
Pharmacological Treatment for Neuroinflammation in Stress-Related Disorder
by
 , , , and
, , , and
Dong-Hun Lee
1,2,† ,
,
Ji-Young Lee
1,†,
Dong-Yong Hong
1,2,
Eun-Chae Lee
1,2,
Sang-Won Park
1,2,
Yun-Kyung Lee
2,*,‡ and
Jae-Sang Oh
1,2,*,‡ 1
Department of Neurosurgery, College of Medicine, Soonchunhyang University, Cheonan Hospital, Cheonan 31151, Korea
2
Soonchunhyang Institute of Medi-bio Science (SIMS), Soon Chun Hyang University, Cheonan 31151, Korea
*
Authors to whom correspondence should be addressed.
†
These authors contributed equally to this work.
‡
These authors contributed equally to this work.
Biomedicines 2022, 10(10), 2518; https://doi.org/10.3390/biomedicines10102518
Submission received: 31 August 2022
/
Revised: 23 September 2022
/
Accepted: 6 October 2022
/
Published: 9 October 2022
(This article belongs to the Special Issue Neuroinflammation in Stress-Related Disorders)
Abstract
:Stress is an organism’s response to a biological or psychological stressor, a method of responding to threats. The autonomic nervous system and hypothalamic–pituitary–adrenal axis (HPA axis) regulate adaptation to acute stress and secrete hormones and excitatory amino acids. This process can induce excessive inflammatory reactions to the central nervous system (CNS) by HPA axis, glutamate, renin-angiotensin system (RAS) etc., under persistent stress conditions, resulting in neuroinflammation. Therefore, in order to treat stress-related neuroinflammation, the improvement effects of several mechanisms of receptor antagonist and pharmacological anti-inflammation treatment were studied. The N-methyl-D-aspartate (NMDA) receptor antagonist, peroxisome proliferator-activated receptor agonist, angiotensin-converting enzyme inhibitor etc., effectively improved neuroinflammation. The interesting fact is that not only can direct anti-inflammation treatment improve neuroinflammation, but so can stress reduction or pharmacological antidepressants. The antidepressant treatments, including selective serotonin reuptake inhibitors (SSRI), also helped improve stress-related neuroinflammation. It presents the direction of future development of stress-related neuroinflammation drugs. Therefore, in this review, the mechanism of stress-related neuroinflammation and pharmacological treatment candidates for it were reviewed. In addition, treatment candidates that have not yet been verified but indicate possibilities were also reviewed.
1. Introduction
Stress is an organism’s response to a physiological, biological, or psychological stressor, a method of responding to conditions such as physical and psychological threats [1]. Acute stress is a cause of psychological changes and causes loss of reality, anxiety, and arousal. On the other hand, chronic stress, although not as strong as acute stressors, can be more negative for health by affecting the body’s daily physiological responses over a long period of time. When a person is under chronic stress, permanent changes in physiological, emotional, and behavioral responses can occur [2]. However, most healthy individuals can still remain disease-free after facing a chronic stressful event, which can be a potential source of stress. This suggests that there are individual differences in the pathogenic effect. There are two main systems for controlling stress in humans or most mammals: the autonomic nervous system and hypothalamic–pituitary–adrenal axis (HPA axis) [3]. The sympathetic nervous system and the parasympathetic nervous system are antagonistic and regulate adaptation to acute stress [4,5,6]. The HPA axis regulates the release of cortisol, which has a great influence on body function, such as immunological function. Through these mechanisms, stress can affect memory, immunity, and metabolic diseases.
Chronic stress in humans can cause physiological, emotional, and behavioral changes in response, and severe acute and chronic stress can cause abnormalities in the serotonin system, catecholamine secretion, and major regulatory systems of the HPA axis [7]. Much evidence has been given that this stress can affect neuroinflammation, and interest in stress, neuroinflammation, and neurological diseases that result from it is increasing [8,9,10].
Inflammation is a protective reaction that helps to eliminate the original cause of cell damage and removes and repairs damaged cell and tissues [11,12,13]. Acute inflammation begins in macrophages and dendritic cells, which have receptors known as pattern regression receptors (PRRs), which recognize submolecules of pathogen-associated molecular pattern (PAMP) and damage-associated molecular pattern (DAMP) [14,15]. Acute inflammation occurs immediately after injury, and pro-inflammatory cytokine and chemokine call up neutrophil and macrophage to the inflammatory site. If this inflammatory reaction persists, chronic inflammation may persist and may develop into cardiovascular disease or chronic disease [16,17,18]. Chronic inflammation is a slow, long-term inflammation that lasts months to years. Infectious organisms remain in tissues for long periods of time or are associated with long-term inflammation-mediated diseases such as diabetes, cardiovascular disease, arthritis, and allergies. Stress and sleep disturbances are also risk factors for chronic inflammation, and mood disorders including chronic fatigue, insomnia, depression, and anxiety are common signs and symptoms that occur during chronic inflammation [19,20,21].
Neuroinflammation occurs when these inflammatory reactions affect the nerve tissue [22]. It may begin in response to infection, traumatic brain injury, toxic metabolites, or autoimmune signals [23,24]. Peripheral inflammatory cells can affect the brain through several mechanisms beyond the blood–brain barrier (BBB) [22,25,26]. In addition, neuronal inflammation can occur due to the activation of microglia and astrocyte in CNS [27,28]. The astrocytes are glial cells abundant in the brain and spinal cord. They perform many functions, including regulating cerebral blood flow, supplying nutrients to nervous tissue, maintaining extracellular ion balance, and post-infection and post-injury processes [29]. The astrocytes sense pro-inflammatory cytokines secreted by the CNS and peripheral immune cells recruited to the CNS and, in response, modulate the responses of neighboring cells throughout the CNS [27]. Activated nuclear factor kappa B (NF-κB) is central to astrocyte activation and contributes to the progression of CNS pathology [27,30]. Nuclear translocation of NF-κB in astrocytes was associated with TNF-α, IL-1β, 1L-17, ROS, Toll-like receptor (TLR), and other factors related to CNS inflammation [31,32]. The microglia are a type of glial cells, resident macrophages that act as a major form of immune defense in the CNS. As for the mechanism by which microglia actively regulate astrocyte function in neuroinflammation, it has been suggested that signaling induces astrocyte glutamate release in the presence of TNF-α [33]. It is a key cell in overall brain care, constantly cleaning out plaque, damaged or unnecessary neurons, synapses, and sources of infection. Astrocyte and microglia are innate immune cells in the CNS, which can over-generate pro-inflammatory cytokines when exposed to stress. Indeed, persistent stressors showed increases in granulocytes, natural killer cells, immunoglobulin A (IgA), and interleukin 6 (IL-6).
To treat this stress-related neuroinflammation, on the other hand, studies have shown that the induction of stress anxiety is suppressed when the release of monocyte from bone marrow and monocyte call-up to the brain are blocked. For example, antidepressants injected in mice prevented stress-derived neuroinflammation, and conversely, mice deficient in C-C chemokine receptor 2 (CCR2) and interleukin-1 receptor (IL-1R) did not experience anxiety after the repeated social defect (RSD) test.
Not only are stress and neuroinflammation related to each other, but the fact that stress reduction and anti-inflammation treatment can improve each other suggests the direction for the development of stress-related neuroinflammation drugs in the future. Therefore, in this review, the mechanism of stress-related neuroinflammation was studied, and pharmacological treatment candidates for it were reviewed.
