
In Silico Screening of the DrugBank Database to Search for Possible Drugs against SARS-CoV-2


Abstract
1. Introduction
2. Results and Discussion
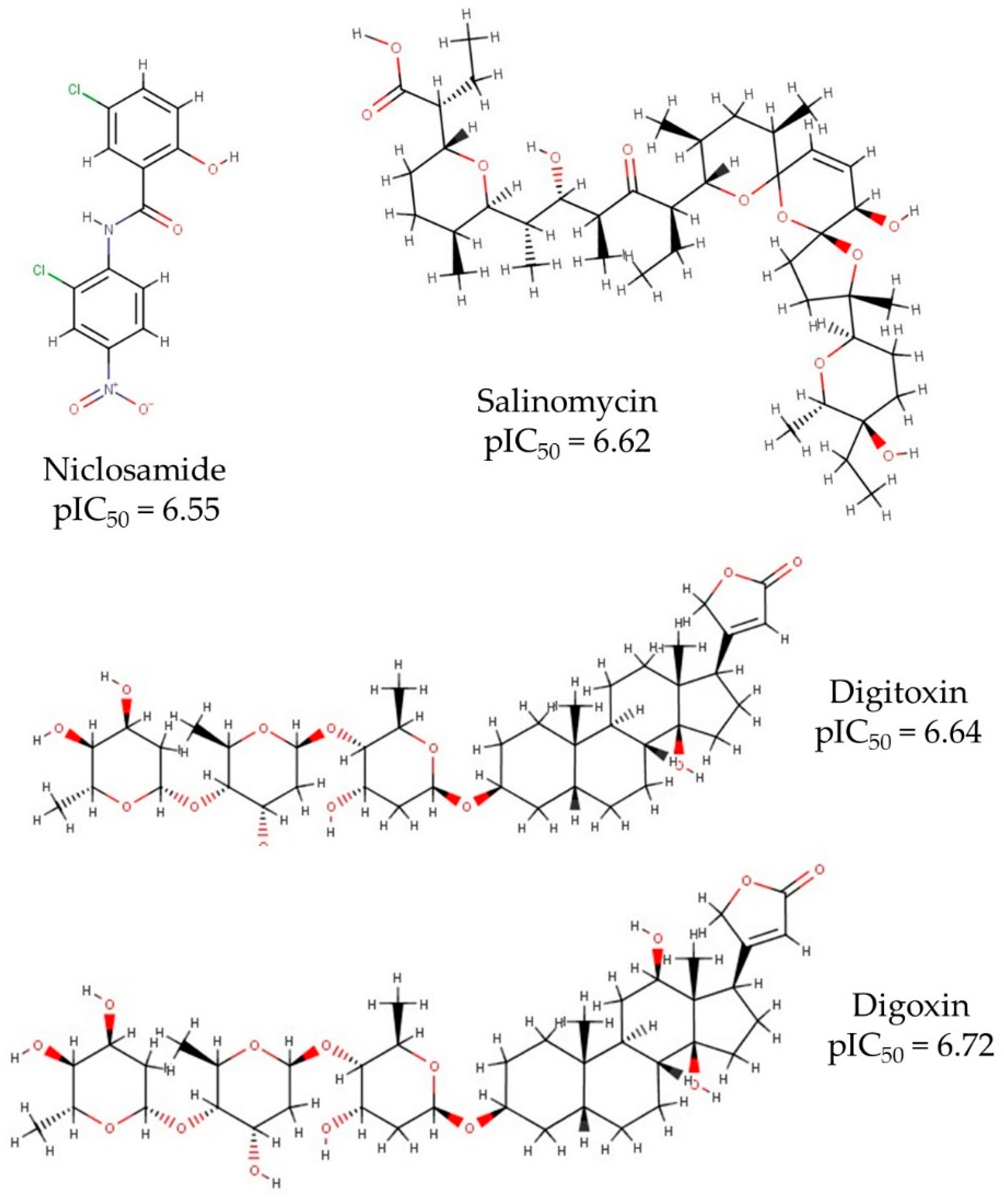
2.1. Data Set
2.2. Dataset Splitting (Training/Test)
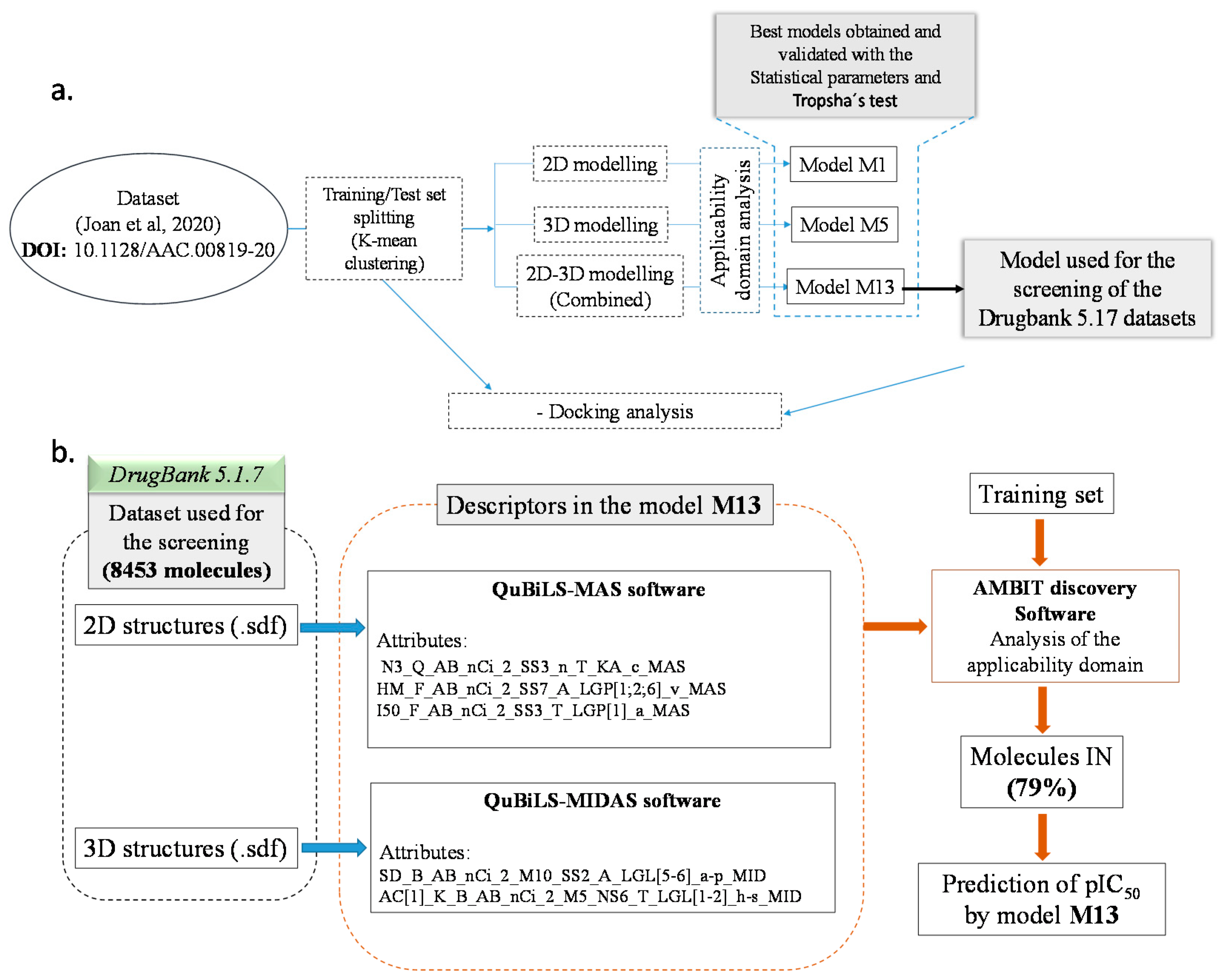
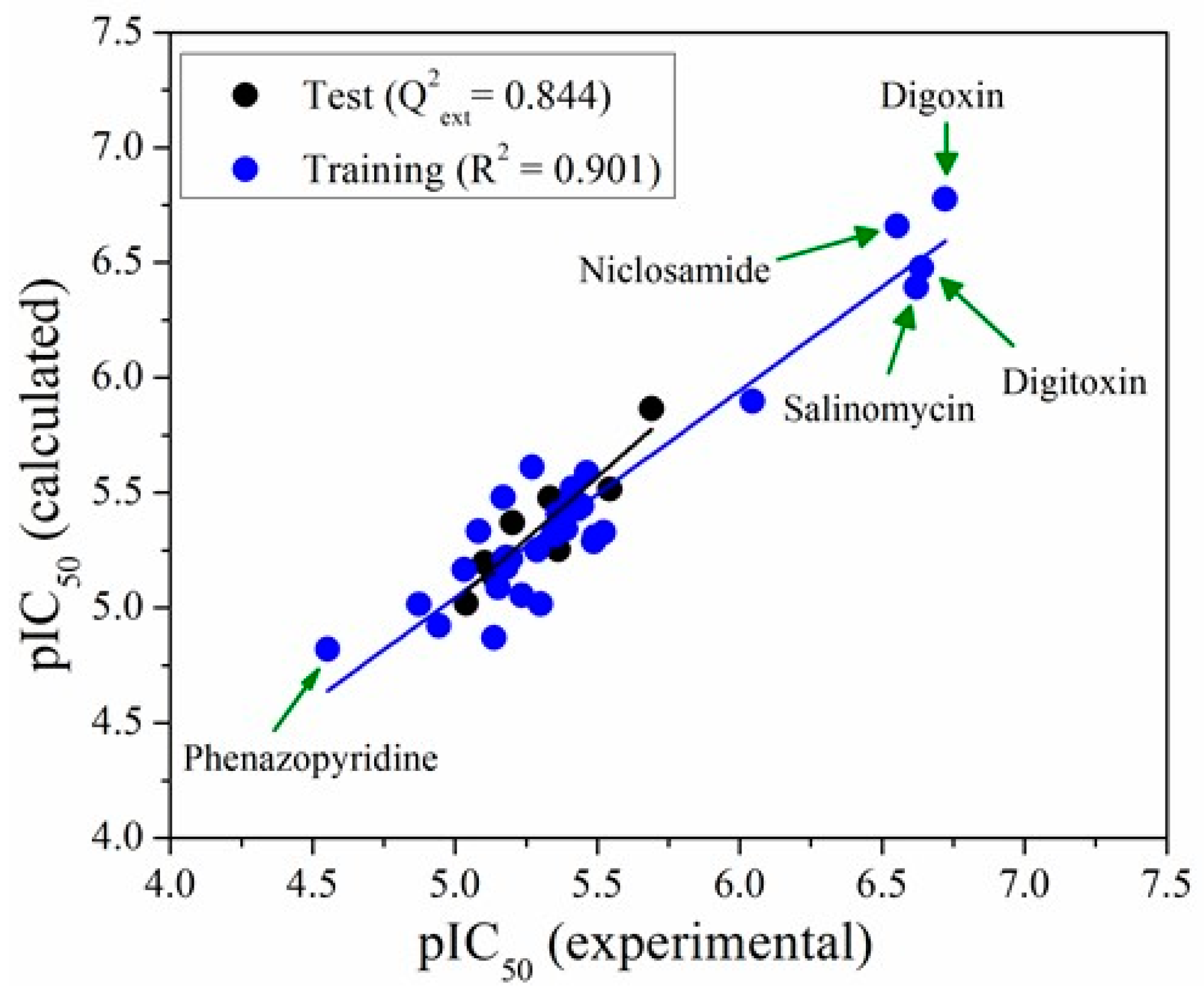
2.3. QSAR Modelling
2.4. DrugBank 5.1.7 Screening
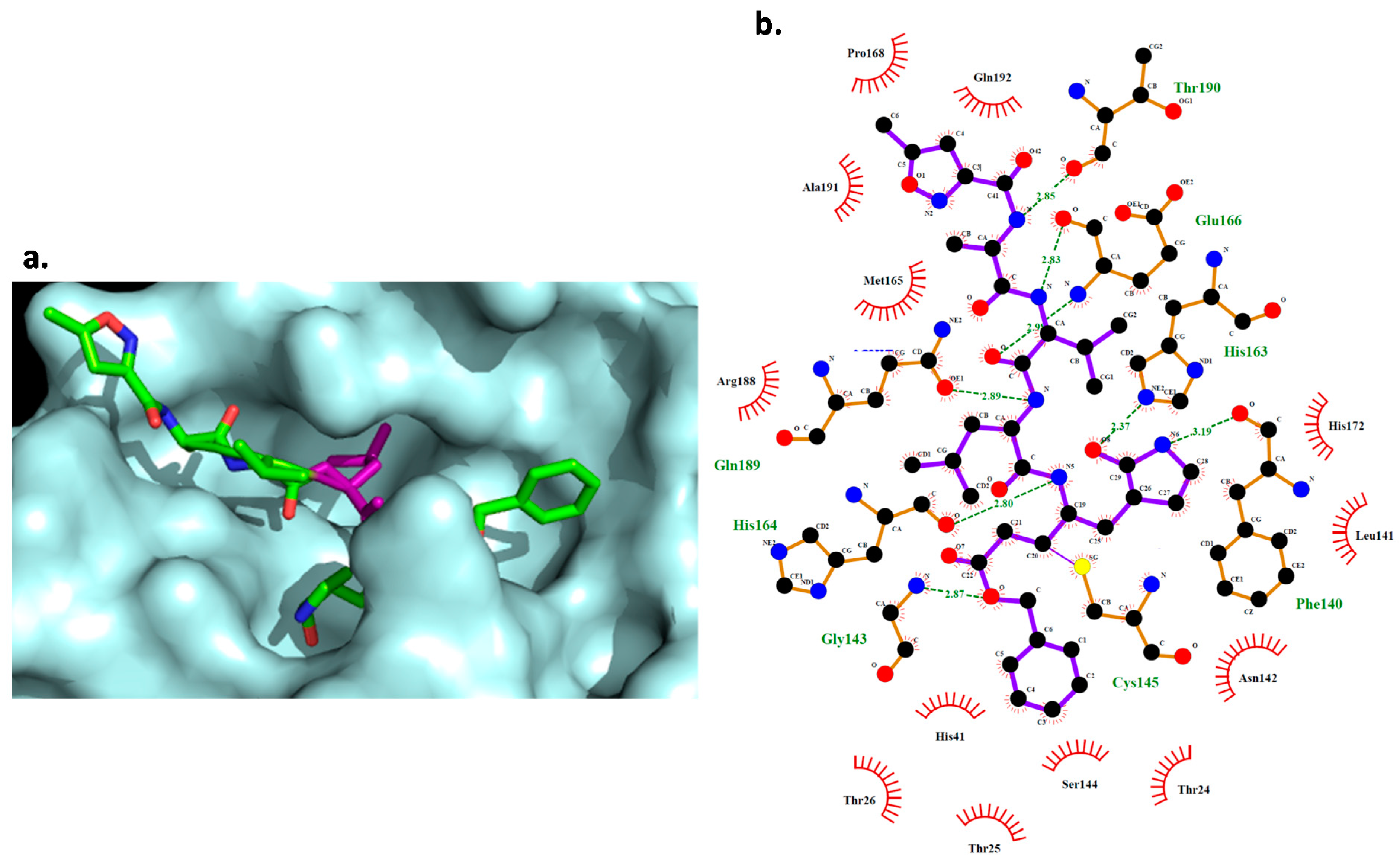
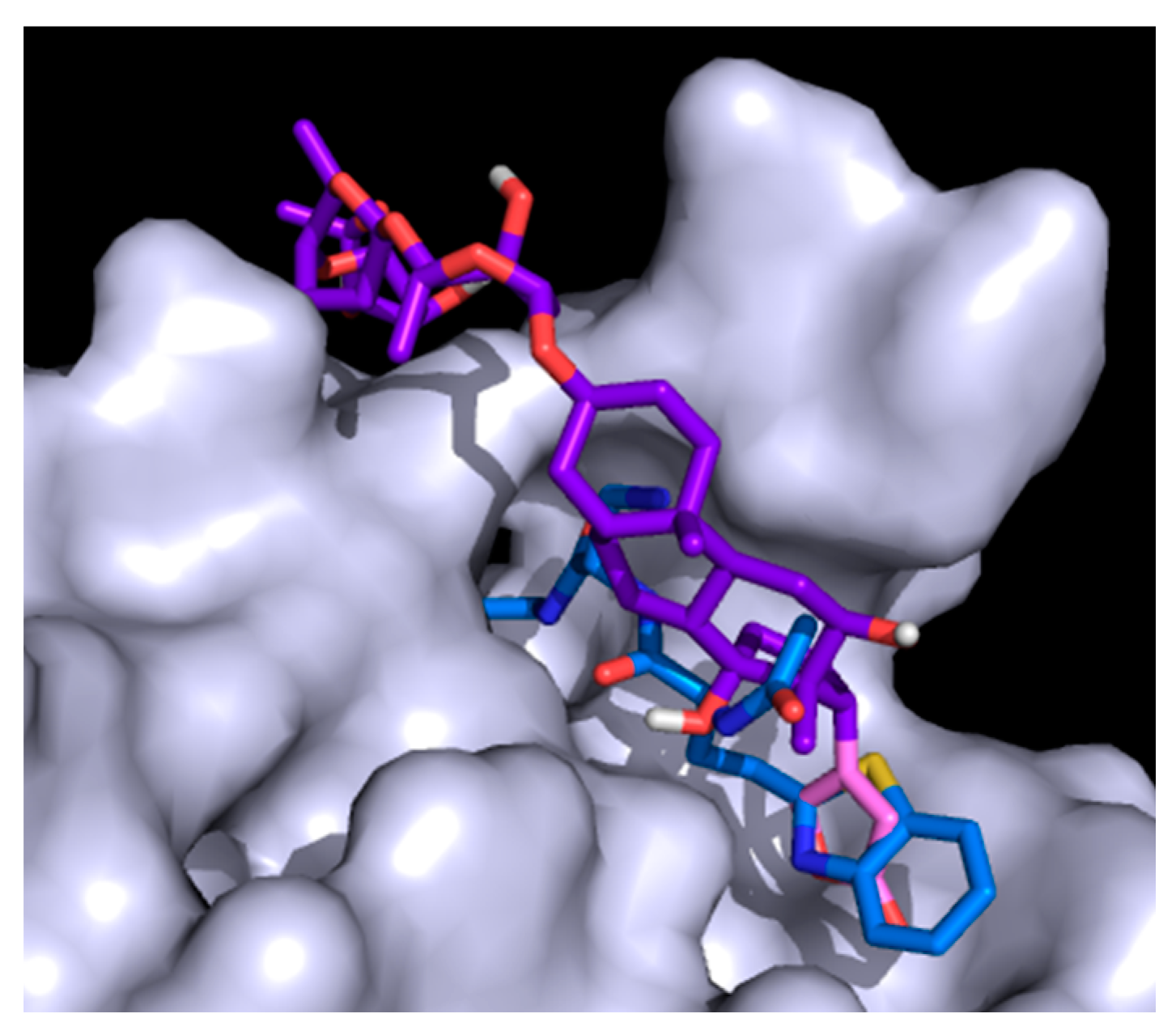
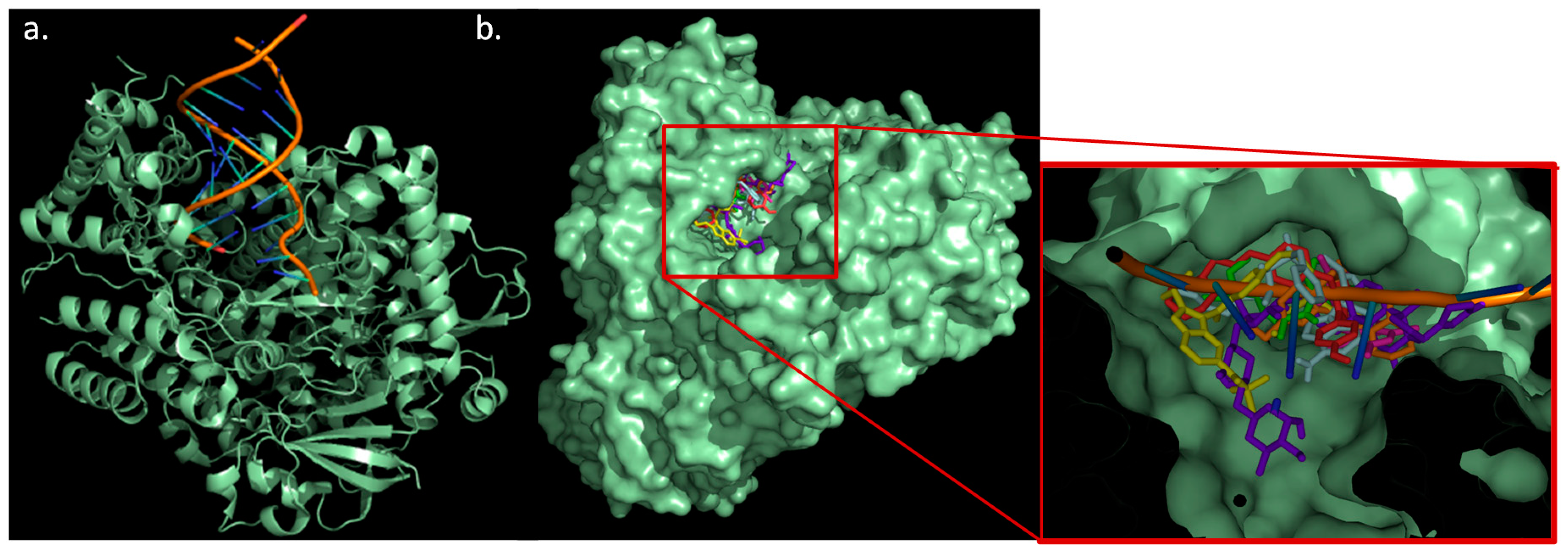
2.5. Docking Study
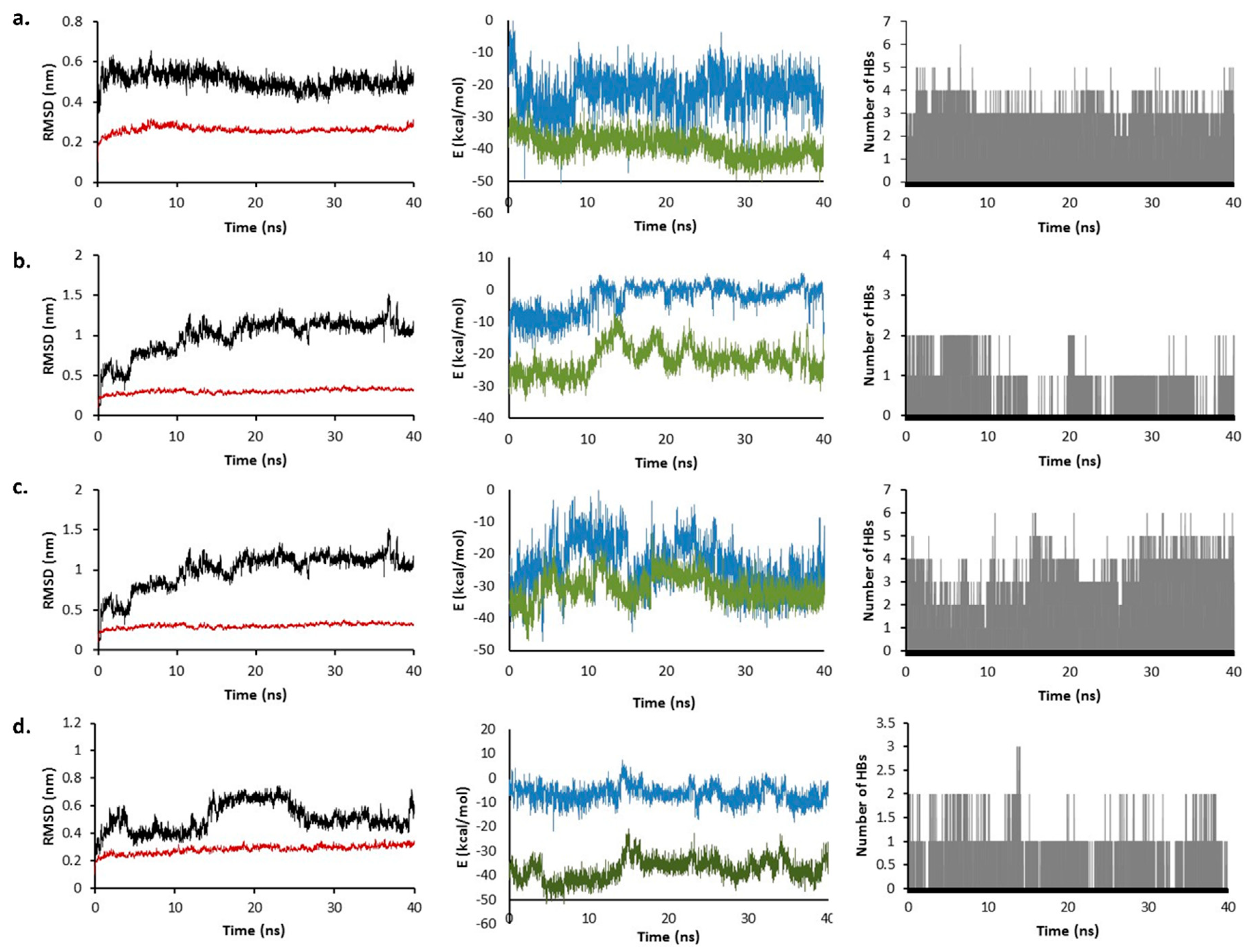
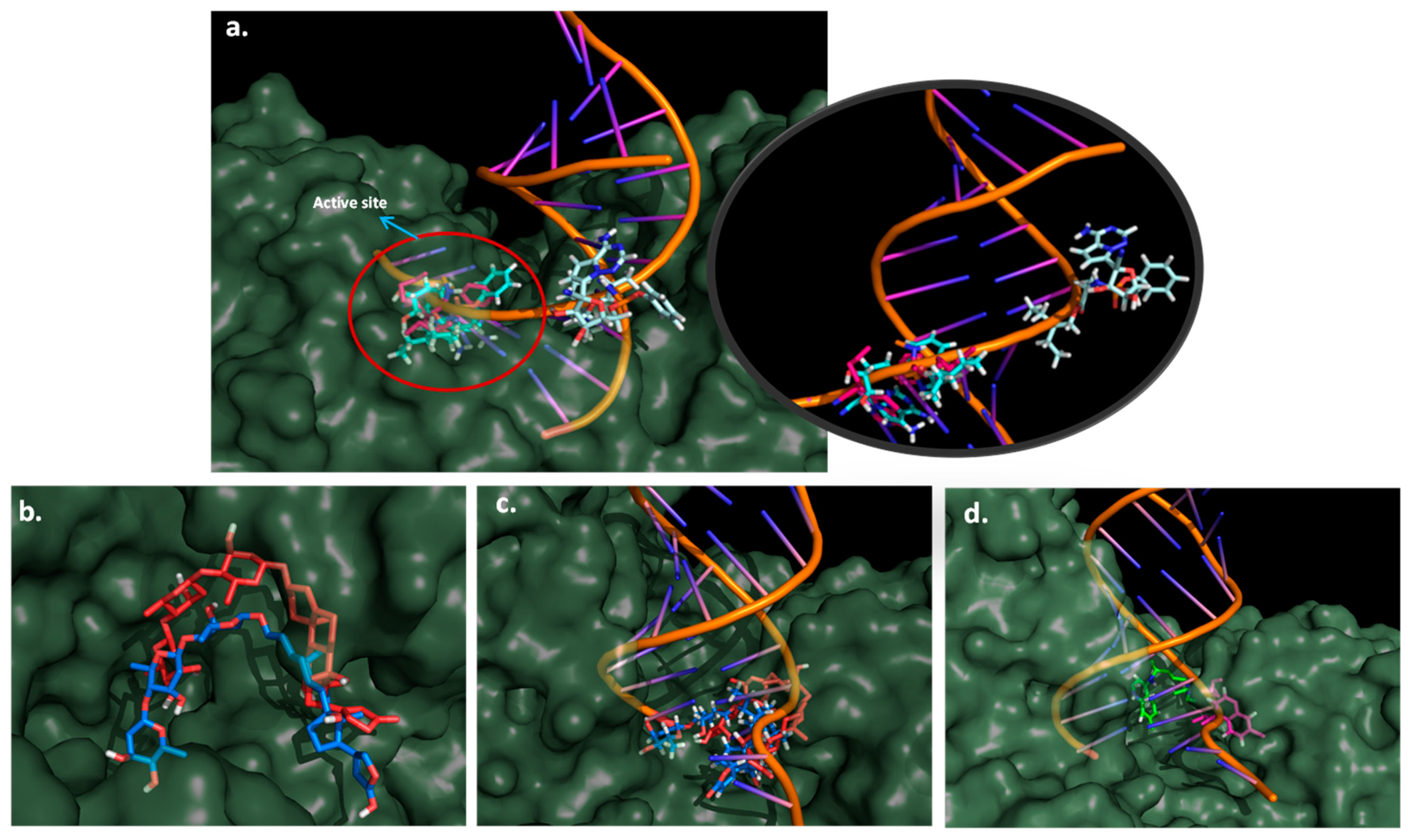
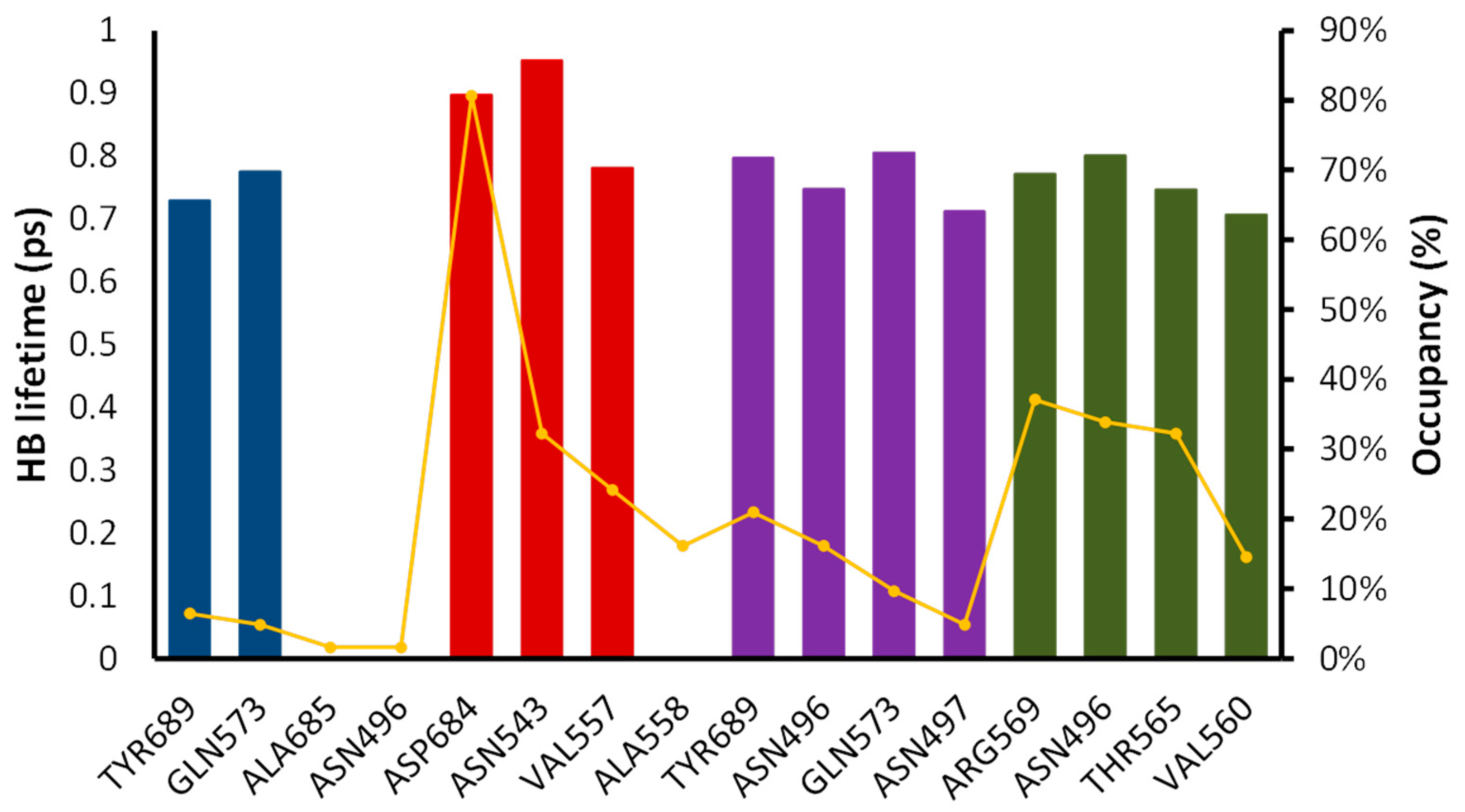
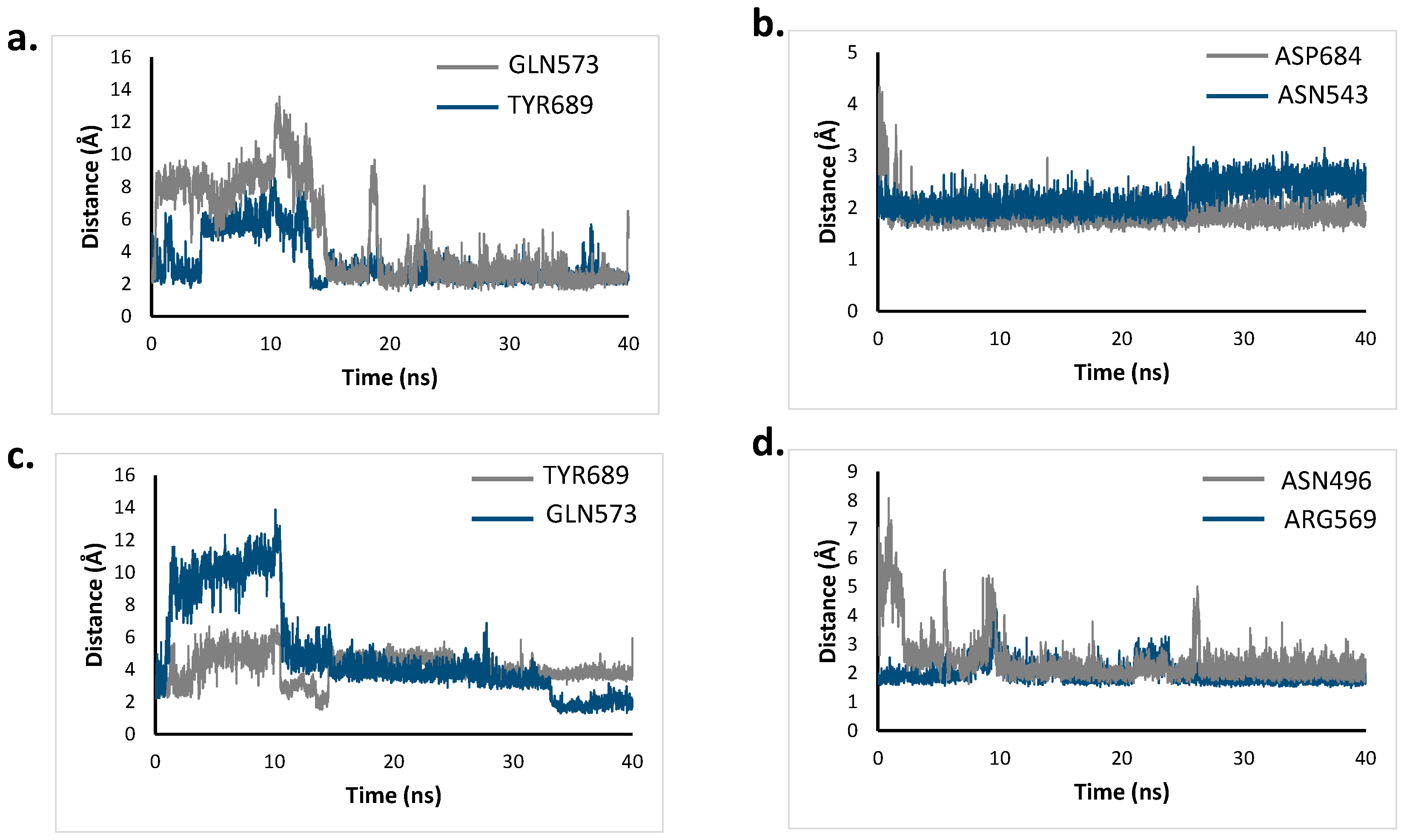
2.6. Molecular Dynamics Simulation
2.7. Total Binding Energy Calculations
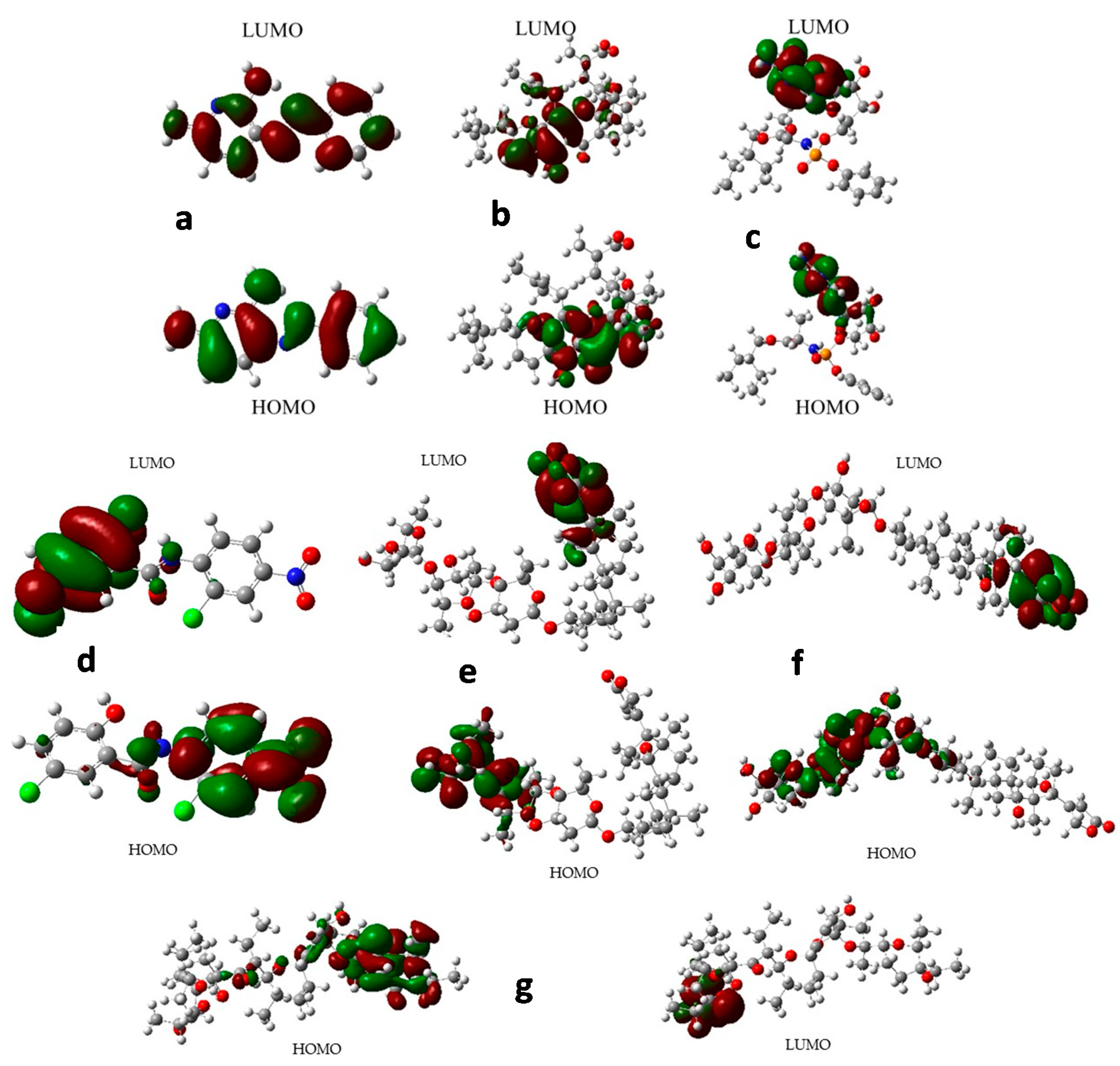
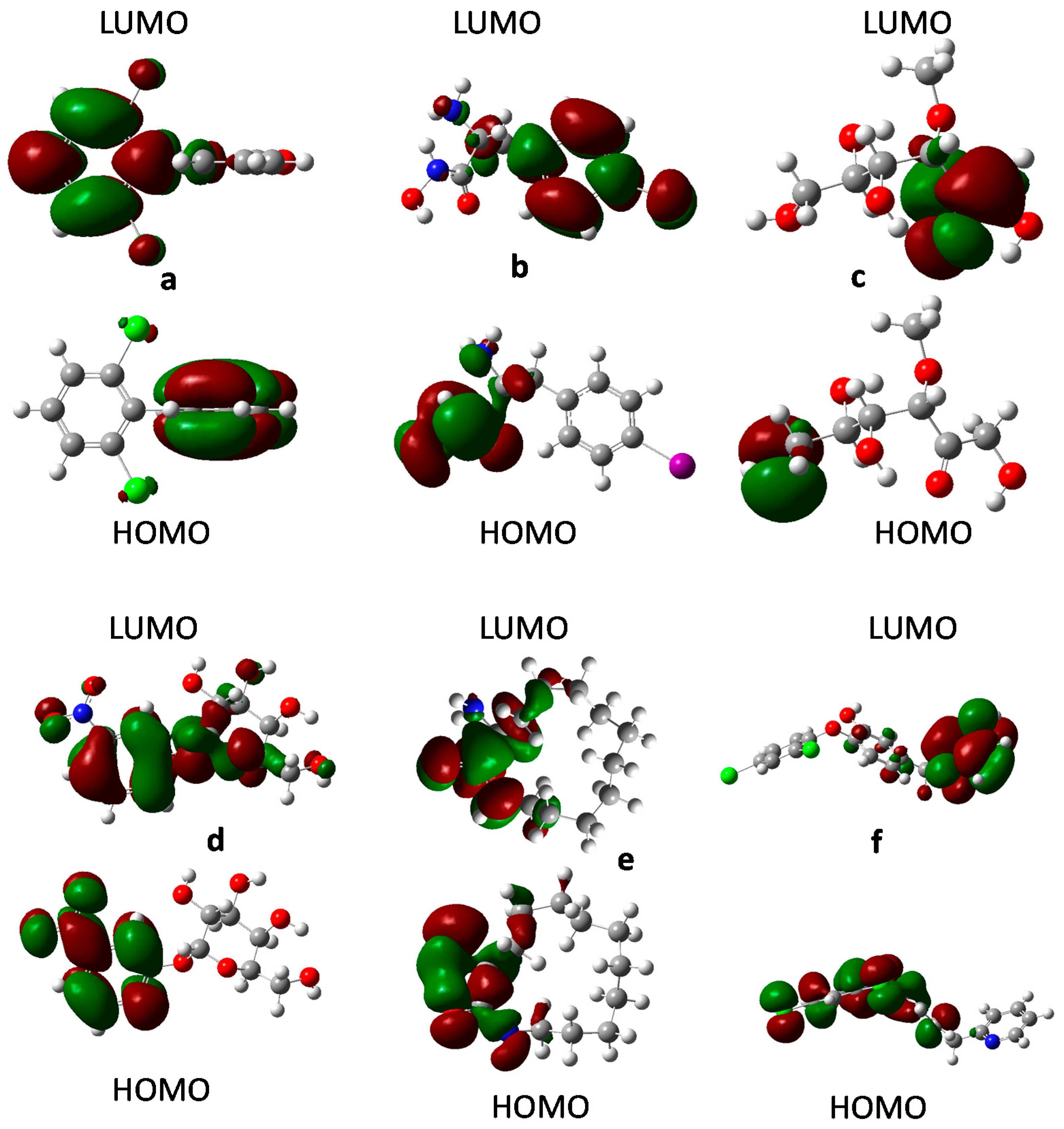
2.8. Frontier Orbital Analysis
3. Materials and Methods
3.1. Datasets Preparation
3.2. Descriptors Calculations and Modelling Process
3.3. Domain of Applicability
3.4. Model Performance
- The collinearity between the descriptors was evaluated by taking into account a Pearson’s correlation coefficient of <0.7.
- The coefficient of determination (R2) is used as a global evaluator of the model.
- The predictability of the model is determined by the leave one out cross-validation coefficient (Q2LOO), the external validation coefficient (Q2ext), and the bootstrapping coefficient (Q2boot).
- The robustness of the model is evaluated in terms of the Y-scrambling analysis (a(R2) and a(Q2)), based on a random perturbation of the Y values.
- Additionally, the linear fitting of the model was evaluated in terms of the Fisher coefficient value (F) and the residual standard deviation (s).
- Tropsha’s test analysis is performed for the leave-one-out and external validation analysis, as suggested by the Organization for Economic Cooperation and Development (OECD) principles [42].
3.5. Pharmacophore Modelling
3.6. Molecular Docking
3.7. Molecular Dynamic Simulations
3.8. Free Energy Calculations
3.9. Frontier Orbital Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Zhu, Y.; Li, J.; Pang, Z. Recent insights for the emerging COVID-19: Drug discovery, therapeutic options and vaccine development. Asian J. Pharm. Sci. 2020, 16, 4–23. [Google Scholar] [CrossRef] [PubMed]
- Tahir ul Qamar, M.; Alqahtani, S.M.; Alamri, M.A.; Chen, L.L. Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J. Pharm. Anal. 2020, 10, 313–319. [Google Scholar] [CrossRef]
- Gil, C.; Ginex, T.; Maestro, I.; Nozal, V.; Barrado-Gil, L.; Cuesta-Geijo, M.Á.; Urquiza, J.; Ramírez, D.; Alonso, C.; Campillo, N.E.; et al. COVID-19: Drug Targets and Potential Treatments. J. Med. Chem. 2020, 6, 1998–2016. [Google Scholar] [CrossRef] [PubMed]
- Hatada, R.; Okuwaki, K.; Mochizuki, Y.; Handa, Y.; Fukuzawa, K.; Komeiji, Y.; Okiyama, Y.; Tanaka, S. Fragment Molecular Orbital Based Interaction Analyses on COVID-19 Main Protease—Inhibitor N3 Complex (PDB ID: 6LU7). J. Chem. Inf. Model. 2020, 60, 3593–3602. [Google Scholar] [CrossRef] [PubMed]
- Eastman, R.T.; Roth, J.S.; Brimacombe, K.R.; Simeonov, A.; Shen, M.; Patnaik, S.; Hall, M.D. Remdesivir: A Review of Its Discovery and Development Leading to Emergency Use Authorization for Treatment of COVID-19. ACS Cent. Sci. 2020, 6, 672–683. [Google Scholar] [CrossRef]
- Xiu, S.; Dick, A.; Ju, H.; Mirzaie, S.; Abdi, F.; Cocklin, S.; Zhan, P.; Liu, X. Inhibitors of SARS-CoV-2 Entry: Current and Future Opportunities. J. Med. Chem. 2020, 63, 12256–12274. [Google Scholar] [CrossRef]
- Ahidjo, B.A.; Loe, M.W.C.; Ng, Y.L.; Mok, C.K.; Chu, J.J.H. Current Perspective of Antiviral Strategies against COVID-19. ACS Infect. Dis. 2020, 6, 1624–1634. [Google Scholar] [CrossRef]
- Korkmaz, B.; Lesner, A.; Marchand-Adam, S.; Moss, C.; Jenne, D.E. Lung Protection by Cathepsin C Inhibition: A New Hope for COVID-19 and ARDS? J. Med. Chem. 2020, 63, 13258–13265. [Google Scholar] [CrossRef]
- Lu, L.; Zhong, W.; Bian, Z.; Li, Z.; Zhang, K.; Liang, B.; Zhong, Y.; Hu, M.; Lin, L.; Liu, J.; et al. A comparison of mortality-related risk factors of COVID-19, SARS, and MERS: A systematic review and meta-analysis: Mortality-related risk factors of COVID-19, SARS, and MERS. J. Infect. 2020, 81, e18–e25. [Google Scholar] [CrossRef]
- Yuan, X.; Yang, C.; He, Q.; Chen, J.; Yu, D.; Li, J.; Zhai, S.; Qin, Z.; Du, K.; Chu, Z.; et al. Current and Perspective Diagnostic Techniques for COVID-19. ACS Infect. Dis. 2020, 6, 1998–2016. [Google Scholar] [CrossRef]
- Barros, R.O.; Junior, F.L.; Pereira, W.S.; Oliveira, N.M.; Ramos, R.M. Interaction of Drug Candidates with Various SARS-CoV-2 Receptors: An in Silico Study to Combat COVID-19. J. Proteome Res. 2020, 19, 4567–4575. [Google Scholar] [CrossRef]
- Gao, K.; Nguyen, D.D.; Chen, J.; Wang, R.; Wei, G.W. Repositioning of 8565 Existing Drugs for COVID-19. J. Phys. Chem. Lett. 2020, 11, 5373–5382. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Song, X.; Ma, T.; Pan, X.; Zhou, Y.; Hou, Y.; Zhang, Z.; Li, K.; Karypis, G.; Cheng, F. Repurpose Open Data to Discover Therapeutics for COVID-19 Using Deep Learning. J. Proteome Res. 2020, 19, 4624–4636. [Google Scholar] [CrossRef] [PubMed]
- Chien, M.; Anderson, T.K.; Jockusch, S.; Tao, C.; Li, X.; Kumar, S.; Russo, J.J.; Kirchdoerfer, R.N.; Ju, J. Nucleotide Analogues as Inhibitors of SARS-CoV-2 Polymerase, a Key Drug Target for COVID-19. J. Proteome Res. 2020, 19, 4690–4697. [Google Scholar] [CrossRef]
- Saul, S.; Einav, S. Old drugs for a new virus: Repurposed approaches for combating COVID-19. ACS Infect. Dis. 2020, 6, 2304–2318. [Google Scholar] [CrossRef]
- Weiss, C.; Carriere, M.; Fusco, L.; Fusco, L.; Capua, I.; Regla-Nava, J.A.; Pasquali, M.; Pasquali, M.; Pasquali, M.; Scott, J.A.; et al. Toward Nanotechnology-Enabled Approaches against the COVID-19 Pandemic. ACS Nano 2020, 14, 6383–6406. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Brindisi, M.; Shahabi, D.; Chapman, M.E.; Mesecar, A.D. Drug Development and Medicinal Chemistry Efforts toward SARS-Coronavirus and Covid-19 Therapeutics. ChemMedChem 2020, 15, 907–932. [Google Scholar] [CrossRef] [PubMed]
- Batra, R.; Chan, H.; Kamath, G.; Ramprasad, R.; Cherukara, M.J.; Sankaranarayanan, S.K. Screening of Therapeutic Agents for COVID-19 Using Machine Learning and Ensemble Docking Studies. J. Phys. Chem. Lett. 2020, 11, 7058–7065. [Google Scholar] [CrossRef]
- Asai, A.; Konno, M.; Ozaki, M.; Otsuka, C.; Vecchione, A.; Arai, T.; Kitagawa, T.; Ofusa, K.; Yabumoto, M.; Hirotsu, T.; et al. COVID-19 drug discovery using intensive approaches. Int. J. Mol. Sci. 2020, 21, 2839. [Google Scholar] [CrossRef]
- Wang, J. Fast Identification of Possible Drug Treatment of Coronavirus Disease-19 (COVID-19) through Computational Drug Repurposing Study. J. Chem. Inf. Model. 2020, 60, 3277–3286. [Google Scholar] [CrossRef]
- Kandeel, M.; Al-Nazawi, M. Virtual screening and repurposing of FDA approved drugs against COVID-19 main protease. Life Sci. 2020, 251, 117627. [Google Scholar] [CrossRef] [PubMed]
- ISRCTN83971151 Public Health Emergency SOLIDARITY Trial of Treatments for COVID-19 Infection in Hospitalized Patients. 2020. Available online: http://www.who.int/trialsearch/Trial2.aspx?TrialID=ISRCTN83971151 (accessed on 17 August 2020).
- WHO. Solidarity Trial Consortium Repurposed Antiviral Drugs for Covid-19—Interim WHO Solidarity Trial Results. N. Engl. J. Med. 2020, 1–15. [Google Scholar] [CrossRef]
- Lou, Y.; Liu, L.; Yao, H.; Hu, X.; Su, J.; Xu, K.; Luo, R.; Yang, X.; He, L.; Lu, X.; et al. Clinical Outcomes and Plasma Concentrations of Baloxavir Marboxil and Favipiravir in COVID-19 Patients: An Exploratory Randomized, Controlled Trial. Eur. J. Pharm. Sci. 2021, 157, 105631. [Google Scholar] [CrossRef] [PubMed]
- Caly, L.; Druce, J.D.; Catton, M.G.; Jans, D.A.; Wagstaff, K.M. The FDA-approved drug ivermectin inhibits the replication of SARS-CoV-2 in vitro. Antivir. Res. 2020, 178, 104787. [Google Scholar] [CrossRef] [PubMed]
- Elnagdy, S.; AlKhazindar, M. The Potential of Antimicrobial Peptides as an Antiviral Therapy against COVID-19. ACS Pharmacol. Transl. Sci. 2020, 3, 780–782. [Google Scholar] [CrossRef]
- Yan, V.C.; Muller, F.L. Advantages of the Parent Nucleoside GS-441524 over Remdesivir for Covid-19 Treatment. ACS Med. Chem. Lett. 2020, 11, 1361–1366. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.; Ko, M.; Lee, J.; Choi, I.; Byun, S.Y.; Park, S.; Shum, D.; Kim, S. Identification of antiviral drug candidates against SARS-CoV-2 from FDA-approved drugs. Antimicrob. Agents Chemother. 2020, 64, e00819-20. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Information, N.C. For B. Digoxin. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Digoxin (accessed on 17 August 2020).
- Johansson, S.; Lindholm, P.; Gullbo, J.; Larsson, R.; Bohlin, L.; Claeson, P. Cytotoxicity of digitoxin and related cardiac glycosides in human tumor cells. Anticancer. Drugs 2001, 12, 475–483. [Google Scholar] [CrossRef]
- Resham, K.; Patel, P.N.; Thummuri, D.; Guntuku, L.; Shah, V.; Bambal, R.B.; Naidu, V.G.M. Preclinical drug metabolism and pharmacokinetics of salinomycin, a potential candidate for targeting human cancer stem cells. Chem. Biol. Interact. 2015, 240, 146–152. [Google Scholar] [CrossRef]
- National Center for Biotechnology Coccidiostats. Available online: https://www.ncbi.nlm.nih.gov/mesh/68003049 (accessed on 17 August 2020).
- Drugbank Niclosamide. Available online: https://www.drugbank.ca/drugs/DB06803 (accessed on 17 August 2020).
- Jaworska, J.; Nikolova-Jeliazkova, N. How can structural similarity analysis help in category formation? SAR QSAR Environ. Res. 2007, 18, 195–207. [Google Scholar] [CrossRef]
- Torrent-Sucarrat, M.; De Proft, F.; Ayers, P.W.; Geerlings, P. On the applicability of local softness and hardness. Phys. Chem. Chem. Phys. 2010, 12, 1072–1080. [Google Scholar] [CrossRef]
- Vandewaterbeemd, H.; Kansy, M. Hydrogen-bonding capacity and brain penetration. CHIMIA Int. J. Chem. 1992, 46, 299–303. [Google Scholar]
- Putz, M.V.; Ionaşcu, C.; Putz, A.M.; Ostafe, V. Alert-QSAR. Implications for electrophilic theory of chemical carcinogenesis. Int. J. Mol. Sci. 2011, 12, 5098–5134. [Google Scholar] [CrossRef] [PubMed]
- Oliferenko, A.A.; Krylenko, P.V.; Palyulin, V.A.; Zefirov, N.S. A new scheme for electronegativity equalization as a source of electronic descriptors: Application to chemical reactivity. SAR QSAR Environ. Res. 2002, 13, 297–305. [Google Scholar] [CrossRef]
- Hajimahdi, Z.; Safizadeh, F.; Zarghi, A. Qsar analysis for some 1, 2-benzisothiazol-3-one derivatives as caspase-3 inhibitors by stepwise mlr method. Iran. J. Pharm. Res. 2016, 15, 439–448. [Google Scholar] [PubMed]
- Gozalbes, R.; Doucet, J.P.; Derouin, F. Application of topological descriptions in QSAR and drug design: History and new trends. Curr. Drug Targets Infect. Disord. 2002, 2, 93–102. [Google Scholar] [CrossRef] [PubMed]
- OECD. Quantitative Structure-Activity Relationships Project [(Q)SARs] OECD. Principles for the Validation, for Regulatory Purposes of (Quantitative) Structure Activity Relationship Models. Available online: http://www.oecd.org/env/ehs/oecdquantitativestructure-activityrelationshipsprojectqsars.htm (accessed on 21 May 2020).
- Kar, S.S.; Bhat, V.G.; Shenoy, V.P.; Bairy, I.; Shenoy, G.G. Design, synthesis, and evaluation of novel diphenyl ether derivatives against drug-susceptible and drug-resistant strains of Mycobacterium tuberculosis. Chem. Biol. Drug Des. 2019, 93, 60–66. [Google Scholar] [CrossRef]
- Schulz, D.; Nachtigall, J.; Geisen, U.; Kalthoff, H.; Imhoff, J.F.; Fiedler, H.P.; Süssmuth, R.D. Silvalactam, a 24-membered macrolactam antibiotic produced by Streptomyces sp. Tudie; 6392. J. Antibiot. 2012, 65, 366–372. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Amagai, K.; Kudo, F.; Eguchi, T. Biosynthetic pathway of macrolactam polyketide antibiotic cremimycin. Tetrahedron 2011, 67, 8559–8563. [Google Scholar] [CrossRef]
- Foss, M.H.; Pou, S.; Davidson, P.M.; Dunaj, J.L.; Winter, R.W.; Pou, S.; Licon, M.H.; Doh, J.K.; Li, Y.; Kelly, J.X.; et al. Diphenylether-Modified 1,2-Diamines with Improved Drug Properties for Development against Mycobacterium tuberculosis. ACS Infect. Dis. 2016, 2, 500–508. [Google Scholar] [CrossRef]
- Fothergill, A.W. Miconazole: A historical perspective. Expert Rev. Anti. Infect. Ther. 2006, 4, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Vandenbosch, D.; Braeckmans, K.; Nelis, H.J.; Coenye, T. Fungicidal activity of miconazole against Candida spp. biofilms. J. Antimicrob. Chemother. 2010, 65, 694–700. [Google Scholar] [CrossRef] [PubMed]
- Mao, K.; Zhang, X.; Ali, E.; Liao, X.; Jin, R.; Ren, Z.; Wan, H.; Li, J. Characterization of nitenpyram resistance in Nilaparvata lugens (Stål). Pestic. Biochem. Physiol. 2019, 157, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Hess, C.; Brockmann, C.; Doberentz, E.; Madea, B.; Musshoff, F. Unintentional lethal overdose with metildigoxin in a 36-week-old infant—Post mortem tissue distribution of metildigoxin and its metabolites by liquid chromatography tandem mass spectrometry. Forensic Sci. Int. 2014, 241, e23–e27. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, A. Issues of Interferences in Therapeutic Drug Monitoring. In Biotin and Other Interferences in Immunoassays; Elsevier: Amsterdam, The Netherlands, 2019; pp. 99–112. [Google Scholar]
- Hauptman, P.J.; Kelly, R.A. Digitalis. Circulation 1999, 99, 1265–1270. [Google Scholar] [CrossRef]
- Drugbank 2′,4′-Dinitrophenyl-2deoxy-2-Fluro-B-D-Cellobioside (CB04086). Available online: https://www.drugbank.ca/drugs/DB04086 (accessed on 20 August 2020).
- González De La Huebra, M.J.; Vincent, U.; Bordin, G.; Rodríguez, A.R. Characterisation of dirithromycin and spiramycin using high performance liquid chromatography and direct infusion mass spectrometry. Anal. Chim. Acta 2004, 503, 247–256. [Google Scholar] [CrossRef]
- Castaldo, R.S.; Celli, B.R.; Gomez, F.; LaVallee, N.; Souhrada, J.; Hanrahan, J.P. A comparison of 5-day courses of dirithromycin and azithromycin in the treatment of acute exacerbations of chronic obstructive pulmonary disease. Clin. Ther. 2003, 25, 542–557. [Google Scholar] [CrossRef]
- Wasilewski, M.M.; Wilson, M.G.; Slides, G.D.; Stotka, J.L. Comparative efficacy of 5 days of dirithromycin and 7 days of erythromycin in skin and soft tissue infections. J. Antimicrob. Chemother. 2000, 46, 255–262. [Google Scholar] [CrossRef][Green Version]
- Chen, T.F.; Chang, Y.C.; Hsiao, Y.; Lee, K.H.; Hsiao, Y.C.; Lin, Y.H.; Tu, Y.C.E.; Huang, H.C.; Chen, C.Y.; Juan, H.F. DockCoV2: A drug database against SARS-CoV-2. Nucleic Acids Res. 2021, 49, D1152–D1159. [Google Scholar] [CrossRef]
- Murugan, N.A.; Kumar, S.; Jeyakanthan, J.; Srivastava, V. Searching for target-specific and multi-targeting organics for Covid-19 in the Drugbank database with a double scoring approach. Sci. Rep. 2020, 10, 19125. [Google Scholar] [CrossRef]
- Chowdhury, K.H.; Chowdhury, M.R.; Mahmud, S.; Tareq, A.M.; Hanif, N.B.; Banu, N.; Ali Reza, A.S.M.; Emran, T.B.; Simal-Gandara, J. Drug repurposing approach against novel coronavirus disease (COVID-19) through virtual screening targeting SARS-CoV-2 main protease. Biology 2021, 10, 2. [Google Scholar] [CrossRef]
- Pantsar, T.; Poso, A. Binding affinity via docking: Fact and fiction. Molecules 2018, 23, 1899. [Google Scholar] [CrossRef]
- Warren, G.L.; Andrews, C.W.; Capelli, A.M.; Clarke, B.; LaLonde, J.; Lambert, M.H.; Lindvall, M.; Nevins, N.; Semus, S.F.; Senger, S.; et al. A critical assessment of docking programs and scoring functions. J. Med. Chem. 2006, 49, 5912–5931. [Google Scholar] [CrossRef]
- Grover, M.; Singh, B.; Bakshi, M.; Singh, S. Quantitative structure-property relationships in pharmaceutical research—Part 1. Pharm. Sci. Technol. Today 2000, 3, 28–35. [Google Scholar] [CrossRef]
- Malhotra, R.; Ravesh, A.; Singh, V. Synthesis, characterization, antimicrobial activities, and QSAR studies of organotin(IV) complexes. Phosphorus Sulfur Silicon Relat. Elem. 2017, 192, 73–80. [Google Scholar] [CrossRef]
- Kumer, A.; Paul, S. The Simulating Study of Homo, Lumo, Thermo Physical and Quantitative Structure of Activity Relationship (QSAR) of some Anticancer Active Ionic Liquids. Eurasian J. Environ. Res. 2019, 3, 1–10. [Google Scholar]
- Kumar, A.; Grewal, A.S.; Singh, V.; Narang, R.; Pandita, D.; Lather, V. Synthesis, Antimicrobial Activity and QSAR Studies of Some New Sparfloxacin Derivatives. Pharm. Chem. J. 2018, 52, 444–454. [Google Scholar] [CrossRef]
- Khodair, A.I.; Awad, M.K.; Gesson, J.P.; Elshaier, Y.A.M.M. New N-ribosides and N-mannosides of rhodanine derivatives with anticancer activity on leukemia cell line: Design, synthesis, DFT and molecular modelling studies. Carbohydr. Res. 2020, 487, 107894. [Google Scholar] [CrossRef]
- Suresh Kumar, S.; Athimoolam, S.; Sridhar, B. Structural, spectral, theoretical and anticancer studies on new co-crystal of the drug 5-fluorouracil. J. Mol. Struct. 2018, 1173, 951–958. [Google Scholar] [CrossRef]
- Kanagamani, K.; Muthukrishnan, P.; Ilayaraja, M.; Shankar, K.; Kathiresan, A. Synthesis, Characterisation and DFT Studies of Stigmasterol Mediated Silver Nanoparticles and Their Anticancer Activity. J. Inorg. Organomet. Polym. Mater. 2018, 28, 702–710. [Google Scholar] [CrossRef]
- Jeyaseelan, S.C.; Premkumar, R.; Kaviyarasu, K.; Franklin Benial, A.M. Spectroscopic, quantum chemical, molecular docking and in vitro anticancer activity studies on 5-Methoxyindole-3-carboxaldehyde. J. Mol. Struct. 2019, 1197, 134–146. [Google Scholar] [CrossRef]
- Sarkar, I.; Goswami, S.; Majumder, P. Quantitative structure–activity relationship (QSAR) study of some DNA-intercalating anticancer drugs. In Computational Advancement in Communication Circuits and Systems; Springer Verlag: Singapore, 2020; Volume 575, pp. 357–366. [Google Scholar]
- Wang, J.; Yun, D.; Yao, J.; Fu, W.; Huang, F.; Chen, L.; Wei, T.; Yu, C.; Xu, H.; Zhou, X.; et al. Design, synthesis and QSAR study of novel isatin analogues inspired Michael acceptor as potential anticancer compounds. Eur. J. Med. Chem. 2018, 144, 493–503. [Google Scholar] [CrossRef]
- Baeten, A.; Tafazoli, M.; Kirsch-Volders, M.; Geerlings, P. Use of the HSAB principle in quantitative structure–activity relationships in toxicological research: Application to the genotoxicity of chlorinated hydrocarbons. Int. J. Quantum Chem. 1999, 74, 351–355. [Google Scholar] [CrossRef]
- Bradbury, S.P.; Mekenyan, O.G.; Ankley, G.T. The role of ligand flexibility in predicting biological activity: Structure-activity relationships for aryl hydrocarbon, estrogen, and androgen receptor binding affinity. Environ. Toxicol. Chem. 1998, 17, 15–25. [Google Scholar]
- Joshi, R.; Pandey, N.; Yadav, S.K.; Tilak, R.; Mishra, H.; Pokharia, S. Synthesis, spectroscopic characterization, DFT studies and antifungal activity of (E)-4-amino-5-[N’-(2-nitro-benzylidene)-hydrazino]-2,4-dihydro-[1,2,4]triazole-3-thione. J. Mol. Struct. 2018, 1164, 386–403. [Google Scholar] [CrossRef]
- Joshi, R.; Kumari, A.; Singh, K.; Mishra, H.; Pokharia, S. Triorganotin(IV) complexes of Schiff base derived from 1,2,4-triazole moiety: Synthesis, spectroscopic investigation, DFT studies, antifungal activity and molecular docking studies. J. Mol. Struct. 2020, 1206, 127639. [Google Scholar] [CrossRef]
- Yan, Z.; Liu, A.; Huang, M.; Liu, M.; Pei, H.; Huang, L.; Yi, H.; Liu, W.; Hu, A. Design, synthesis, DFT study and antifungal activity of the derivatives of pyrazolecarboxamide containing thiazole or oxazole ring. Eur. J. Med. Chem. 2018, 149, 170–181. [Google Scholar] [CrossRef]
- Ali, M.S.; Farah, M.A.; Al-Lohedan, H.A.; Al-Anazi, K.M. Comprehensive exploration of the anticancer activities of procaine and its binding with calf thymus DNA: A multi spectroscopic and molecular modelling study. RSC Adv. 2018, 8, 9083–9093. [Google Scholar] [CrossRef]
- Rachedi, K.O.; Ouk, T.S.; Bahadi, R.; Bouzina, A.; Djouad, S.E.; Bechlem, K.; Zerrouki, R.; Ben Hadda, T.; Almalki, F.; Berredjem, M. Synthesis, DFT and POM analyses of cytotoxicity activity of α-amidophosphonates derivatives: Identification of potential antiviral O,O-pharmacophore site. J. Mol. Struct. 2019, 1197, 196–203. [Google Scholar] [CrossRef]
- Da Costa, R.M.; Bastos, J.K.; Costa, M.C.A.; Ferreira, M.M.C.; Mizuno, C.S.; Caramori, G.F.; Nagurniak, G.R.; Simão, M.R.; dos Santos, R.A.; Veneziani, R.C.S.; et al. In vitro cytotoxicity and structure-activity relationship approaches of ent-kaurenoic acid derivatives against human breast carcinoma cell line. Phytochemistry 2018, 156, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Soffers, A.E.M.F.; Boersma, M.G.; Vaes, W.H.J.; Vervoort, J.; Tyrakowska, B.; Hermens, J.L.M.; Rietjens, I.M.C.M. Computer-modeling-based QSARs for analyzing experimental data on biotransformation and toxicity. In Toxicology In Vitro; Elsevier: Pergamon, Turkey, 2001; Volume 15, pp. 539–551. [Google Scholar]
- Flores, M.C.; Márquez, E.A.; Mora, J.R. Molecular modeling studies of bromopyrrole alkaloids as potential antimalarial compounds: A DFT approach. Med. Chem. Res. 2018, 27, 844–856. [Google Scholar] [CrossRef]
- Cortes, E.; Mora, J.R.; Márquez, E. Modelling the Anti-Methicillin-Resistant Staphylococcus Aureus (MRSA) Activity of Cannabinoids: A QSAR and Docking Study. Crystals 2020, 10, 692. [Google Scholar] [CrossRef]
- Mizukami, Y. Character of Frontier Orbitals of Antiviral Drugs: Candidate Drugs against Covid-19. Open J. Phys. Chem. 2020, 10, 158–165. [Google Scholar] [CrossRef]
- Hagar, M.; Ahmed, H.A.; Aljohani, G.; Alhaddad, O.A. Investigation of some antiviral N-heterocycles as COVID 19 drug: Molecular docking and DFT calculations. Int. J. Mol. Sci. 2020, 21, 3922. [Google Scholar] [CrossRef]
- Valdés-Martiní, J.R.; Marrero-Ponce, Y.; García-Jacas, C.R.; Martinez-Mayorga, K.; Barigye, S.J.; D’Almeida, Y.S.V.; Pham-The, H.; Pérez-Giménez, F.; Morell, C.A. QuBiLS-MAS, open source multi-platform software for atom- and bond-based topological (2D) and chiral (2.5D) algebraic molecular descriptors computations. J. Cheminform. 2017, 9, 35. [Google Scholar] [CrossRef]
- Leonard, J.T.; Roy, K. On selection of training and test sets for the development of predictive QSAR models. QSAR Comb. Sci. 2006, 25, 235–251. [Google Scholar] [CrossRef]
- García-Jacas, C.R.; Marrero-Ponce, Y.; Acevedo-Martínez, L.; Barigye, S.J.; Valdés-Martiní, J.R.; Contreras-Torres, E. QuBiLS-MIDAS: A parallel free-software for molecular descriptors computation based on multilinear algebraic maps. J. Comput. Chem. 2014, 35, 1395–1409. [Google Scholar] [CrossRef] [PubMed]
- García-Jacas, C.R.; Marrero-Ponce, Y.; Cortés-Guzmán, F.; Suárez-Lezcano, J.; Martinez-Rios, F.O.; García-González, L.A.; Pupo-Meriño, M.; Martinez-Mayorga, K. Enhancing Acute Oral Toxicity Predictions by using Consensus Modeling and Algebraic Form-Based 0D-to-2D Molecular Encodes. Chem. Res. Toxicol. 2019, 32, 1178–1192. [Google Scholar] [CrossRef]
- Mora, J.R.; Marrero-Ponce, Y.; García-Jacas, C.R.; Suarez Causado, A. Ensemble Models Based on QuBiLS-MAS Features and Shallow Learning for the Prediction of Drug-Induced Liver Toxicity: Improving Deep Learning and Traditional Approaches. Chem. Res. Toxicol. 2020, 33, 1855–1873. [Google Scholar] [CrossRef] [PubMed]
- Marrero-Ponce, Y.; Iyarreta-Veitía, M.; Montero-Torres, A.; Romero-Zaldivar, C.; Brandt, C.A.; Ávila, P.E.; Kirchgatter, K.; Machado, Y. Ligand-based virtual screening and in silico design of new antimalarial compounds using nonstochastic and stochastic total and atom-type quadratic maps. J. Chem. Inf. Model. 2005, 45, 1082–1100. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, N.; Mora, J.R.; Marquez, E.A. Computational Molecular Modeling of Pin1 Inhibition Activity of Quinazoline, Benzophenone, and Pyrimidine Derivatives. J. Chem. 2019, 2019, 2954250. [Google Scholar] [CrossRef]
- Mora, J.R.; Márquez, E.A.; Calle, L. Computational molecular modelling of N-cinnamoyl and hydroxycinnamoyl amides as potential α-glucosidase inhibitors. Med. Chem. Res. 2018, 27, 2214–2223. [Google Scholar] [CrossRef]
- Márquez, E.; Mora, J.R.; Flores-Morales, V.; Insuasty, D.; Calle, L. Modeling the antileukemia activity of ellipticine-related compounds: QSAR and molecular docking study. Molecules 2020, 25, 24. [Google Scholar] [CrossRef] [PubMed]
- Edraki, N.; Das, U.; Hemateenejad, B.; Dimmock, J.R.; Miri, R. Comparative QSAR analysis of 3,5-bis (arylidene)-4-piperidone derivatives: The development of predictive cytotoxicity models. Iran. J. Pharm. Res. 2016, 15, 425–437. [Google Scholar]
- Matlab, version 8.4 (R2014b); The MathWorks Inc.: Natick, MA, USA, 2014.
- Gramatica, P. Principles of QSAR Modeling: Comments and Suggestions from Personal Experience. Int. J. Quant. Struct. Relationships 2020, 5, 61–97. [Google Scholar] [CrossRef]
- Schneidman-Duhovny, D.; Dror, O.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. Deterministic pharmacophore detection via multiple flexible alignment of drug-like molecules. J. Comput. Biol. 2008, 15, 737–754. [Google Scholar] [CrossRef] [PubMed]
- Schneidman-Duhovny, D.; Dror, O.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PharmaGist: A webserver for ligand-based pharmacophore detection. Nucleic Acids Res. 2008, 36 (Suppl. 2), W223–W228. [Google Scholar] [CrossRef]
- Sunseri, J.; Koes, D.R. Pharmit: Interactive exploration of chemical space. Nucleic Acids Res. 2016, 44, W442–W448. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The protein data bank. Acta Crystallogr. Sect. D Biol. Crystallogr. 2002, 28, 235–242. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef]
- The PyMol Molecular Graphics System, version 1.8; Schrödinger, LLC: New York, NY, USA, 2015.
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2010, 16, 2785–2791. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- Discovery Studio Visualizer, version 16.1; Biovia, Dassault Systèmes: San Diego, CA, USA, 2020.
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; Dinola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Consortium, O.S.D.D.; Lynn, A. g_mmpbsa—A GROMACS tool for MM-PBSA and its optimization for high-throughput binding energy calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Adolfo Cuesta, S.; Cordova-Sintjago, T.; Ramón Mora, J. Sulfonylation of Five-Membered Aromatic Heterocycles Compounds through Nucleophilic Aromatic Substitution: Concerted or Stepwise Mechanism? ChemistrySelect 2020, 5, 4515–4524. [Google Scholar] [CrossRef]
- Cervantes, C.; Mora, J.R.; Marquez, E.; Torres, J.; Rincón, L.; Mendez, M.A.; Alcázar, J.J. Theoretical calculations of the multistep reaction mechanism involved in asparagine pyrolysis supported by degree of rate control and thermodynamic control analyses. Appl. Sci. 2019, 9, 4847. [Google Scholar] [CrossRef]
- Frisch, M.J.G.; Trucks, W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 16; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N° of Attributes | R2 | Q2LOO | Q2LMO | Q2boot | Q2ext | |||
|---|---|---|---|---|---|---|---|---|
| 5 | 0.897 | 0.854 | 0.834 | 0.829 | 0.876 | |||
| a(R2) | a(Q2) | Kx | Kxy | F | s | |||
| 0.104 | −0.328 | 0.259 | 0.287 | 50.61 | 0.160 | |||
| Tropsha’s Test | ||||||||
| Leave-One-Out Validation | External Validation | |||||||
| Criterion | Result | Assessment | Result | Assessment | ||||
| R2 > 0.6 | 0.897 | PASS | 0.897 | PASS | ||||
| R2Val * > 0.5 | 0.854 | PASS | 0.876 | PASS | ||||
| (R2Val − R02)/R2Val < 0.1 | 0.001 | PASS | 0.002 | PASS | ||||
| (R2Val − R0′2)/R2Val < 0.1 | 0.021 | PASS | 0.020 | PASS | ||||
| abs(R02 − R0′2) < 0.1 | 0.018 | PASS | 0.016 | PASS | ||||
| 0.85 < k < 1.15 | 1.001 | PASS | 1.017 | PASS | ||||
| 0.85 < k′ < 1.15 | 0.998 | PASS | 0.982 | PASS | ||||
| Database | N° Total of Molecules | N° Molecules IN | Coverage (%) | pIC50 Range (Only Molecules IN) |
|---|---|---|---|---|
| Approved | 2265 | 1882 | 83 | 3.68–7.10 |
| Experimental | 5858 | 4519 | 77 | 3.58–7.51 |
| Nutraceutical | 102 | 55 | 54 | 3.99–5.85 |
| Withdrawn | 228 | 208 | 91 | 4.24–7.15 |
| Model | Features | Number of Hits |
|---|---|---|
| Model 1 | HBA1, HBA2, HBA3, HPH4 | 1 |
| Model 2 | HBA2, HBA3, HPH4 | 11 |
| Model 3 | HBA1, HBA3, HPH4 | 16 |
| Model 4 | HBA1, HBA2, HPH4 | 48 |
| Model 5 | HBA1, HBA2, HBA3 | 45 |
| Drug | Interactions |
|---|---|
| Digoxin | Thr24, Thr25, Thr26, Thr45, Met49, Leu141, Asn142, Gly143, Cys145, His164, Met165, Glu166, Pro168, Gln189, Thr190 |
| Hexachlorophene | Met49, Cys145, His164, Met165, Glu166 |
| Bazedoxifene | Thr25, Thr26, Leu27, His41, Met49, Tyr54, Asn142, Gly143, Cys145, His164, Met165, Gln166, Asp187, Arg188, Gln189, |
| Dronedarone | Thr25, Thr26, Met49, Phe140, Leu141, Asn142, Gly143, Cys145, His163, Met165, Glu166, Pro168, Gln189, Thr190, Ala191 |
| Thioridazine | His41, Asn142, Met49, Tyr54, Leu141, His164, Met165, Glu166, Asp187, Arg188, Gln189, Thr190 |
| Chloroquine | His41, Met49, Tyr54, Cys145, His164, Met165, Glu166, Asp187, Arg188, Gln189, Thr190, Gln192 |
| Remdesivir | His41, Thr25, Thr26, Leu27, Met49, Phe140, Leu141, Asn142, Gly143, Ser144, Cys145, His163, His164, Met165, Glu166, Asp187, Arg188, Gln189 |
| DB03259 | His41, Asn142, Gly143, Ser144, His163, Cys145, His164, Met165, Gln189 |
| DB01980 | His41, Asn142, Cys145, His164, Met165, Glu166, Arg188, Gln189 |
| DB02438 | Phe140, Leu141, Asn142, Gly143, Ser144, Cys145, His163, His164, Met165, Glu166 |
| DB02213 | His41, Leu141, Asn142, Gly143, Cys145, His164, Met165, Glu166, Asp187, Arg188, Gln189 |
| DB08476 | His41, Cys145, His164, Met165, Glu166, Asp187, Arg188, Gln189 |
| DB07287 | His41, Met49, Tyr54, His164, Met165, Glu166, Pro168, Asp187, Gln189, Thr190, Ala191 |
| Drug | Interactions |
|---|---|
| Digoxin | Asp108, Asn109, Gly160, Glu161, Leu162, Gly163, Asp164, Pro248, Tyr264, Tyr268, Tyr273, Thr301 |
| Hexachlorophene | Leu162, Asp164, Gly163, Pro248, Tyr264, Tyr268, Gln269, Gly271, Tyr273 |
| Bazedoxifene | Leu162, Gly163, Asp164, Pro247, Pro248, Tyr264, Asn267, Tyr268, Gln269, Tyr273 |
| Dronedarone | Leu162, Gly163, Asp164, Arg166, Met208, Ser245, Ala246, Pro248, Gly266, Tyr264, Tyr268, Tyr273, Thr301, Asp302 |
| Thioridazine | Leu162, Gly163, Asp164, Pro247, Pro248, Tyr264, Gly266, Asn267, Tyr268, Tyr273, Thr301 |
| Chloroquine | Leu162, Gly163, Asp164, Pro248, Tyr264, Tyr268, Gln269, Tyr273, Thr301 |
| Remdesivir | Leu162, Gly163, Asp164, Arg166, Glu167, Met208, Pro248, Tyr264, Tyr268, Gly271, Tyr273 |
| DB03259 | Gly163, Asp164, Arg166, Pro248, Tyr264, Tyr268, Tyr273, Thr301 |
| DB01980 | Asp164, Arg166, Pro247, Pro248, Tyr264, Tyr268, Tyr273, Thr301, Asp302 |
| DB02438 | Asp164, Arg166, Pro247, Pro248, Tyr264, Tyr273, Thr301, Asp302 |
| DB02213 | Leu162, Gly163, Asp164, Glu167, Pro248, Tyr264, Tyr268, Tyr273, Thr301 |
| DB08476 | Asp164, Met208, Pro247, Pro248, Tyr264, Tyr268, Tyr273, Thr301 |
| DB07287 | Leu162, Gly163, Asp164, Arg166, Tyr264, Tyr268, Tyr273, Thr301, Asp302 |
| Drug | Interactions |
|---|---|
| Digoxin | Lys500, Ser501, Asn543, Arg555, Val557, Ala558, Gly559, Val560, Thr565, Arg569, Ser682, Gly683, Asp684, Ala685, Thr687, Ala688, Asn691, Ser759, Thr868 |
| Hexachlorophene | Ser501, Thr565, Gly559, Val560, Arg 569, Gly683, |
| Bazedoxifene | Asn496, Asn497, Lys500, Ser501, Val560, Ser561, Ile562, Thr565, Arg569, Gln573, Leu576, Lys577, Gly683, Tyr689 |
| Dronedarone | Asn496, Asn497, Ser501, Val560, Thr565, Arg569, Gln573, Leu576, Lys577, Gly683, Asp684, Ala685, Thr687 Ala688, Tyr689 |
| Thioridazine | Lys500, Ser501, Ala512, Asn543, Val557, Ala558, Gly559, Ile562, Thr565, Arg569, Gly683, Asp684, Ala685 |
| Chloroquine | Asn496, Asn497, Ser501, Ala502, Val560, Ser561, Ile562, Thr565, Agr569, Gly683, Ala685, |
| Remdesivir | Asn497, Val495, Asn496, Lys500, Ser501, Val560, Thr565, Arg569, Gln573, Leu576, Lys577, Gly683, Ala685 |
| DB03259 | Val410, Gln444, Ala547, Ile548, Ser549, Arg553, Ala554, Arg555 |
| DB01980 | Thr565, Arg569, Gln573, Leu576, Lys577, Ala685, Tyr689 |
| DB02438 | Lys500, Ser501, Gly559, Val560, Ser561, Ile562, Thr565, Arg569 |
| DB02213 | Val410, Phe440, Phe441, Gln444, Lys545, Ile548, Ser549, Lys551, Arg553, Ala554, Arg555 |
| DB08476 | Asp452, Arg553, Arg555, Thr556, Asp623, Arg624, Thr680, Ser682, Asn691 |
| DB07287 | Lys500, Ser501, Ala512, Thr565, Arg569, Gln573, Leu576, Lys577, Gly683, Ala685, Tyr689 |
| Protein | Compound | van der Waals (kcal/mol) | Electrostatics (kcal/mol) | SASA (kcal/mol) | Total Binding Energy (kcal/mol) | pIC50 experimental | pIC50 predicted |
|---|---|---|---|---|---|---|---|
| MPro | Chloroquine | −26.89 | −1.71 | −3.10 | −17.55 | 5.14 | 5.15 |
| Digoxin | −43.41 | −15.82 | −5.47 | −16.64 | 6.72 | 6.68 | |
| Dronedarone | −11.21 | −1.30 | −1.28 | −4.92 | 5.41 | 5.46 | |
| Remdesivir | −14.17 | −3.05 | −1.81 | −2.77 | 4.94 | 4.87 | |
| N3 | −44.96 | −8.74 | −5.67 | −15.99 | -- | -- | |
| PLpro | Bazedoxifene | −16.72 | −1.43 | −1.89 | −10.96 | 5.46 | 5.57 |
| RdRp | Chloroquine | −24.21 | −1.49 | −3.37 | −17.70 | 5.14 | 5.15 |
| Digoxin | −46.28 | −18.53 | −6.06 | −23.79 | 6.72 | 6.68 | |
| Dronedarone | −41.18 | −1.88 | −5.82 | −20.97 | 5.41 | 5.46 | |
| Remdesivir | −35.98 | −16.80 | −4.85 | −15.79 | 4.94 | 4.87 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cuesta, S.A.; Mora, J.R.; Márquez, E.A. In Silico Screening of the DrugBank Database to Search for Possible Drugs against SARS-CoV-2. Molecules 2021, 26, 1100. https://doi.org/10.3390/molecules26041100
Cuesta SA, Mora JR, Márquez EA. In Silico Screening of the DrugBank Database to Search for Possible Drugs against SARS-CoV-2. Molecules. 2021; 26(4):1100. https://doi.org/10.3390/molecules26041100
Chicago/Turabian StyleCuesta, Sebastián A., José R. Mora, and Edgar A. Márquez. 2021. "In Silico Screening of the DrugBank Database to Search for Possible Drugs against SARS-CoV-2" Molecules 26, no. 4: 1100. https://doi.org/10.3390/molecules26041100
APA StyleCuesta, S. A., Mora, J. R., & Márquez, E. A. (2021). In Silico Screening of the DrugBank Database to Search for Possible Drugs against SARS-CoV-2. Molecules, 26(4), 1100. https://doi.org/10.3390/molecules26041100