
Dissection of the Genetic Association between Anorexia Nervosa and Obsessive–Compulsive Disorder at the Network and Cellular Levels


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Collection
2.2. Weighted Gene Co-Expression Network Analysis (WGCNA)
2.3. Gene Ontology (GO) Analysis
2.4. Spatiotemporal Expression Analysis
2.5. Network Analysis
2.6. Differential Expression Analysis
2.7. Gene Set Enrichment Analysis
3. Results
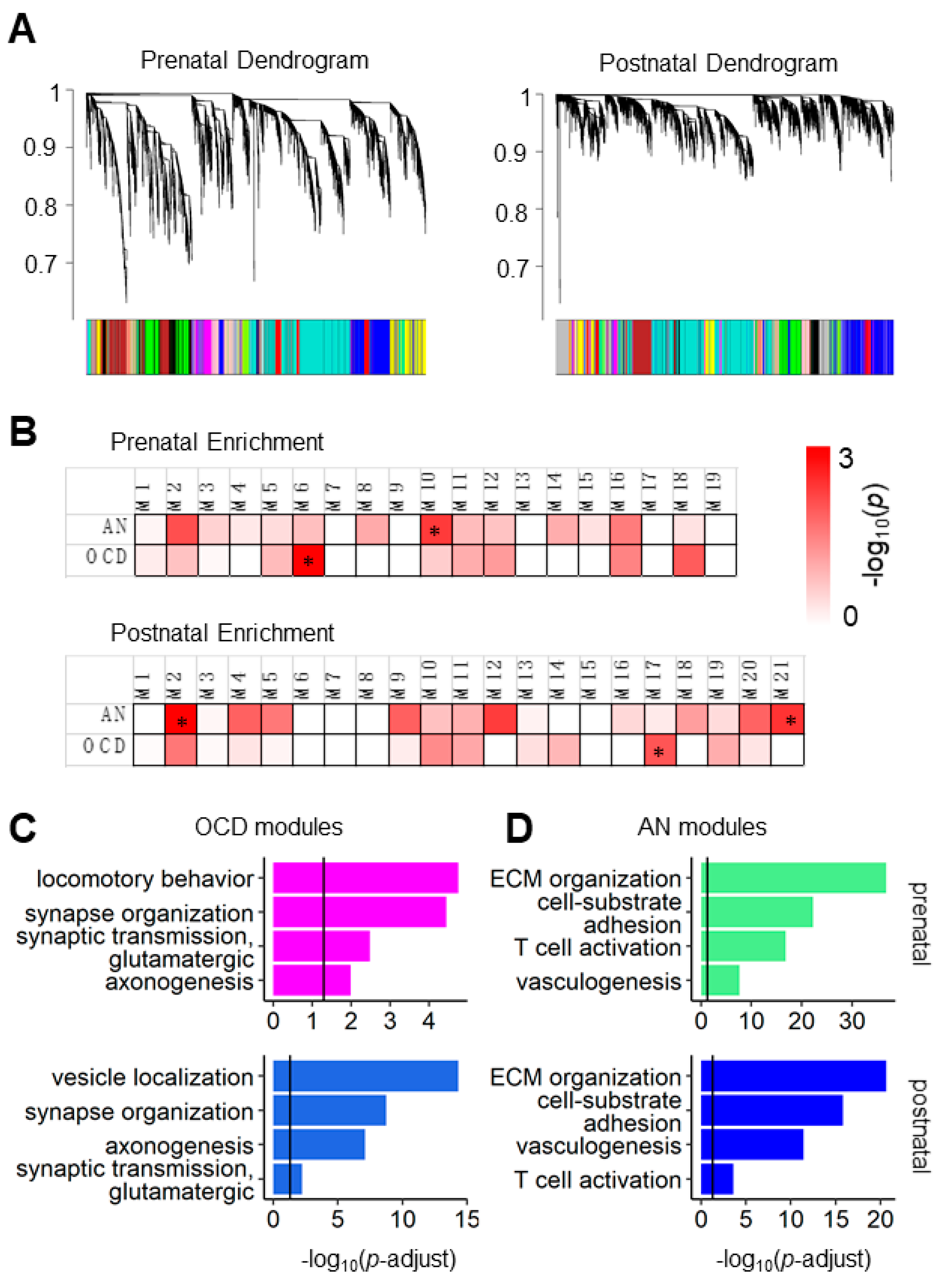
3.1. AN and OCD Risk Genes Coexpression Modules Show Different Biological Functions in Prenatal and Postnatal Periods
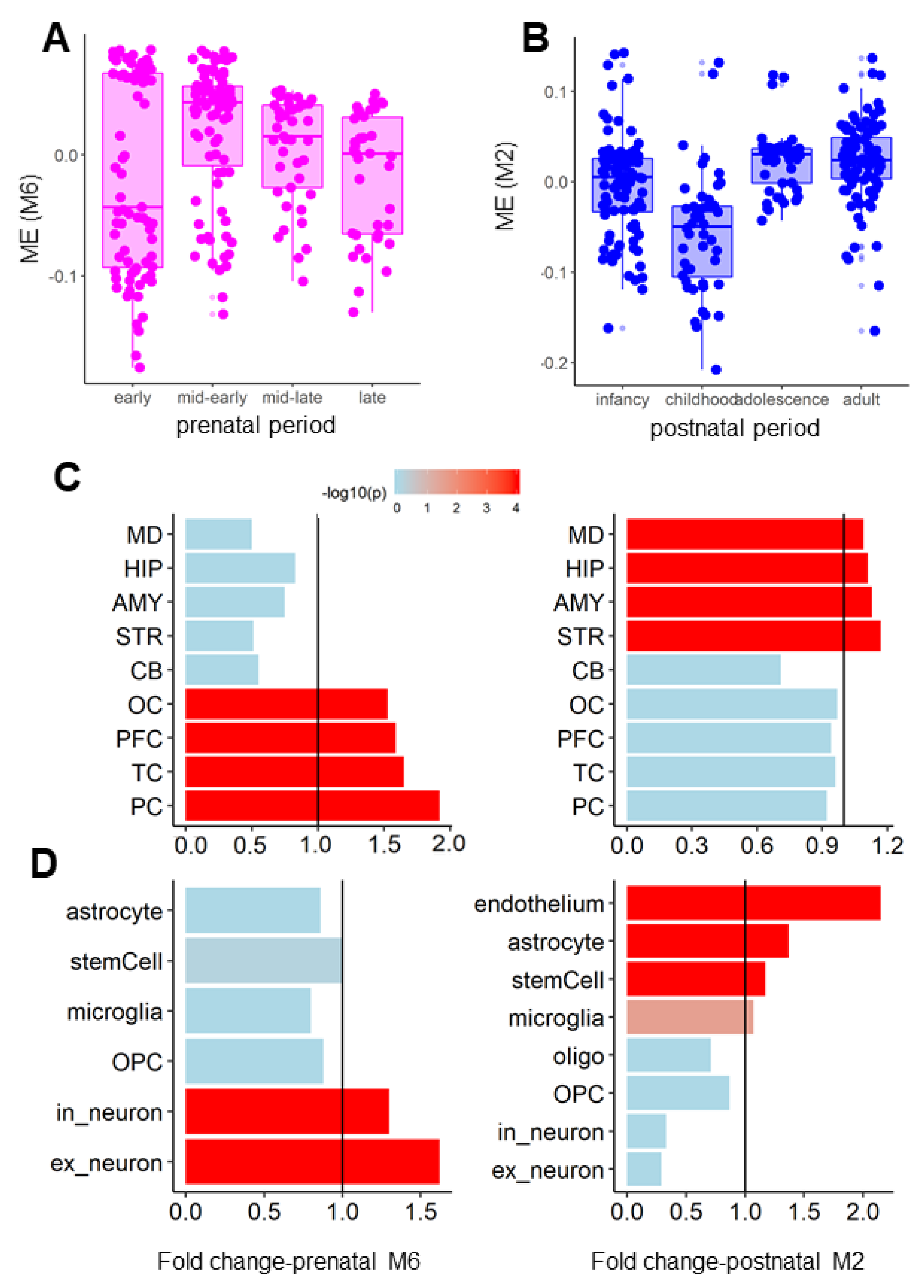
3.2. Coexpression Modules of the AN and OCD Risk Genes Exhibited Distinct Spatiotemporal and Cell-Type-Specific Expression Patterns
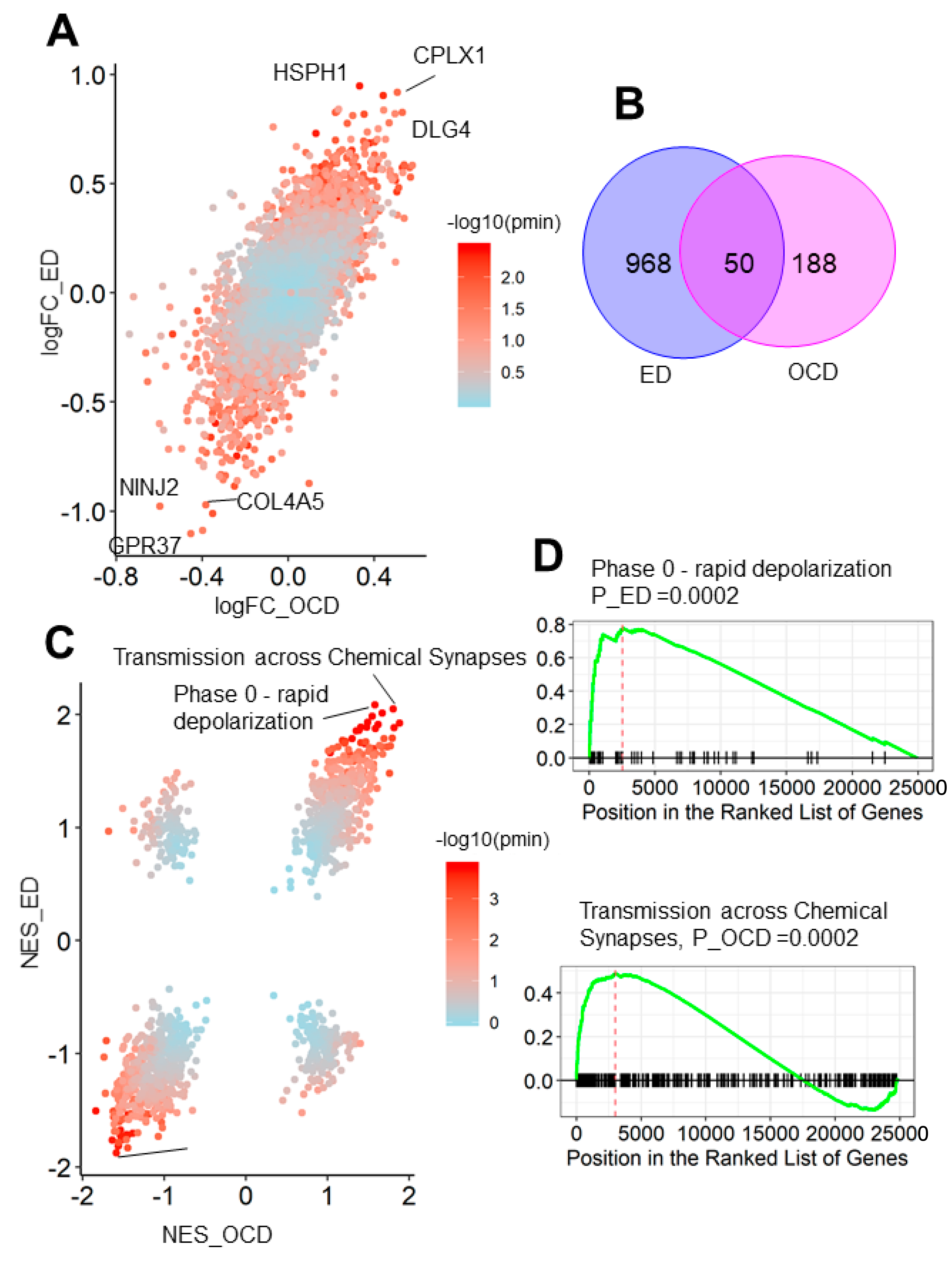
3.3. OCD and Eating Disorder Led to a Similar Brain Transcriptome Alteration Pattern
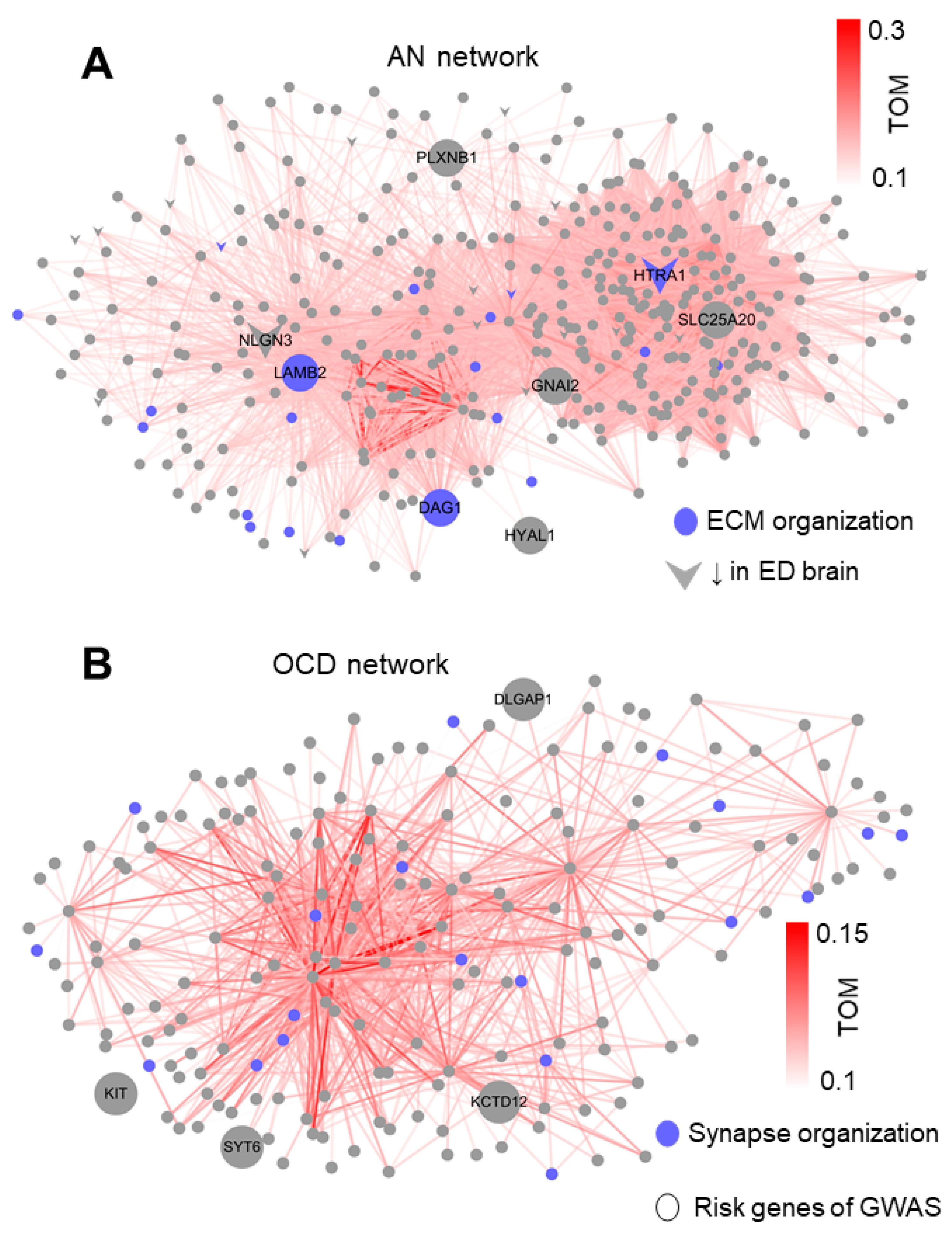
3.4. LAMB2, HTRA1, and KCTD12 as Hubs for AN and OCD Networks
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Trace, S.E.; Baker, J.H.; Peñas-Lledó, E.; Bulik, C.M. The genetics of eating disorders. Annu. Rev. Clin. Psychol. 2013, 9, 589–620. [Google Scholar] [CrossRef]
- Pauls, D.L.; Abramovitch, A.; Rauch, S.L.; Geller, D.A. Obsessive-compulsive disorder: An integrative genetic and neurobiological perspective. Nat. Publ. Group 2014, 15, 410–424. [Google Scholar] [CrossRef]
- Pettersson, E.; Lichtenstein, P.; Larsson, H.; Song, J.; Agrawal, A.; Borglum, A.D.; Bulik, C.M.; Daly, M.J.; Davis, L.K.; Demontis, D.; et al. Genetic influences on eight psychiatric disorders based on family data of 4 408 646 full and half-siblings, and genetic data of 333 748 cases and controls. Psychol. Med. 2019, 49, 1166–1173. [Google Scholar] [CrossRef]
- Cederlöf, M.; Thornton, L.M.; Baker, J.; Lichtenstein, P.; Larsson, H.; Rück, C.; Bulik, C.M.; Mataix-Cols, D. Etiological overlap between obsessive-compulsive disorder and anorexia nervosa: A longitudinal cohort, multigenerational family and twin study. World Psychiatry 2015, 14, 333–338. [Google Scholar] [CrossRef]
- Swinbourne, J.M.; Touyz, S.W. The co-morbidity of eating disorders and anxiety disorders: A review. Eur. Eat. Disord. Rev. 2007, 15, 253–274. [Google Scholar] [CrossRef]