2. Stress-Related Neuroinflammatory Mechanism
2.1. HPA Axis and Glucocorticoids
The HPA axis is a complex interaction axis of the hypothalamic–pituitary–adrenal axis. It is a major neuroendocrine system that controls responses to stress and regulates many body processes [34]. Under stress, the hippocampus secretes corticotropin [35]. The corticotropin is adrenocorticotropic hormone (ACTH), which stimulates the adrenal cortex to induce the production of catecholamines and glucocorticoids in the systemic circulatory system. Therefore, high levels of glucocorticoids in blood plasma can be identified as typical characteristics of stress reactions. The glucocorticoids are activated by binding to glucocorticoid receptors (GR). The activated glucocorticoid complex regulates the expression of anti-inflammatory proteins and inhibits the expression of pro-inflammatory proteins [36]. However, if the stress state persists, glucocorticoid remains at a high level, resulting in fatigue in GR, which causes GR resistance, leading to high levels of glucocorticoid maintenance and impairment in anti-inflammatory action [37]. In addition, GR resistance causes a negative feedback failure, so the glucocorticoid level is not downregulated. As a result, stress-induced HPA axis hyperactivation leads to neuroinflammation (Figure 1) [7,38].
Some studies have shown that excessive production of stress-derived glucocorticoids damages the hippocampus and reduces long-term potentiation (LTP). As a mechanism for this, the hypothesis that changes in glucose carrier potential and decreases in mRNA levels cause problems in glucose transport in glial cells, resulting in problems in ATP production, is based on reduced ATP levels in the brain cortex of rats exposed to stress [39,40].
2.2. Glutamate (Excitatory Amino Acids)
Under stress conditions, excitatory amino acids increase rapidly, one of which is glutamate. The glutamate is used as a neurotransmitter for all major excitatory functions of the vertebrate brain and is involved in cognitive functions such as learning and memory in the brain because of its role in synaptic plasticity [41]. However, excessive glutamate accumulates outside the cell and can contribute to brain damage caused by neuroinflammation. The glucocorticoid described above increases intracellular calcium levels, causing glutamate to enter the cell through the NMDA glutamate receptor, resulting in nerve cell damage and eventual cell death, causing excitatory toxicity. In other words, increased intracellular glutamate concentration can induce apoptosis of nerve cells and nitric oxide (NO)-mediated neuroinflammation [42]. In addition, excessive glutamate mediates the promotion of pro-apoptotic transcription factors and the downregulation of transcription factors for anti-apoptotic genes [43]. Toxicity from excessive glutamate release and absorption disorders is associated with brain-related diseases such as stroke, autism, intellectual disability, and Alzheimer’s disease [44]. In fact, in the stressed rat model, it was confirmed that the excitatory amino acids transporters (EAAT) responsible for glutamate transport exhibited dysfunction, EAAT expression itself decreased, and glutamate aspiration decreased and remained at a high level [45,46].
2.3. Renin–Angiotensin System
Angiotensin II (Ang II) is an active principle of the renin–angiotensin system (RAS), originally involved in the regulation of blood pressure and body fluid metabolism, vasoconstriction, aldosterone release, sodium, and water retention and ingestion [47]. However, under stress conditions, the activity of Ang II can cause neuroinflammation. Acute stress upregulates Ang II production through sympathetic nerve activation of β-adrenergic receptors and increases levels of Ang II in the circulatory system [48,49]. These high levels of Ang II promote the production of reactive oxygen species (ROS) and pro-inflammatory cytokines [50]. The circulating pro-inflammatory cytokines, TNF-α and IL-1β, increase cyclooxygenase-2 (COX-2) activity and cause prostaglandin E2 (PGE2) production [51]. The PGE2, which occurs in response to physiological and psychological stress, is involved in several inflammatory pathways [52,53]. The PGE2 contributes to inflammation by improving leukocyte infiltration due to increased vascular projectivity when acting on receptors. As a result, Ang II causes neuroinflammation. In addition, increased circulation of Ang II can stimulate Ang II receptors in the pituitary anterior lobe and adrenal cortex, contributing to the upregulation of the pituitary anterior lobe, adrenocorticotropic hormone (ACTH), and glucocorticoids during stress [54,55]. It has been previously explained that glucocorticoids can contribute to neuroinflammation caused by stress (Figure 2).
2.4. Neuroinflammation by Stress-Related Oxidative and Nitric Oxide Products
Neuroinflammation can also be caused by oxidative and NO-mediated inflammatory reactions [56]. In the paragraph describing glutamate above, it was explained that an increase in glutamate induced by stress may cause NO-mediated neuroinflammation. Inducible nitric oxide (iNOS) is an enzyme that catalyzes NO production. The NO is involved in synaptic plasticity [57,58], smooth muscle relaxation [59], and vasodilation in CNS [60]. However, under sustained stress conditions, excessive production of NO is associated with a variety of diseases, along with causing neuroinflammation. Exposure to stress increases iNOS activity, expression at the brain and end of the body, and increases NO production [46,61,62]. Increased NO is also associated with NF-κB under stress. Stress-exposed rats caused NF-κB activation. When NF-κB inhibitors were administered just before stress, inhibition of iNOS expression, along with inhibition of NF-κB, was also shown, but the exact mechanism was not revealed [61]. The excessive NO production has also been proven to be associated with other diseases associated with neuroinflammation of the CNS, such as ischemic damage, Alzheimer’s disease, and Parkinson’s disease [63].
COX, also known as prostaglandin synthetase, is at the heart of anti-inflammation therapy for neuropathology, including neuroinflammation and neurodegenerative diseases. The COX has been identified as both COX-1 and COX-2 isoforms [64], among which COX-2 is rapidly expressed in multiple cells as a function of cytokines and pro-inflammatory molecules. In particular, COX-2 is prominently expressed in the hippocampus [65,66]. COX-2 is induced and expressed by macrophages and mononuclear cells involved in inflammation [67]. It is expressed in brain cells and is upregulated in various neurological diseases such as stroke and Alzheimer’s dementia [68]. It has also been explained through the renin–angiotensin system that COX-2 can biosynthesize prostaglandin to produce free radicals and activate PGE2 to contribute to neuroinflammation [67,69,70]. It can also contribute to neuroinflammation by inducing glutamate release and apoptosis from astrocytes rich in the brain [71]. The mRNA level of COX-2 was increased in mice who experienced forced swimming stress, and it was confirmed that it was mediated by glutamate and NF-κB [46,72].
3. NMDA Glutamate Receptor Inhibition for Treatment of Neuroinflammation
Upregulated glutamate due to stress can cause nerve cell damage and NO-mediated neuroinflammation due to excitatory toxicity. This is because glutamate is excessively introduced into the cell through the NMDA glutamate receptor, maintaining a high level of glutamate environment in the cell. The NMDA receptor is an ion channel protein that accepts glutamate in nerve cells. When glutamate and glycine are combined and activated, ions flow through the cell membrane, regulating cell signaling and synaptic plasticity by calcium, and are involved in memory cell activation [73]. The NMDA receptor is divided into the protein domain to which glutamate binds, the transmembrane domain responsible for transporting subunits, and the cytoplasmic domain directly involved in cell signal regulation.
It is possible to reduce oxidative and NO damage by inhibiting the NMDA glutamate receptor and preventing neuroinflammation and cell damage [74,75,76,77]. Competitive NMDA receptor antagonists to inhibit these NMDA glutamate receptors compete with the agent, glutamate, and bind to the same site of the receptor to block glutamate. However, this can cause problems because it also blocks the normal functioning of glutamate [78,79]. Blocking all NMDA receptor activity can lead to side effects, such as hallucinations and anesthesia. An alternative is the non-competitive NMDA receptor antagonist. Non-competitive NMDA receptor antagonists prevent excessive inflow of calcium. They block excess activity while preserving physiological NMDA receptor activity, and only blocks receptor ion channels when the channel is excessively opened [76].
The effectiveness of these NMDA glutamate receptors on neuroinflammation has been confirmed in animal experiments. Although increased TNF-α and increased activity of a TNF-α converting enzyme (TACE) in the brain cortex of rats that experienced stress, the administration of MK-801 (dizocilpine), a non-competitive NMDA receptor antagonist, reduced stress-derived activity, and expression of TNF-α [80].
4. Peroxisome Proliferator-Activated Receptor (PPAR) agonist for Treatment Neuroinflammation
A peroxisome proliferator-activated receptor (PPAR) is a protein that functions as a transcription factor that regulates gene expression [81]. PPAR plays an essential role in cell differentiation, development, and regulation of metabolism [82,83,84]. There are four types of PPAR: PPARα, PPARβ/δ, and PPARγ [83].
Among them, PPARα may exhibit an anti-inflammatory effect by inhibiting NF-κB activated in the PPAR-dependent pathway. In agonist studies acting as ligand on PPAR, it has been demonstrated to prevent multiple post-stress neuroinflammation and oxidative/NO damage, including iNOS inhibition, NF-κB blocking, TNF-α emission inhibition, and COX-2 expression reduction. In addition, PPARα activation plays an important role in the development of glucose homeostasis and insulin resistance as well as anti-inflammation, helping to reduce the risk of coronary heart disease and hypertension. As a ligand of PPARα, natural agents are typically omega-3 fatty acids.
PPARγ uses the prostaglandin mentioned earlier as ligand. It is expressed in microglia and astrocyte and is a target of anti-inflammatory activity in inflammatory nervous system diseases [85,86]. The expression of PPARγ was found to improve in the cortex of rats’ brains which underwent acute stress and was clearly identified in neurons and astrocytes [87]. When natural and synthetic PPARγ ligand was administered to rats, it was proven to prevent stress-derived CNS inflammation, and oxidation and nitrification reactions [88]. It can also activate antioxidant pathways such as nuclear factor erythroid 2-related factor 2 (NRF2) [89]. Synthetic agents include rosiglitazone, troglitazone, pioglitazone, and ciglitazone.
5. Angiotensin-Converting Enzyme (ACE) Inhibition and Angiotensin Receptor Blocking (ARB) for Treatment of Neuroinflammation
Ang II can cause neuroinflammation under stress [90]. It can increase pro-inflammatory cytokine and COX-2 activity, induce PGE2 production, and induce glucocorticoid secretion. Angiotensin 1 (Ang I) is a precursor of Ang II activated as a substrate of the RAS system. The ACE converts Ang I to Ang II [91]. ACE inhibitors can block this RAS system and contribute to improving neuroinflammation. It reduces ROS, decreases CRP that is upstream in the inflammatory response, and activates the complement system [92], and inhibits NF-κB. One clinical study suggested that ACE inhibitor and ARB-prescribed patients had a protective effect on stress-related disorders [93].
Some studies have shown that angiotensin(1–7) [Ang(1–7)] has the opposite effect of Ang II. The Ang II is hydrolyzed into Ang(1–7) through the action of ACE2. The Ang(1–7) binds and activates the Mas receptor to activate ACE2/Ang(1–7)/MasR axis, which weakens the hypertension induced by Ang II and weakens the inflammatory response [94]. Treatment with Ang(1–7) inhibited inflammatory markers in several disease models [95,96,97,98,99,100,101,102,103]. Angiotensin II receptor type 1 (AT1R) over-activates RAS by accepting Ang II [104,105,106,107]. It was shown that telmisartan, an AT1R blocker, reduced the synthesis of TNF-α and IL-1β of microglia, and significantly reduced iNOS [108]. Therefore, it contributes to beneficial effects as an ACE inhibitor and AT1R antagonist [109]. In addition, NF-κB was inhibited in the ischemic stroke rat model and cell death of white blood cells was induced [110,111,112].
6. Other Pharmacological Treatments for Neuroinflammation
In addition to the method of suppressing receptors, much effort is being devoted to developing pharmacological treatments to improve stress-related neuroinflammations. Several studies have also alleviated neuroinflammation by directly inhibiting inflammatory cytokines or by caring for pharmacological mental stress (Table 1).
NSAIDs are competitive inhibitors of COX, an enzyme that converts arachidonic acid to the inflammatory prostaglandin. It directly targets and inhibits COX-2 [113]. It has been found to be effective in inhibiting neuroinflammatory pathways and has helped improve symptoms in animal studies on depression, schizophrenia, affective disorder, and obsessive-compulsive disorder [114]. Studies have been conducted to support the association between chronic inflammation and mental disorders, which are reduced by the use of COX-2 inhibitors. However, in clinical trials, COX-2 inhibitors have been found to significantly increase the risk of heart attack and stroke, requiring a careful approach to use the drug, and some drugs have been revoked [115,116]. Typical COX-2 inhibitors include celecoxib, rofecoxib, and etoricoxib.
On the one hand, more and more studies are showing that antidepressants have immunosuppression and anti-inflammatory properties by regulating neurotransmitters [117]. According to some meta-analysis studies and clinical studies, antidepressant treatment showed a decrease in TNF-α, IL-1, IL-6, and a decrease in mRNA expression of both TNF-α and IL-1β in microglia in rats treated with antidepressants [118,119,120]. These improvements are explained by the discovery that antidepressant therapy induces upregulation of Treg cell, which can downregulate cytokine production [121].
7. New Candidates for Treatment
In addition to the drugs described in Table 1, efforts are being made to improve the neuroinflammation in stress-related disorders.
A study by Hanhai Zeng (2021) reported weakening of neuroinflammation with nuclear receptor binding factor 2 (NRBF2) [122]. The NRBF2 is involved in several diseases and stress conditions [123,124,125], especially autophagosome. Activation of autophagy reduces neuroinflammation [126].
There are also efforts using stem cells to alleviate neuroinflammation. Sandra A. (2015)’s study alleviated chronic inflammation of the stroke animal model with bone marrow stem cell therapy [127]. Jung (2016) also reported anti-inflammatory properties of CNS with mesenchymal stem cell therapy [128]. Several other studies have shown that stem cell therapy appears to help alleviate neuroinflammation. However, publications suggest that further investigation is needed in preparation for side effects commonly found in stem cells.
A relatively recent study is also paying attention to microbiome. This axis, called “gut–brain axis”, indicates that the health of the brain and gut is deeply related. Importantly, this gut microbiome (GM) imbalance can affect neuroinflammation in the brain. Sudo (2004)’s study investigated the effect of GM on HPA axis reactions on stress and found that HPA axis reactions were over-activated in mice with unbalanced GM than those with normal GM [129]. In addition, GM imbalances can transform the expression of tight junction proteins that maintain the BBB’s integrity, destroying the BBB’s integrity and helping to migrate inflammatory cells. Indeed, in preclinical studies, it has been reported that GM-deficient mice easily destroy the BBB permeability by regulating claudin-5 and occluding expression [130,131,132]. However, there are not enough studies using GM as a strategy to treat neuroinflammation at present. There have been several animal model studies attempting to intervene in GM composition, but they have not been followed in the long run [133,134]. Although not a pharmacological treatment, diet such as intermittent fasting can cause long-term changes in GM composition. Calorie restriction through intermittent fasting can positively affect GM composition in favor of the growth of beneficial anti-inflammatory microbial systems.
8. Conclusions
The brain was once considered to have immune privileges, but research into neuroinflammation and inflammatory cells passing through the BBB is now widely known. In addition, neuroinflammation in CNS is activated not only by response to infection or trauma, but also by psychological stress. Stress stimulates the HPA axis to upregulate glucocorticoids and induce the release of excitatory amino acids, such as glutamate. Continuous stress induces resistance of GR, which hinders the anti-inflammatory action of glucocorticoids. Elevated glutamate induces apoptosis due to excessive intracellular concentration of calcium and contributes to NO production, resulting in NO-mediated neuroinflammation. In fact, a study on rats identified elevated EAAT and glutamate levels and found that they were associated with many neurological diseases. Ang II is hyperactivated from sympathetic nerves under stress conditions, inducing ROS and pro-inflammatory cytokines. It can also contribute to the upregulation of ATCH and glucocorticoids mentioned above, which can also affect neuroinflammatory induction by other mechanisms. Oxidative/NO products produced through these processes are associated with NO activity and stimulate COX-2 activity.
Because each of these mechanisms is hyperactive through the receptor, an antagonist to the receptor can be intervened to improve stress-derived neuroinflammation. In fact, in many studies using animals, NMDA glutamate receptor antagonists reduced the activity of TNF-α, reduced oxidative/NO damage of astrocytes and neurons with PPAR agents, and activated antioxidant mechanisms such as NRF2. ACE inhibitor also inhibited inflammatory activity such as COX-2 in clinical and animal studies. However, the improvement effect has been confirmed, but the mechanism is not clear, and excessive blocking of the receptor may interfere with normal functions, so continuous research is needed.
In addition, it is very interesting that stress-induced neuroinflammation can be responded to with anti-inflammatory treatment, and pharmacological treatment methods that treat psychological stress, such as antidepressants, can lower the level of neuroinflammation. Indeed, treatment of neuroinflammation with antidepressants showed decreased expression of pro-inflammatory cytokines, such as TNF-a, IL-1, and IL-6, in experimental animals. Therefore, it is expected that research will become active on whether not only direct pharmacological treatments for neuroinflammation, but also pharmacological or psychological treatment methods that improve psychological stress, can help to improve neuroinflammation.
Moreover, beyond well-known pharmacological treatments, neuroinflammation treatment using NRBF2, stem cell therapy, and GM balance has not been studied well in the past, but it is expected to be a new treatment strategy in the future. In particular, since GM also affects the HPA axis and BBB, which are the main mechanisms of stress-related inflammations, further investigation is expected.
Author Contributions
Conceptualization, J.-S.O. and Y.-K.L.; methodology, D.-H.L., J.-Y.L. and D.-Y.H., E.-C.L.; software, J.-S.O. and S.-W.P.; validation, Y.-K.L. and J.-S.O.; formal analysis, D.-H.L., J.-Y.L., D.-Y.H. and E.-C.L.; investigation, J.-S.O.; resources, J.-S.O. and Y.-K.L.; data curation, J.-S.O. and Y.-K.L.; writing—original draft preparation, D.-H.L., J.-S.O. and Y.-K.L.; writing—review and editing, D.-H.L., J.-Y.L., J.-S.O. and Y.-K.L.; visualization, D.-H.L., J.-Y.L., J.-S.O. and Y.-K.L.; supervision, J.-S.O. and Y.-K.L.; project administration, J.-S.O. and Y.-K.L.; funding acquisition, J.-S.O. and Y.-K.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by a grant from the Soonchunhyang University Fund, by the Bio & Medical Technology Development Program of the National Research Foundation funded by the Korean government (NRF-2019M3E5D1A02069061 and 2020R1F1A1066362).
Institutional Review Board Statement
This research was performed in compliance with the Institutional Animal Care and Use Committee of Soon Chun Hyang University (IACUC No. SCH 20-0065).
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare that they have no conflict of interest.
References
- Muthukumar, K.; Nachiappan, V. Cadmium-induced oxidative stress in Saccharomyces cerevisiae. Indian J. Biochem. Biophys. 2010, 47, 383–387. [Google Scholar]
- Cohen, S.; Janicki-Deverts, D.; Miller, G.E. Psychological stress and disease. JAMA 2007, 298, 1685–1687. [Google Scholar] [CrossRef]
- Ulrich-Lai, Y.M.; Herman, J.P. Neural regulation of endocrine and autonomic stress responses. Nat. Rev. Neurosci. 2009, 10, 397–409. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, D.S. Stress-induced activation of the sympathetic nervous system. Baillieres Clin. Endocrinol. Metab. 1987, 1, 253–278. [Google Scholar] [CrossRef]
- Miki, K.; Yoshimoto, M. Sympathetic nerve activity during sleep, exercise, and mental stress. Auton. Neurosci. 2013, 174, 15–20. [Google Scholar] [CrossRef]
- Won, E.; Kim, Y.K. Stress, the Autonomic Nervous System, and the Immune-kynurenine Pathway in the Etiology of Depression. Curr. Neuropharmacol. 2016, 14, 665–673. [Google Scholar] [CrossRef] [Green Version]
- Herman, J.P.; McKlveen, J.M.; Ghosal, S.; Kopp, B.; Wulsin, A.; Makinson, R.; Scheimann, J.; Myers, B. Regulation of the Hypothalamic-Pituitary-Adrenocortical Stress Response. Compr. Physiol. 2016, 6, 603–621. [Google Scholar] [CrossRef] [Green Version]
- Woodburn, S.C.; Bollinger, J.L.; Wohleb, E.S. The semantics of microglia activation: Neuroinflammation, homeostasis, and stress. J. Neuroinflamm. 2021, 18, 258. [Google Scholar] [CrossRef]
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139 (Suppl. S2), 136–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, V.X.; Patel, S.; Jones, H.F.; Dale, R.C. Maternal immune activation and neuroinflammation in human neurodevelopmental disorders. Nat. Rev. Neurol. 2021, 17, 564–579. [Google Scholar] [CrossRef]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Ferrero-Miliani, L.; Nielsen, O.H.; Andersen, P.S.; Girardin, S.E. Chronic inflammation: Importance of NOD2 and NALP3 in interleukin-1beta generation. Clin. Exp. Immunol. 2007, 147, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204–7218. [Google Scholar] [CrossRef] [Green Version]
- Maverakis, E.; Kim, K.; Shimoda, M.; Gershwin, M.E.; Patel, F.; Wilken, R.; Raychaudhuri, S.; Ruhaak, L.R.; Lebrilla, C.B. Glycans in the immune system and The Altered Glycan Theory of Autoimmunity: A critical review. J. Autoimmun. 2015, 57, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seong, S.Y.; Matzinger, P. Hydrophobicity: An ancient damage-associated molecular pattern that initiates innate immune responses. Nat. Rev. Immunol. 2004, 4, 469–478. [Google Scholar] [CrossRef]
- Zhong, J.; Shi, G. Editorial: Regulation of Inflammation in Chronic Disease. Front. Immunol. 2019, 10, 737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cobo, G.; Lindholm, B.; Stenvinkel, P. Chronic inflammation in end-stage renal disease and dialysis. Nephrol. Dial. Transplant. 2018, 33, iii35–iii40. [Google Scholar] [CrossRef] [Green Version]
- Zhao, R.; Liang, H.; Clarke, E.; Jackson, C.; Xue, M. Inflammation in Chronic Wounds. Int. J. Mol. Sci. 2016, 17, 2085. [Google Scholar] [CrossRef]
- Michels da Silva, D.; Langer, H.; Graf, T. Inflammatory and Molecular Pathways in Heart Failure-Ischemia, HFpEF and Transthyretin Cardiac Amyloidosis. Int. J. Mol. Sci. 2019, 20, 2322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milenkovic, V.M.; Stanton, E.H.; Nothdurfter, C.; Rupprecht, R.; Wetzel, C.H. The Role of Chemokines in the Pathophysiology of Major Depressive Disorder. Int. J. Mol. Sci. 2019, 20, 2283. [Google Scholar] [CrossRef] [Green Version]
- Needham, E.J.; Helmy, A.; Zanier, E.R.; Jones, J.L.; Coles, A.J.; Menon, D.K. The immunological response to traumatic brain injury. J. Neuroimmunol. 2019, 332, 112–125. [Google Scholar] [CrossRef]
- Lyman, M.; Lloyd, D.G.; Ji, X.; Vizcaychipi, M.P.; Ma, D. Neuroinflammation: The role and consequences. Neurosci. Res. 2014, 79, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ebert, S.E.; Jensen, P.; Ozenne, B.; Armand, S.; Svarer, C.; Stenbaek, D.S.; Moeller, K.; Dyssegaard, A.; Thomsen, G.; Steinmetz, J.; et al. Molecular imaging of neuroinflammation in patients after mild traumatic brain injury: A longitudinal (123) I-CLINDE single photon emission computed tomography study. Eur. J. Neurol. 2019, 26, 1426–1432. [Google Scholar] [CrossRef] [PubMed]
- Gendelman, H.E. Neural immunity: Friend or foe? J. Neurovirol. 2002, 8, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Dantzer, R.; O’Connor, J.C.; Freund, G.G.; Johnson, R.W.; Kelley, K.W. From inflammation to sickness and depression: When the immune system subjugates the brain. Nat. Rev. Neurosci. 2008, 9, 46–56. [Google Scholar] [CrossRef] [Green Version]
- Hong, D.Y.; Lee, D.H.; Lee, J.Y.; Lee, E.C.; Park, S.W.; Lee, M.R.; Oh, J.S. Relationship between Brain Metabolic Disorders and Cognitive Impairment: LDL Receptor Defect. Int. J. Mol. Sci. 2022, 23, 8384. [Google Scholar] [CrossRef]
- Linnerbauer, M.; Wheeler, M.A.; Quintana, F.J. Astrocyte Crosstalk in CNS Inflammation. Neuron 2020, 108, 608–622. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [Green Version]
- Freeman, M.R.; Rowitch, D.H. Evolving concepts of gliogenesis: A look way back and ahead to the next 25 years. Neuron 2013, 80, 613–623. [Google Scholar] [CrossRef] [Green Version]
- Rothhammer, V.; Quintana, F.J. The aryl hydrocarbon receptor: An environmental sensor integrating immune responses in health and disease. Nat. Rev. Immunol. 2019, 19, 184–197. [Google Scholar] [CrossRef]
- Qian, Y.; Liu, C.; Hartupee, J.; Altuntas, C.Z.; Gulen, M.F.; Jane-Wit, D.; Xiao, J.; Lu, Y.; Giltiay, N.; Liu, J.; et al. The adaptor Act1 is required for interleukin 17-dependent signaling associated with autoimmune and inflammatory disease. Nat. Immunol. 2007, 8, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; Trzyna, W.C.; McClintock, D.S.; Schumacker, P.T. Role of oxidants in NF-kappa B activation and TNF-alpha gene transcription induced by hypoxia and endotoxin. J. Immunol. 2000, 165, 1013–1021. [Google Scholar] [CrossRef] [Green Version]
- Bezzi, P.; Domercq, M.; Brambilla, L.; Galli, R.; Schols, D.; De Clercq, E.; Vescovi, A.; Bagetta, G.; Kollias, G.; Meldolesi, J.; et al. CXCR4-activated astrocyte glutamate release via TNFalpha: Amplification by microglia triggers neurotoxicity. Nat. Neurosci. 2001, 4, 702–710. [Google Scholar] [CrossRef]
- Lee, D.H.; Lee, J.Y.; Hong, D.Y.; Lee, E.C.; Park, S.W.; Lee, M.R.; Oh, J.S. Neuroinflammation in Post-Traumatic Stress Disorder. Biomedicines 2022, 10, 953. [Google Scholar] [CrossRef]
- Raadsheer, F.C.; van Heerikhuize, J.J.; Lucassen, P.J.; Hoogendijk, W.J.; Tilders, F.J.; Swaab, D.F. Corticotropin-releasing hormone mRNA levels in the paraventricular nucleus of patients with Alzheimer’s disease and depression. Am. J. Psychiatry 1995, 152, 1372–1376. [Google Scholar] [CrossRef] [PubMed]
- Rhen, T.; Cidlowski, J.A. Antiinflammatory action of glucocorticoids--new mechanisms for old drugs. N. Engl. J. Med. 2005, 353, 1711–1723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, P.J.; Adcock, I.M. Glucocorticoid resistance in inflammatory diseases. Lancet 2009, 373, 1905–1917. [Google Scholar] [CrossRef]
- Gjerstad, J.K.; Lightman, S.L.; Spiga, F. Role of glucocorticoid negative feedback in the regulation of HPA axis pulsatility. Stress 2018, 21, 403–416. [Google Scholar] [CrossRef] [Green Version]
- McEwen, B.S.; Sapolsky, R.M. Stress and cognitive function. Curr. Opin. Neurobiol. 1995, 5, 205–216. [Google Scholar] [CrossRef]
- De Cristobal, J.; Madrigal, J.L.; Lizasoain, I.; Lorenzo, P.; Leza, J.C.; Moro, M.A. Aspirin inhibits stress-induced increase in plasma glutamate, brain oxidative damage and ATP fall in rats. Neuroreport 2002, 13, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Meldrum, B.S. Glutamate as a neurotransmitter in the brain: Review of physiology and pathology. J. Nutr. 2000, 130, 1007S–1015S. [Google Scholar] [CrossRef]
- Murphy, M.P. Nitric oxide and cell death. Biochim. Biophys. Acta 1999, 1411, 401–414. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.X.; Wang, Y.; Qin, Z.H. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 2009, 30, 379–387. [Google Scholar] [CrossRef] [Green Version]
- Shinohe, A.; Hashimoto, K.; Nakamura, K.; Tsujii, M.; Iwata, Y.; Tsuchiya, K.J.; Sekine, Y.; Suda, S.; Suzuki, K.; Sugihara, G.; et al. Increased serum levels of glutamate in adult patients with autism. Prog. Neuropsychopharmacol. Biol. Psychiatry 2006, 30, 1472–1477. [Google Scholar] [CrossRef] [Green Version]
- Leza, J.C.; Salas, E.; Sawicki, G.; Russell, J.C.; Radomski, M.W. The effects of stress on homeostasis in JCR-LA-cp rats: The role of nitric oxide. J. Pharmacol. Exp. Ther. 1998, 286, 1397–1403. [Google Scholar]
- Madrigal, J.L.; Garcia-Bueno, B.; Caso, J.R.; Perez-Nievas, B.G.; Leza, J.C. Stress-induced oxidative changes in brain. CNS Neurol. Disord. Drug Targets 2006, 5, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Rauf, A.; Khan, H.; Abu-Izneid, T. Renin-angiotensin-aldosterone (RAAS): The ubiquitous system for homeostasis and pathologies. Biomed. Pharmacother. 2017, 94, 317–325. [Google Scholar] [CrossRef]
- Yang, G.; Xi, Z.X.; Wan, Y.; Wang, H.; Bi, G. Changes in circulating and tissue angiotensin II during acute and chronic stress. Biol. Signals 1993, 2, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Wan, Y.; Zhu, Y. Angiotensin II—An important stress hormone. Biol. Signals 1996, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, J.P.; Sriramula, S.; Mariappan, N.; Agarwal, D.; Francis, J. Angiotensin II-induced hypertension is modulated by nuclear factor-kappaBin the paraventricular nucleus. Hypertension 2012, 59, 113–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Zhang, Z.H.; Wei, S.G.; Serrats, J.; Weiss, R.M.; Felder, R.B. Brain perivascular macrophages and the sympathetic response to inflammation in rats after myocardial infarction. Hypertension 2010, 55, 652–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawahara, K.; Hohjoh, H.; Inazumi, T.; Tsuchiya, S.; Sugimoto, Y. Prostaglandin E2-induced inflammation: Relevance of prostaglandin E receptors. Biochim. Biophys. Acta 2015, 1851, 414–421. [Google Scholar] [CrossRef]
- Tsuge, K.; Inazumi, T.; Shimamoto, A.; Sugimoto, Y. Molecular mechanisms underlying prostaglandin E2-exacerbated inflammation and immune diseases. Int. Immunol. 2019, 31, 597–606. [Google Scholar] [CrossRef]
- Saavedra, J.M. Brain and pituitary angiotensin. Endocr. Rev. 1992, 13, 329–380. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, G. Factors controlling steroid biosynthesis in the zona glomerulosa of the adrenal. J. Steroid Biochem. Mol. Biol. 1993, 45, 147–151. [Google Scholar] [CrossRef]
- Yuste, J.E.; Tarragon, E.; Campuzano, C.M.; Ros-Bernal, F. Implications of glial nitric oxide in neurodegenerative diseases. Front. Cell. Neurosci. 2015, 9, 322. [Google Scholar] [CrossRef] [Green Version]
- Zorumski, C.F.; Izumi, Y. Nitric oxide and hippocampal synaptic plasticity. Biochem. Pharmacol. 1993, 46, 777–785. [Google Scholar] [CrossRef]
- Huang, E.P. Synaptic plasticity: A role for nitric oxide in LTP. Curr. Biol. 1997, 7, R141–R143. [Google Scholar] [CrossRef] [Green Version]
- Schini, V.B.; Busse, R.; Vanhoutte, P.M. Inducible nitric oxide synthase in vascular smooth muscle. Arzneimittelforschung 1994, 44, 432–435. [Google Scholar] [PubMed]
- Ferrari, A.U.; Radaelli, A.; Mori, T.; Mircoli, L.; Perlini, S.; Meregalli, P.; Fedele, L.; Mancia, G. Nitric oxide-dependent vasodilation and the regulation of arterial blood pressure. J. Cardiovasc. Pharmacol. 2001, 38 (Suppl. S2), S19–S22. [Google Scholar] [CrossRef]
- Madrigal, J.L.; Moro, M.A.; Lizasoain, I.; Lorenzo, P.; Castrillo, A.; Bosca, L.; Leza, J.C. Inducible nitric oxide synthase expression in brain cortex after acute restraint stress is regulated by nuclear factor kappaB-mediated mechanisms. J. Neurochem. 2001, 76, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.W.; Kashiwabara, Y.; Nathan, C. Role of transcription factor NF-kappa B/Rel in induction of nitric oxide synthase. J. Biol. Chem. 1994, 269, 4705–4708. [Google Scholar] [CrossRef]
- Moncada, S.; Palmer, R.M.; Higgs, E.A. Nitric oxide: Physiology, pathophysiology, and pharmacology. Pharmacol. Rev. 1991, 43, 109–142. [Google Scholar] [PubMed]
- Hinz, B.; Brune, K. Cyclooxygenase-2—10 years later. J. Pharmacol. Exp. Ther. 2002, 300, 367–375. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, G.P.; Ford-Hutchinson, A.W. Expression of mRNA for cyclooxygenase-1 and cyclooxygenase-2 in human tissues. FEBS Lett. 1993, 330, 156–160. [Google Scholar] [CrossRef] [Green Version]
- Yasojima, K.; Schwab, C.; McGeer, E.G.; McGeer, P.L. Distribution of cyclooxygenase-1 and cyclooxygenase-2 mRNAs and proteins in human brain and peripheral organs. Brain Res. 1999, 830, 226–236. [Google Scholar] [CrossRef]
- Simon, L.S. Role and regulation of cyclooxygenase-2 during inflammation. Am. J. Med. 1999, 106, 37S–42S. [Google Scholar] [CrossRef]
- Nogawa, S.; Zhang, F.; Ross, M.E.; Iadecola, C. Cyclo-oxygenase-2 gene expression in neurons contributes to ischemic brain damage. J. Neurosci. 1997, 17, 2746–2755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minghetti, L.; Pocchiari, M. Cyclooxygenase-2, prostaglandin E2, and microglial activation in prion diseases. Int. Rev. Neurobiol. 2007, 82, 265–275. [Google Scholar] [PubMed]
- Muller-Decker, K.; Furstenberger, G. The cyclooxygenase-2-mediated prostaglandin signaling is causally related to epithelial carcinogenesis. Mol. Carcinog. 2007, 46, 705–710. [Google Scholar] [CrossRef]
- Vesce, S.; Rossi, D.; Brambilla, L.; Volterra, A. Glutamate release from astrocytes in physiological conditions and in neurodegenerative disorders characterized by neuroinflammation. Int. Rev. Neurobiol. 2007, 82, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Yamagata, K.; Andreasson, K.I.; Kaufmann, W.E.; Barnes, C.A.; Worley, P.F. Expression of a mitogen-inducible cyclooxygenase in brain neurons: Regulation by synaptic activity and glucocorticoids. Neuron 1993, 11, 371–386. [Google Scholar] [CrossRef]
- Furukawa, H.; Singh, S.K.; Mancusso, R.; Gouaux, E. Subunit arrangement and function in NMDA receptors. Nature 2005, 438, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.S.; Lipton, S.A. The chemical biology of clinically tolerated NMDA receptor antagonists. J. Neurochem. 2006, 97, 1611–1626. [Google Scholar] [CrossRef]
- Kemp, J.A.; McKernan, R.M. NMDA receptor pathways as drug targets. Nat. Neurosci. 2002, 5, 1039–1042. [Google Scholar] [CrossRef]
- Lipton, S.A. Paradigm shift in neuroprotection by NMDA receptor blockade: Memantine and beyond. Nat. Rev. Drug Discov. 2006, 5, 160–170. [Google Scholar] [CrossRef]
- Koch, H.J.; Szecsey, A.; Haen, E. NMDA-antagonism (memantine): An alternative pharmacological therapeutic principle in Alzheimer’s and vascular dementia. Curr. Pharm. Des. 2004, 10, 253–259. [Google Scholar] [CrossRef]
- Lipton, S.A. Failures and successes of NMDA receptor antagonists: Molecular basis for the use of open-channel blockers like memantine in the treatment of acute and chronic neurologic insults. NeuroRx 2004, 1, 101–110. [Google Scholar] [CrossRef]
- Sonkusare, S.K.; Kaul, C.L.; Ramarao, P. Dementia of Alzheimer’s disease and other neurodegenerative disorders—Memantine, a new hope. Pharmacol. Res. 2005, 51, 1–17. [Google Scholar] [CrossRef]
- Madrigal, J.L.; Hurtado, O.; Moro, M.A.; Lizasoain, I.; Lorenzo, P.; Castrillo, A.; Bosca, L.; Leza, J.C. The increase in TNF-alpha levels is implicated in NF-kappaB activation and inducible nitric oxide synthase expression in brain cortex after immobilization stress. Neuropsychopharmacology 2002, 26, 155–163. [Google Scholar] [CrossRef]
- Michalik, L.; Auwerx, J.; Berger, J.P.; Chatterjee, V.K.; Glass, C.K.; Gonzalez, F.J.; Grimaldi, P.A.; Kadowaki, T.; Lazar, M.A.; O’Rahilly, S.; et al. International Union of Pharmacology. LXI. Peroxisome proliferator-activated receptors. Pharmacol. Rev. 2006, 58, 726–741. [Google Scholar] [CrossRef] [PubMed]
- Jenum, P.A. Rapid identification of Escherichia coli from routine urine specimens based on macroscopic criteria. Acta Pathol. Microbiol. Immunol. Scand. B 1985, 93, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.; Moller, D.E. The mechanisms of action of PPARs. Annu. Rev. Med. 2002, 53, 409–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feige, J.N.; Gelman, L.; Michalik, L.; Desvergne, B.; Wahli, W. From molecular action to physiological outputs: Peroxisome proliferator-activated receptors are nuclear receptors at the crossroads of key cellular functions. Prog. Lipid Res. 2006, 45, 120–159. [Google Scholar] [CrossRef] [PubMed]
- Feinstein, D.L. Therapeutic potential of peroxisome proliferator-activated receptor agonists for neurological disease. Diabetes Technol. Ther. 2003, 5, 67–73. [Google Scholar] [CrossRef]
- Luna-Medina, R.; Cortes-Canteli, M.; Alonso, M.; Santos, A.; Martinez, A.; Perez-Castillo, A. Regulation of inflammatory response in neural cells in vitro by thiadiazolidinones derivatives through peroxisome proliferator-activated receptor gamma activation. J. Biol. Chem. 2005, 280, 21453–21462. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Bueno, B.; Madrigal, J.L.; Lizasoain, I.; Moro, M.A.; Lorenzo, P.; Leza, J.C. Peroxisome proliferator-activated receptor gamma activation decreases neuroinflammation in brain after stress in rats. Biol. Psychiatry 2005, 57, 885–894. [Google Scholar] [CrossRef]
- Garcia-Bueno, B.; Madrigal, J.L.; Lizasoain, I.; Moro, M.A.; Lorenzo, P.; Leza, J.C. The anti-inflammatory prostaglandin 15d-PGJ2 decreases oxidative/nitrosative mediators in brain after acute stress in rats. Psychopharmacology 2005, 180, 513–522. [Google Scholar] [CrossRef]
- Park, E.Y.; Cho, I.J.; Kim, S.G. Transactivation of the PPAR-responsive enhancer module in chemopreventive glutathione S-transferase gene by the peroxisome proliferator-activated receptor-gamma and retinoid X receptor heterodimer. Cancer Res. 2004, 64, 3701–3713. [Google Scholar] [CrossRef] [Green Version]
- Gaddam, R.R.; Chambers, S.; Bhatia, M. ACE and ACE2 in inflammation: A tale of two enzymes. Inflamm. Allergy Drug Targets 2014, 13, 224–234. [Google Scholar] [CrossRef]
- Coates, D. The angiotensin converting enzyme (ACE). Int. J. Biochem. Cell Biol. 2003, 35, 769–773. [Google Scholar] [CrossRef]
- Thompson, D.; Pepys, M.B.; Wood, S.P. The physiological structure of human C-reactive protein and its complex with phosphocholine. Structure 1999, 7, 169–177. [Google Scholar] [CrossRef]
- Khoury, N.M.; Marvar, P.J.; Gillespie, C.F.; Wingo, A.; Schwartz, A.; Bradley, B.; Kramer, M.; Ressler, K.J. The renin-angiotensin pathway in posttraumatic stress disorder: Angiotensin-converting enzyme inhibitors and angiotensin receptor blockers are associated with fewer traumatic stress symptoms. J. Clin. Psychiatry 2012, 73, 849–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, R.A.; Simoes e Silva, A.C.; Maric, C.; Silva, D.M.; Machado, R.P.; de Buhr, I.; Heringer-Walther, S.; Pinheiro, S.V.; Lopes, M.T.; Bader, M.; et al. Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc. Natl. Acad. Sci. USA 2003, 100, 8258–8263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da Silveira, K.D.; Coelho, F.M.; Vieira, A.T.; Sachs, D.; Barroso, L.C.; Costa, V.V.; Bretas, T.L.; Bader, M.; de Sousa, L.P.; da Silva, T.A.; et al. Anti-inflammatory effects of the activation of the angiotensin-(1-7) receptor, MAS, in experimental models of arthritis. J. Immunol. 2010, 185, 5569–5576. [Google Scholar] [CrossRef] [Green Version]
- Feltenberger, J.D.; Andrade, J.M.; Paraiso, A.; Barros, L.O.; Filho, A.B.; Sinisterra, R.D.; Sousa, F.B.; Guimaraes, A.L.; de Paula, A.M.; Campagnole-Santos, M.J.; et al. Oral formulation of angiotensin-(1-7) improves lipid metabolism and prevents high-fat diet-induced hepatic steatosis and inflammation in mice. Hypertension 2013, 62, 324–330. [Google Scholar] [CrossRef] [Green Version]
- Khajah, M.A.; Fateel, M.M.; Ananthalakshmi, K.V.; Luqmani, Y.A. Anti-Inflammatory Action of Angiotensin 1-7 in Experimental Colitis. PLoS ONE 2016, 11, e0150861. [Google Scholar] [CrossRef] [Green Version]
- Meng, Y.; Li, T.; Zhou, G.S.; Chen, Y.; Yu, C.H.; Pang, M.X.; Li, W.; Li, Y.; Zhang, W.Y.; Li, X. The angiotensin-converting enzyme 2/angiotensin (1-7)/Mas axis protects against lung fibroblast migration and lung fibrosis by inhibiting the NOX4-derived ROS-mediated RhoA/Rho kinase pathway. Antioxid. Redox Signal. 2015, 22, 241–258. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Li, J.; Hao, P.; Chen, W.; Meng, X.; Li, H.; Zhang, Y.; Zhang, C.; Yang, J. Imbalance between angiotensin II and angiotensin-(1-7) in human coronary atherosclerosis. J. Renin Angiotensin Aldosterone Syst. 2016, 17, 1470320316659618. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zeng, Z.; Cao, Y.; Liu, Y.; Ping, F.; Liang, M.; Xue, Y.; Xi, C.; Zhou, M.; Jiang, W. Angiotensin-converting enzyme 2 prevents lipopolysaccharide-induced rat acute lung injury via suppressing the ERK1/2 and NF-kappaB signaling pathways. Sci. Rep. 2016, 6, 27911. [Google Scholar] [CrossRef] [Green Version]
- Magalhaes, G.S.; Rodrigues-Machado, M.G.; Motta-Santos, D.; Silva, A.R.; Caliari, M.V.; Prata, L.O.; Abreu, S.C.; Rocco, P.R.; Barcelos, L.S.; Santos, R.A.; et al. Angiotensin-(1-7) attenuates airway remodelling and hyperresponsiveness in a model of chronic allergic lung inflammation. Br. J. Pharmacol. 2015, 172, 2330–2342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues-Machado, M.G.; Magalhaes, G.S.; Cardoso, J.A.; Kangussu, L.M.; Murari, A.; Caliari, M.V.; Oliveira, M.L.; Cara, D.C.; Noviello, M.L.; Marques, F.D.; et al. AVE 0991, a non-peptide mimic of angiotensin-(1-7) effects, attenuates pulmonary remodelling in a model of chronic asthma. Br. J. Pharmacol. 2013, 170, 835–846. [Google Scholar] [CrossRef] [PubMed]
- Magalhaes, G.S.; Barroso, L.C.; Reis, A.C.; Rodrigues-Machado, M.G.; Gregorio, J.F.; Motta-Santos, D.; Oliveira, A.C.; Perez, D.A.; Barcelos, L.S.; Teixeira, M.M.; et al. Angiotensin-(1-7) Promotes Resolution of Eosinophilic Inflammation in an Experimental Model of Asthma. Front. Immunol. 2018, 9, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ena, P.; Madeddu, P.; Glorioso, N.; Cerimele, D.; Rappelli, A. High prevalence of cardiovascular diseases and enhanced activity of the renin-angiotensin system in psoriatic patients. Acta Cardiol. 1985, 40, 199–205. [Google Scholar]
- Kawajiri, M.; Mogi, M.; Higaki, N.; Matsuoka, T.; Ohyagi, Y.; Tsukuda, K.; Kohara, K.; Horiuchi, M.; Miki, T.; Kira, J.I. Angiotensin-converting enzyme (ACE) and ACE2 levels in the cerebrospinal fluid of patients with multiple sclerosis. Mult. Scler. 2009, 15, 262–265. [Google Scholar] [CrossRef]
- Sagawa, K.; Nagatani, K.; Komagata, Y.; Yamamoto, K. Angiotensin receptor blockers suppress antigen-specific T cell responses and ameliorate collagen-induced arthritis in mice. Arthritis Rheum. 2005, 52, 1920–1928. [Google Scholar] [CrossRef]
- Timmermans, S.; Bogie, J.F.; Vanmierlo, T.; Lutjohann, D.; Stinissen, P.; Hellings, N.; Hendriks, J.J. High fat diet exacerbates neuroinflammation in an animal model of multiple sclerosis by activation of the Renin Angiotensin system. J. Neuroimmune Pharmacol. 2014, 9, 209–217. [Google Scholar] [CrossRef]
- Torika, N.; Asraf, K.; Danon, A.; Apte, R.N.; Fleisher-Berkovich, S. Telmisartan Modulates Glial Activation: In Vitro and In Vivo Studies. PLoS ONE 2016, 11, e0155823. [Google Scholar] [CrossRef] [Green Version]
- Benter, I.F.; Yousif, M.H.; Al-Saleh, F.M.; Raghupathy, R.; Chappell, M.C.; Diz, D.I. Angiotensin-(1-7) blockade attenuates captopril- or hydralazine-induced cardiovascular protection in spontaneously hypertensive rats treated with NG-nitro-L-arginine methyl ester. J. Cardiovasc. Pharmacol. 2011, 57, 559–567. [Google Scholar] [CrossRef] [Green Version]
- Fujihara, S.; Jaffray, E.; Farrow, S.N.; Rossi, A.G.; Haslett, C.; Hay, R.T. Inhibition of NF-kappa B by a cell permeable form of I kappa B alpha induces apoptosis in eosinophils. Biochem. Biophys. Res. Commun. 2005, 326, 632–637. [Google Scholar] [CrossRef]
- Sousa, L.P.; Carmo, A.F.; Rezende, B.M.; Lopes, F.; Silva, D.M.; Alessandri, A.L.; Bonjardim, C.A.; Rossi, A.G.; Teixeira, M.M.; Pinho, V. Cyclic AMP enhances resolution of allergic pleurisy by promoting inflammatory cell apoptosis via inhibition of PI3K/Akt and NF-kappaB. Biochem. Pharmacol. 2009, 78, 396–405. [Google Scholar] [CrossRef]
- Sousa, L.P.; Lopes, F.; Silva, D.M.; Tavares, L.P.; Vieira, A.T.; Rezende, B.M.; Carmo, A.F.; Russo, R.C.; Garcia, C.C.; Bonjardim, C.A.; et al. PDE4 inhibition drives resolution of neutrophilic inflammation by inducing apoptosis in a PKA-PI3K/Akt-dependent and NF-kappaB-independent manner. J. Leukoc. Biol. 2010, 87, 895–904. [Google Scholar] [CrossRef]
- Vane, J.R. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat. New. Biol. 1971, 231, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Sethi, R.; Gomez-Coronado, N.; Walker, A.J.; Robertson, O.D.; Agustini, B.; Berk, M.; Dodd, S. Neurobiology and Therapeutic Potential of Cyclooxygenase-2 (COX-2) Inhibitors for Inflammation in Neuropsychiatric Disorders. Front. Psychiatry 2019, 10, 605. [Google Scholar] [CrossRef] [Green Version]
- Antman, E.M.; Bennett, J.S.; Daugherty, A.; Furberg, C.; Roberts, H.; Taubert, K.A.; American Heart, A. Use of nonsteroidal antiinflammatory drugs: An update for clinicians: A scientific statement from the American Heart Association. Circulation 2007, 115, 1634–1642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philp, R.B.; Arora, P.; McIver, D.J. Effects of gaseous anesthetics and ultrashort and short-acting barbiturates on human blood platelet free cytosolic calcium: Relevance to their effects on platelet aggregation. Can. J. Physiol. Pharmacol. 1992, 70, 1161–1166. [Google Scholar] [CrossRef]
- Schmidt, F.M.; Kirkby, K.C.; Lichtblau, N. Inflammation and Immune Regulation as Potential Drug Targets in Antidepressant Treatment. Curr. Neuropharmacol. 2016, 14, 674–687. [Google Scholar] [CrossRef] [Green Version]
- Strawbridge, R.; Arnone, D.; Danese, A.; Papadopoulos, A.; Herane Vives, A.; Cleare, A.J. Inflammation and clinical response to treatment in depression: A meta-analysis. Eur. Neuropsychopharmacol. 2015, 25, 1532–1543. [Google Scholar] [CrossRef]
- Hannestad, J.; DellaGioia, N.; Bloch, M. The effect of antidepressant medication treatment on serum levels of inflammatory cytokines: A meta-analysis. Neuropsychopharmacology 2011, 36, 2452–2459. [Google Scholar] [CrossRef] [PubMed]
- Obuchowicz, E.; Bielecka, A.M.; Paul-Samojedny, M.; Pudelko, A.; Kowalski, J. Imipramine and fluoxetine inhibit LPS-induced activation and affect morphology of microglial cells in the rat glial culture. Pharmacol. Rep. 2014, 66, 34–43. [Google Scholar] [CrossRef]
- Himmerich, H.; Milenovic, S.; Fulda, S.; Plumakers, B.; Sheldrick, A.J.; Michel, T.M.; Kircher, T.; Rink, L. Regulatory T cells increased while IL-1beta decreased during antidepressant therapy. J. Psychiatr. Res. 2010, 44, 1052–1057. [Google Scholar] [CrossRef]
- Zeng, H.; Chen, H.; Li, M.; Zhuang, J.; Peng, Y.; Zhou, H.; Xu, C.; Yu, Q.; Fu, X.; Cao, S.; et al. Autophagy protein NRBF2 attenuates endoplasmic reticulum stress-associated neuroinflammation and oxidative stress via promoting autophagosome maturation by interacting with Rab7 after SAH. J. Neuroinflamm. 2021, 18, 210. [Google Scholar] [CrossRef]
- Lu, J.; He, L.; Behrends, C.; Araki, M.; Araki, K.; Jun Wang, Q.; Catanzaro, J.M.; Friedman, S.L.; Zong, W.X.; Fiel, M.I.; et al. NRBF2 regulates autophagy and prevents liver injury by modulating Atg14L-linked phosphatidylinositol-3 kinase III activity. Nat. Commun. 2014, 5, 3920. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.Y.; Liu, L.; Wang, E.J.; Xiao, H.T.; Cai, C.Z.; Wang, J.; Su, H.; Wang, Y.; Tan, J.; Zhang, Z.; et al. PI3KC3 complex subunit NRBF2 is required for apoptotic cell clearance to restrict intestinal inflammation. Autophagy 2021, 17, 1096–1111. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Cai, C.Z.; Song, J.X.; Tan, J.Q.; Durairajan, S.S.K.; Iyaswamy, A.; Wu, M.Y.; Chen, L.L.; Yue, Z.; Li, M.; et al. NRBF2 is involved in the autophagic degradation process of APP-CTFs in Alzheimer disease models. Autophagy 2017, 13, 2028–2040. [Google Scholar] [CrossRef]
- Sinha, P.; Verma, B.; Ganesh, S. Trehalose Ameliorates Seizure Susceptibility in Lafora Disease Mouse Models by Suppressing Neuroinflammation and Endoplasmic Reticulum Stress. Mol. Neurobiol. 2021, 58, 1088–1101. [Google Scholar] [CrossRef]
- Acosta, S.A.; Tajiri, N.; Hoover, J.; Kaneko, Y.; Borlongan, C.V. Intravenous Bone Marrow Stem Cell Grafts Preferentially Migrate to Spleen and Abrogate Chronic Inflammation in Stroke. Stroke 2015, 46, 2616–2627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, H.S.; Jeong, S.Y.; Yang, J.; Kim, S.D.; Zhang, B.; Yoo, H.S.; Song, S.U.; Jeon, M.S.; Song, Y.S. Neuroprotective effect of mesenchymal stem cell through complement component 3 downregulation after transient focal cerebral ischemia in mice. Neurosci. Lett. 2016, 633, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Sudo, N.; Chida, Y.; Aiba, Y.; Sonoda, J.; Oyama, N.; Yu, X.N.; