossRef]
- Chen, C.H.; Ferreira, J.C.; Gross, E.R.; Mochly-Rosen, D. Targeting Aldehyde Dehydrogenase 2: New Therapeutic Opportunities. Physiol. Rev. 2014, 94, 1–34. [Google Scholar] [CrossRef]
- Kaludercic, N.; Di Lisa, F. Mitochondrial ROS Formation in the Pathogenesis of Diabetic Cardiomyopathy. Front. Cardiovasc. Med. 2020, 7, 12. [Google Scholar] [CrossRef]
- Brownlee, M. The Pathobiology of Diabetic Complications: A Unifying Mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef]
- Calabrese, V.; Mancuso, C.; Sapienza, M.; Puleo, E.; Calafato, S.; Cornelius, C.; Finocchiaro, M.; Mangiameli, A.; Di Mauro, M.; Stella, A.M.; et al. Oxidative stress and cellular stress response in diabetic nephropathy. Cell Stress Chaperon. 2007, 12, 299–306. [Google Scholar] [CrossRef]
- Chen, J.; Henderson, G.I.; Freeman, G.L. Role of 4-Hydroxynonenal in Modification of Cytochrome c Oxidase in Ischemia/Reperfused Rat Heart. J. Mol. Cell. Cardiol. 2001, 33, 1919–1927. [Google Scholar] [CrossRef]
- Jia, G.; Demarco, V.G.; Sowers, J.R. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat. Rev. Endocrinol. 2016, 12, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Hölscher, M.E.; Bode, C.; Bugger, H. Diabetic Cardiomyopathy: Does the Type of Diabetes Matter? Int. J. Mol. Sci. 2016, 17, 2136. [Google Scholar] [CrossRef]
- Raev, D.C. Which Left Ventricular Function Is Impaired Earlier in the Evolution of Diabetic Cardiomyopathy?: An echocardiographic study of young type I diabetic patients. Diabetes Care 1994, 17, 633–639. [Google Scholar] [CrossRef]
- Pan, G.; Deshpande, M.; Thandavarayan, R.A.; Palaniyandi, S.S. ALDH2 Inhibition Potentiates High Glucose Stress-Induced Injury in Cultured Cardiomyocytes. J. Diabetes Res. 2016, 2016, 1390861. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.J.; Kang, P.F.; Wu, W.J.; Tang, Y.; Pan, Q.Q.; Ye, H.W.; Tang, B.; Li, Z.H.; Gao, Q. Changes in cardiac mitochondrial aldehyde dehydrogenase 2 activity in relation to oxidative stress and inflammatory injury in diabetic rats. Mol. Med. Rep. 2013, 8, 686–690. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Stewart, M.J.; Malek, K.; Crabb, D.W. Distribution of messenger RNAs for aldehyde dehydrogenase 1, aldehyde dehydrogenase 2, and aldehyde dehydrogenase 5 in human tissues. J. Investig. Med. 1996, 44, 42–46. [Google Scholar] [PubMed]
- Esterbauer, H.; Schaur, R.J.; Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 1991, 11, 81–128. [Google Scholar] [CrossRef]
- Westermeier, F.; Riquelme, J.A.; Pavez, M.; Garrido, V.; Díaz, A.; Verdejo, H.E.; Castro, P.F.; García, L.; Lavandero, S. New Molecular Insights of Insulin in Diabetic Cardiomyopathy. Front. Physiol. 2016, 7, 125. [Google Scholar] [CrossRef]
- Ding, Y.; Wang, Y.; Zhang, W.; Jia, Q.; Wang, X.; Li, Y.; Lv, S.; Zhang, J. Roles of Biomarkers in Myocardial Fibrosis. Aging Dis. 2020, 11, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Wu, H.; Xia, W.; Chen, X.; Zhu, S.; Zhang, S.; Shao, Y.; Ma, W.; Yang, D.; Zhang, J. Periostin expression is upregulated and associated with myocardial fibrosis in human failing hearts. J. Cardiol. 2014, 63, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Tang, Y.; Wang, F. The Protective Effect of HIF-1α in T Lymphocytes on Cardiac Damage in Diabetic Mice. Ann. Clin. Lab. Sci. 2016, 46, 32–43. [Google Scholar] [PubMed]
- Lennon, G.P.; Bettini, M.; Burton, A.R.; Vincent, E.; Arnold, P.Y.; Santamaria, P.; Vignali, D.A. T Cell Islet Accumulation in Type 1 Diabetes Is a Tightly Regulated, Cell-Autonomous Event. Immunity 2009, 31, 643–653. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, O.; Picatoste, B.; Ares-Carrasco, S.; Ramírez, E.; Egido, J.; Tuñón, J. Potential role of nuclear factor κB in diabetic cardiomyopathy. Mediat. Inflamm. 2011, 2011, 652097. [Google Scholar] [CrossRef]
- Ramezani, N.; Vanaky, B.; Shakeri, N.; Soltanian, Z.; Rad, F.F.; Shams, Z. Evaluation of Bcl-2 and Bax Expression in the Heart of Diabetic Rats after Four Weeks of High Intensity Interval Training. Med Lab. J. 2019, 13, 15–20. [Google Scholar] [CrossRef]
- Golstein, P. Controlling Cell Death. Science 1997, 275, 1081–1082. [Google Scholar] [CrossRef]
- Bhuvaneshwari, P.; Riswana, N.A. Apoptotic cell death in heart failure associated with diabetes. Int. J. Clin. Correl. 2019, 3, 12. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, H.; Zhang, J.; Xia, Z.; Chen, W. Streptozotocin-induced diabetic cardiomyopathy in rats: Ameliorative effect of PIPERINE via Bcl2, Bax/Bcl2, and caspase-3 pathways. Biosci. Biotechnol. Biochem. 2020, 84, 2533–2544. [Google Scholar] [CrossRef]
- Li, B.; Zheng, Z.; Wei, Y.; Wang, M.; Peng, J.; Kang, T.; Huang, X.; Xiao, J.; Li, Y.; Li, Z. Therapeutic effects of neuregulin-1 in diabetic cardiomyopathy rats. Cardiovasc. Diabetol. 2011, 10, 69. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.V.; Sandeep, N.; Paige, S.L.; Ranjbarvaziri, S.; Hu, D.-Q.; Zhao, M.; Lan, I.S.; Coronado, M.; Kooiker, K.B.; Wu, S.M.; et al. 4HNE Impairs Myocardial Bioenergetics in Congenital Heart Disease-Induced Right Ventricular Failure. Circulation 2020, 142, 1667–1683. [Google Scholar] [CrossRef] [PubMed]
- Hill, B.G.; Dranka, B.P.; Zou, L.; Chatham, J.C.; Darley-Usmar, V.M. Importance of the bioenergetic reserve capacity in response to cardiomyocyte stress induced by 4-hydroxynonenal. Biochem. J. 2009, 424, 99–107. [Google Scholar] [CrossRef]
- Mak, S.; Lehotay, S.C.; Yazdanpanah, M.; Azevedo, E.R.; Liu, P.P.; Newton, G.E. Unsaturated aldehydes including 4-OH-nonenal are elevated in patients with congestive heart failure. J. Card. Fail. 2000, 6, 108–114. [Google Scholar] [CrossRef]
- Lashina, O.M.; Szweda, P.A.; Szweda, L.I.; Romania, A.M.P. Decreased complex II respiration and HNE-modified SDH subunit in diabetic heart. Free Radic. Biol. Med. 2006, 40, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Ahmed, S.; Grupp, I.L.; Matlib, M.A. Altered SR protein expression associated with contractile dysfunction in diabetic rat hearts. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H1137–H1147. [Google Scholar] [CrossRef] [PubMed]
- Kanamori, H.; Takemura, G.; Goto, K.; Tsujimoto, A.; Mikami, A.; Ogino, A.; Watanabe, T.; Morishita, K.; Okada, H.; Kawasaki, M.; et al. Autophagic adaptations in diabetic cardiomyopathy differ between type 1 and type 2 diabetes. Autophagy 2015, 11, 1146–1160. [Google Scholar] [CrossRef]
- Xie, Z.; He, C.; Zou, M.-H. AMP-activated protein kinase modulates cardiac autophagy in diabetic cardiomyopathy. Autophagy 2011, 7, 1254–1255. [Google Scholar] [CrossRef]
- Shimomura, H.; Terasaki, F.; Hayashi, T.; Kitaura, Y.; Isomura, T.; Suma, H. Autophagic Degeneration as a Possible Mechanism of Myocardial Cell Death in Dilated Cardiomyopathy. Jpn. Circ. J. 2001, 65, 965–968. [Google Scholar] [CrossRef]
- Xie, Z.; Lau, K.; Eby, B.; Lozano, P.; He, C.; Pennington, B.; Li, H.; Rathi, S.; Dong, Y.; Tian, R.; et al. Improvement of Cardiac Functions by Chronic Metformin Treatment Is Associated With Enhanced Cardiac Autophagy in Diabetic OVE26 Mice. Diabetes 2011, 60, 1770–1778. [Google Scholar] [CrossRef]
- Chapple, S.J.; Cheng, X.; Mann, G.E. Effects of 4-hydroxynonenal on vascular endothelial and smooth muscle cell redox signaling and function in health and disease. Redox Biol. 2013, 1, 319–331. [Google Scholar] [CrossRef]
- Leonarduzzi, G.; Chiarpotto, E.; Biasi, F.; Poli, G. 4-Hydroxynonenal and cholesterol oxidation products in atherosclerosis. Mol. Nutr. Food Res. 2005, 49, 1044–1049. [Google Scholar] [CrossRef] [PubMed]
- Vladykovskaya, E.; Sithu, S.D.; Haberzettl, P.; Wickramasinghe, N.S.; Merchant, M.L.; Hill, B.G.; McCracken, J.; Agarwal, A.; Dougherty, S.; Gordon, S.A.; et al. Lipidperoxidationproduct4-hydroxy-trans-2-nonenalcauses endothelial activation by inducing endoplasmic reticulum stress. J. Biol. Chem. 2021, 287, 11398–11409. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Zampetaki, A.; Margariti, A.; Pepe, A.E.; Alam, S.; Martin, D.; Xiao, Q.; Wang, W.; Jin, Z.-G.; Cockerill, G.; et al. Sustained activation of XBP1 splicing leads to endothelial apoptosis and atherosclerosis development in response to disturbed flow. Proc. Natl. Acad. Sci. USA 2009, 106, 8326–8331. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.-L.; Hee, S.-W.; Hsuan, C.-F.; Yang, W.; Huang, J.-Y.; Lin, Y.-L.; Hsu, C.-N.; Hwang, J.-J.; Chen, S.-M.; Ding, Z.-Z.; et al. A Novel ALDH2 Activator AD-9308 Improves Diastolic and Systolic Myocardial Functions in Streptozotocin-Induced Diabetic Mice. Antioxidants 2021, 10, 450. https://doi.org/10.3390/antiox10030450
Lee H-L, Hee S-W, Hsuan C-F, Yang W, Huang J-Y, Lin Y-L, Hsu C-N, Hwang J-J, Chen S-M, Ding Z-Z, et al. A Novel ALDH2 Activator AD-9308 Improves Diastolic and Systolic Myocardial Functions in Streptozotocin-Induced Diabetic Mice. Antioxidants. 2021; 10(3):450. https://doi.org/10.3390/antiox10030450
Chicago/Turabian StyleLee, Hsiao-Lin, Siow-Wey Hee, Chin-Feng Hsuan, Wenjin Yang, Jing-Yong Huang, Ya-Ling Lin, Chih-Neng Hsu, Juey-Jen Hwang, Shiau-Mei Chen, Zhi-Zhong Ding, and et al. 2021. "A Novel ALDH2 Activator AD-9308 Improves Diastolic and Systolic Myocardial Functions in Streptozotocin-Induced Diabetic Mice" Antioxidants 10, no. 3: 450. https://doi.org/10.3390/antiox10030450
APA StyleLee, H.-L., Hee, S.-W., Hsuan, C.-F., Yang, W., Huang, J.-Y., Lin, Y.-L., Hsu, C.-N., Hwang, J.-J., Chen, S.-M., Ding, Z.-Z., Lee, T.-Y., Lin, Y.-C., Tsai, F.-C., Su, W.-L., Chueh, L.-Y., Hsieh, M.-L., Chen, C.-H., Mochly-Rosen, D., Chang, Y.-C., & Chuang, L.-M. (2021). A Novel ALDH2 Activator AD-9308 Improves Diastolic and Systolic Myocardial Functions in Streptozotocin-Induced Diabetic Mice. Antioxidants, 10(3), 450. https://doi.org/10.3390/antiox10030450