- Micali, N.; Hilton, K.; Natatani, E.; Heyman, I.; Turner, C.; Mataix-Cols, D. Is childhood OCD a risk factor for eating disorders later in life? A longitudinal study. Psychol. Med. 2011, 41, 2507–2513. [Google Scholar] [CrossRef]
- Levinson, C.A.; Brosof, L.C.; Ram, S.S.; Pruitt, A.; Russell, S.; Lenze, E.J. Obsessions are strongly related to eating disorder symptoms in anorexia nervosa and atypical anorexia nervosa. Eat. Behav. 2019, 34. [Google Scholar] [CrossRef] [PubMed]
- Naylor, H.; Mountford, V.; Brown, G. Beliefs about excessive exercise in eating disorders: The role of obsessions and compulsions. Eur. Eat. Disord. Rev. 2011, 19, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Sunday, S.R.; Halmi, K.A.; Einhorn, A. The Yale-Brown-Cornell Eating Disorder Scale: A new scale to assess eating disorder symptomatology. Int. J. Eat. Disord. 1995, 18, 237–245. [Google Scholar] [CrossRef]
- Levinson, C.A.; Sala, M.; Fewell, L.; Brosof, L.C.; Fournier, L.; Lenze, E.J. Meal and snack-time eating disorder cognitions predict eating disorder behaviors and vice versa in a treatment seeking sample: A mobile technology based ecological momentary assessment study. Behav. Res. Ther. 2018, 105, 36–42. [Google Scholar] [CrossRef]
- Altman, S.E.; Shankman, S.A. What is the association between obsessive-compulsive disorder and eating disorders? Clin. Psychol. Rev. 2009, 29, 638–646. [Google Scholar] [CrossRef]
- Strober, M.; Freeman, R.; Lampert, C.; Diamond, J. The association of anxiety disorders and obsessive compulsive personality disorder with anorexia nervosa: Evidence from a family study with discussion of nosological and neurodevelopmental implications. Int. J. Eat. Disord. 2007, 40. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, Z.; Halvorsen, M.; Bryois, J.; Yu, D.; Thornton, L.M.; Zerwas, S.; Micali, N.; Moessner, R.; Burton, C.L.; Zai, G.; et al. Examination of the shared genetic basis of anorexia nervosa and obsessive-compulsive disorder. Mol. Psychiatry 2018. [Google Scholar] [CrossRef] [PubMed]
- Anttila, V.; Bulik-Sullivan, B.; Finucane, H.K.; Walters, R.K.; Bras, J.; Duncan, L.; Escott-Price, V.; Falcone, G.J.; Gormley, P.; Malik, R.; et al. Analysis of shared heritability in common disorders of the brain. Science 2018, 360. [Google Scholar] [CrossRef]
- Cross-Disorder Group of the Psychiatric Genomics Consortium; Won, H.; Feng, Y.-C.A.; Rosenthal, J.; Zhu, Z.; Tucker-Drob, E.M.; Nivard, M.G.; Grotzinger, A.D.; Posthuma, D.; Wang, M.M.-J.; et al. Genomic relationships, novel loci, and pleiotropic mechanisms across eight psychiatric disorders. Cell 2019, 179, 1469–1482. [Google Scholar] [CrossRef] [PubMed]
- Boisseau, C.L.; Thompson-Brenner, H.; Caldwell-Harris, C.; Pratt, E.; Farchione, T.; Harrison Barlow, D. Behavioral and cognitive impulsivity in obsessive-compulsive disorder and eating disorders. Psychiatry Res. 2012, 200, 1062–1066. [Google Scholar] [CrossRef] [PubMed]
- Van Passel, B.; Danner, U.N.; Dingemans, A.E.; Aarts, E.; Sternheim, L.C.; Becker, E.S.; Van Elburg, A.A.; Van Furth, E.F.; Hendriks, G.J.; Cath, D.C. Cognitive remediation therapy does not enhance treatment effect in obsessive-compulsive disorder and anorexia nervosa: A randomized controlled trial. Psychother. Psychosom. 2020, 89, 228–241. [Google Scholar] [CrossRef]
- Fischer, M.S.; Baucom, D.H.; Baucom, B.R.; Abramowitz, J.S.; Kirby, J.S.; Bulik, C.M. Disorder-specific patterns of emotion coregulation in couples: Comparing obsessive compulsive disorder and anorexia nervosa. J. Fam. Psychol. 2017, 31, 304–315. [Google Scholar] [CrossRef]
- Schneier, F.R.; Kimeldorf, M.B.; Choo, T.H.; Steinglass, J.E.; Wall, M.M.; Fyer, A.J.; Simpson, H.B. Attention bias in adults with anorexia nervosa, obsessive-compulsive disorder, and social anxiety disorder. J. Psychiatr. Res. 2016, 79, 61–69. [Google Scholar] [CrossRef]
- Young, S.; Rhodes, P.; Touyz, S.; Hay, P. The relationship between obsessive-compulsive personality disorder traits, obsessive-compulsive disorder and excessive exercise in patients with anorexia nervosa: A systematic review. J. Eat. Disord. 2013, 1, 16. [Google Scholar] [CrossRef]
- Flamarique, I.; Plana, M.T.; Castro-Fornieles, J.; Borràs, R.; Moreno, E.; Lázaro, L. Comparison of perfectionism dimensions in adolescents with anorexia nervosa or obsessive-compulsive disorder. J. Can. Acad. Child Adolesc. Psychiatry 2019, 28, 45–54. [Google Scholar]
- Beszłej, J.A.; Wieczorek, T.; Kobyłko, A.; Piotrowski, P.; Siwicki, D.; Weiser, A.; Fila-Witecka, K.; Rymaszewska, J.; Tabakow, P. Deep brain stimulation: New possibilities for the treatment of mental disorders. Psychiatr. Pol. 2019, 53, 789–806. [Google Scholar] [CrossRef]
- Mayer, L.E.; Walsh, B.T. The use of selective serotonin reuptake inhibitors in eating disorders. J. Clin. Psychiatry 1998, 59, 28–34. [Google Scholar] [PubMed]
- Simpson, H.B.; Wetterneck, C.T.; Cahill, S.P.; Steinglass, J.E.; Franklin, M.E.; Leonard, R.C.; Weltzin, T.E.; Riemann, B.C. Treatment of obsessive-compulsive disorder complicated by comorbid eating disorders. Cogn. Behav. Ther. 2013, 42, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Watson, H.J.; Yilmaz, Z.; Thornton, L.M.; Hübel, C.; Coleman, J.R.I.; Gaspar, H.A.; Bryois, J.; Hinney, A.; Leppä, V.M.; Mattheisen, M.; et al. Genome-wide association study identifies eight risk loci and implicates metabo-psychiatric origins for anorexia nervosa. Nat. Genet. 2019, 51, 1207–1214. [Google Scholar] [CrossRef] [PubMed]
- Arnold, P.D.; Askland, K.D.; Barlassina, C.; Bellodi, L.; Bienvenu, O.J.; Black, D.; Bloch, M.; Brentani, H.; Burton, C.L.; Camarena, B.; et al. Revealing the complex genetic architecture of obsessive-compulsive disorder using meta-analysis. Mol. Psychiatry 2018, 23, 1181–1188. [Google Scholar] [CrossRef]
- Miller, J.A.; Ding, S.-L.; Sunkin, S.M.; Smith, K.A.; Ng, L.; Szafer, A.; Ebbert, A.; Riley, Z.L.; Royall, J.J.; Aiona, K.; et al. Transcriptional landscape of the prenatal human brain. Nature 2014, 508, 199–206. [Google Scholar] [CrossRef]
- Jaffe, A.E.; Deep-Soboslay, A.; Tao, R.; Hauptman, D.T.; Kaye, W.H.; Arango, V.; Weinberger, D.R.; Hyde, T.M.; Kleinman, J.E. Genetic neuropathology of obsessive psychiatric syndromes. Transl. Psychiatry 2014, 4. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Skene, N.G.; Grant, S.G.N. Identification of vulnerable cell types in major brain disorders using single cell transcriptomes and expression weighted cell type enrichment. Front. Neurosci. 2016, 10, 16. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.N.; Corominas, R.; Lemmens, I.; Yang, X.; Tavernier, J.; Hill, D.E.E.; Vidal, M.; Sebat, J.; Iakoucheva, L.M.M. Spatiotemporal 16p11.2 protein network implicates cortical late mid-fetal brain development and KCTD13-Cul3-RhoA pathway in psychiatric diseases. Neuron 2015, 85, 742–754. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef]
- Yu, G.; He, Q.Y. ReactomePA: An R/Bioconductor package for reactome pathway analysis and visualization. Mol. Biosyst. 2016, 12, 477–479. [Google Scholar] [CrossRef]
- Gandal, M.J.; Zhang, P.; Hadjimichael, E.; Walker, R.L.; Chen, C.; Liu, S.; Won, H.; van Bakel, H.; Varghese, M.; Wang, Y.; et al. Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 2018, 362, eaat8127. [Google Scholar] [CrossRef]
- Gallagher, M.D.; Chen-Plotkin, A.S. The post-GWAS era: From association to function. Am. J. Hum. Genet. 2018, 102, 717–730. [Google Scholar] [CrossRef]
- Lin, G.N.; Guo, S.; Tan, X.; Wang, W.; Qian, W.; Song, W.; Wang, J.; Yu, S.; Wang, Z.; Cui, D.; et al. PsyMuKB: An integrative de novo variant knowledge base for developmental disorders. Genom. Proteom. Bioinform. 2019. [Google Scholar] [CrossRef]
- Gandal, M.J.; Haney, J.R.; Parikshak, N.N.; Leppa, V.; Ramaswami, G.; Hartl, C.; Schork, A.J.; Appadurai, V.; Buil, A.; Werge, T.M.; et al. Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science 2018, 359, 693–697. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.J. New approaches to psychiatric diagnostic classification. Neuron 2014, 84, 564–571. [Google Scholar] [CrossRef]
- Shohat, S.; Ben-David, E.; Shifman, S. Varying intolerance of gene pathways to mutational classes explain genetic convergence across neuropsychiatric disorders. Cell Rep. 2017, 18, 2217–2227. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Santpere, G.; Kawasawa, Y.I.; Evgrafov, O.V.; Gulden, F.O.; Pochareddy, S.; Sunkin, S.M.; Li, Z.; Shin, Y.; Zhu, Y.; et al. Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science 2018, 362, eaat7615. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, M.; Bundo, M.; Iwamoto, K.; Kato, T. Somatic mutations in the human brain: Implications for psychiatric research. Mol. Psychiatry 2019, 24, 839–856. [Google Scholar] [CrossRef]
- Crossley, N.A.; Scott, J.; Ellison-Wright, I.; Mechelli, A. Neuroimaging distinction between neurological and psychiatric disorders. Br. J. Psychiatry 2015, 207. [Google Scholar] [CrossRef]
- Zhan, X.; Yu, R. A window into the brain: Advances in psychiatric fMRI. BioMed Res. Int. 2015, 2015, 542467. [Google Scholar] [CrossRef]
- Bassett, D.S.; Xia, C.H.; Satterthwaite, T.D. Understanding the emergence of neuropsychiatric disorders with network neuroscience. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2018, 3, 742–753. [Google Scholar] [CrossRef]
- McCarthy, S.E.; Gillis, J.; Kramer, M.; Lihm, J.; Yoon, S.; Berstein, Y.; Mistry, M.; Pavlidis, P.; Solomon, R.; Ghiban, E.; et al. De novo mutations in schizophrenia implicate chromatin remodeling and support a genetic overlap with autism and intellectual disability. Mol. Psychiatry 2014, 19, 652–658. [Google Scholar] [CrossRef] [PubMed]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Cicek, A.E.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Hamdan, F.F.; Srour, M.; Capo-Chichi, J.-M.; Daoud, H.; Nassif, C.; Patry, L.; Massicotte, C.; Ambalavanan, A.; Spiegelman, D.; Diallo, O.; et al. De novo mutations in moderate or severe intellectual disability. PLoS Genet. 2014, 10, e1004772. [Google Scholar] [CrossRef] [PubMed]
- Cappi, C.; Oliphant, M.E.; Péter, Z.; Zai, G.; do Rosário, M.C.; Sullivan, C.A.W.; Gupta, A.R.; Hoffman, E.J.; Virdee, M.; Olfson, E.; et al. De novo damaging DNA coding mutations are associated with obsessive-compulsive disorder and overlap with Tourette’s disorder and autism. Biol. Psychiatry 2019. [Google Scholar] [CrossRef]
- Huckins, L.M.; Hatzikotoulas, K.; Southam, L.; Thornton, L.M.; Steinberg, J.; Aguilera-Mckay, F.; Treasure, J.; Schmidt, U.; Gunasinghe, C.; Romero, A.; et al. Investigation of common, low-frequency and rare genome-wide variation in anorexia nervosa. Mol. Psychiatry 2018, 23, 1169–1180. [Google Scholar] [CrossRef]
- Lawrence, M.; Daujat, S.; Schneider, R. Lateral thinking: How histone modifications regulate gene expression. Trends Genet. 2016, 32, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Thomson, C.A.; McColl, A.; Cavanagh, J.; Graham, G.J. Peripheral inflammation is associated with remote global gene expression changes in the brain. J. Neuroinflamm. 2014, 11. [Google Scholar] [CrossRef]
- McEwen, B.S.; Bowles, N.P.; Gray, J.D.; Hill, M.N.; Hunter, R.G.; Karatsoreos, I.N.; Nasca, C. Mechanisms of stress in the brain. Nat. Neurosci. 2015, 18, 1353–1363. [Google Scholar] [CrossRef] [PubMed]
- Carr, C.P.; Martins, C.M.S.; Stingel, A.M.; Lemgruber, V.B.; Juruena, M.F. The role of early life stress in adult psychiatric disorders: A systematic review according to childhood trauma subtypes. J. Nerv. Ment. Dis. 2013, 201, 1007–1020. [Google Scholar] [CrossRef]
- Bauer, M.E.; Teixeira, A.L. Inflammation in psychiatric disorders: What comes first? Ann. N. Y. Acad. Sci. 2019, 1437, 57–67. [Google Scholar] [CrossRef]
- Solmi, M.; Veronese, N.; Favaro, A.; Santonastaso, P.; Manzato, E.; Sergi, G.; Correll, C.U. Inflammatory cytokines and anorexia nervosa: A meta-analysis of cross-sectional and longitudinal studies. Psychoneuroendocrinology 2015, 51, 237–252. [Google Scholar] [CrossRef] [PubMed]
- Malik, R.; Chauhan, G.; Traylor, M.; Sargurupremraj, M.; Okada, Y.; Mishra, A.; Rutten-Jacobs, L.; Giese, A.K.; Van Der Laan, S.W.; Gretarsdottir, S.; et al. Multiancestry genome-wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat. Genet. 2018, 50, 524–537. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef]
- Huang, N.; Lee, I.; Marcotte, E.M.; Hurles, M.E. Characterising and predicting haploinsufficiency in the human genome. PLoS Genet. 2010, 6, e1001154. [Google Scholar] [CrossRef]
- Favaro, A.; Caregaro, L.; Tenconi, E.; Bosello, R.; Santonastaso, P. Time trends in age at onset of anorexia nervosa and bulimia nervosa. J. Clin. Psychiatry 2009, 70, 1715–1721. [Google Scholar] [CrossRef] [PubMed]
- Volpe, U.; Tortorella, A.; Manchia, M.; Monteleone, A.M.; Albert, U.; Monteleone, P. Eating disorders: What age at onset? Psychiatry Res. 2016, 238, 225–227. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S. Early versus late onset obsessive–compulsive disorder: Evidence for distinct subtypes. Clin. Psychol. Rev. 2011, 31, 1083–1100. [Google Scholar] [CrossRef] [PubMed]
- Scharner, S.; Stengel, A. Alterations of brain structure and functions in anorexia nervosa. Clin. Nutr. Exp. 2019, 28, 22–32. [Google Scholar] [CrossRef]
- Li, B.; Mody, M. Cortico-striato-thalamo-cortical circuitry, working memory, and obsessive-compulsive disorder. Front. Psychiatry 2016, 7, 78. [Google Scholar] [CrossRef]
- Sinopoli, V.M.; Burton, C.L.; Kronenberg, S.; Arnold, P.D. A review of the role of serotonin system genes in obsessive-compulsive disorder. Neurosci. Biobehav. Rev. 2017, 80, 372–381. [Google Scholar] [CrossRef]
- Riva, G. Neurobiology of anorexia nervosa: Serotonin dysfunctions link self-starvation with body image disturbances through an impaired body memory. Front. Hum. Neurosci. 2016, 10. [Google Scholar] [CrossRef] [PubMed]
- Barbarich, N.C.; Kaye, W.H.; Jimerson, D. Neurotransmitter and imaging studies in anorexia nervosa: New targets for treatment. Curr. Drug Targets CNS Neurol. Disord. 2003, 2, 61–72. [Google Scholar] [CrossRef]
- Kaye, W.H.; Fudge, J.L.; Paulus, M. New insights into symptoms and neurocircuit function of anorexia nervosa. Nat. Rev. Neurosci. 2009, 10, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Barros, C.S.; Franco, S.J.; Müller, U. Extracellular Matrix: Functions in the nervous system. Cold Spring Harb. Perspect. Biol. 2011, 3, 1–24. [Google Scholar] [CrossRef]
- Auld, D.S.; Robitaille, R. Glial cells and neurotransmission: An inclusive view of synaptic function. Neuron 2003, 40, 389–400. [Google Scholar] [CrossRef]
- Kesselmeier, M.; Pütter, C.; Volckmar, A.L.; Baurecht, H.; Grallert, H.; Illig, T.; Ismail, K.; Ollikainen, M.; Silén, Y.; Keski-Rahkonen, A.; et al. High-throughput DNA methylation analysis in anorexia nervosa confirms TNXB hypermethylation. World J. Biol. Psychiatry 2018, 19, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Frintrop, L.; Trinh, S.; Liesbrock, J.; Leunissen, C.; Kempermann, J.; Etdöger, S.; Kas, M.J.; Tolba, R.; Heussen, N.; Neulen, J.; et al. The reduction of astrocytes and brain volume loss in anorexia nervosa—The impact of starvation and refeeding in a rodent model. Transl. Psychiatry 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Cappi, C.; Brentani, H.; Lima, L.; Sanders, S.J.; Zai, G.; Diniz, B.J.; Reis, V.N.S.; Hounie, A.G.; Conceição do Rosário, M.; Mariani, D.; et al. Whole-exome sequencing in obsessive-compulsive disorder identifies rare mutations in immunological and neurodevelopmental pathways. Transl. Psychiatry 2016, 6, e764. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, W.; Wang, W.; Yu, S.; Lin, G.N. Dissection of the Genetic Association between Anorexia Nervosa and Obsessive–Compulsive Disorder at the Network and Cellular Levels. Genes 2021, 12, 491. https://doi.org/10.3390/genes12040491
Song W, Wang W, Yu S, Lin GN. Dissection of the Genetic Association between Anorexia Nervosa and Obsessive–Compulsive Disorder at the Network and Cellular Levels. Genes. 2021; 12(4):491. https://doi.org/10.3390/genes12040491
Chicago/Turabian StyleSong, Weichen, Weidi Wang, Shunying Yu, and Guan Ning Lin. 2021. "Dissection of the Genetic Association between Anorexia Nervosa and Obsessive–Compulsive Disorder at the Network and Cellular Levels" Genes 12, no. 4: 491. https://doi.org/10.3390/genes12040491
APA StyleSong, W., Wang, W., Yu, S., & Lin, G. N. (2021). Dissection of the Genetic Association between Anorexia Nervosa and Obsessive–Compulsive Disorder at the Network and Cellular Levels. Genes, 12(4), 491. https://doi.org/10.3390/genes12040491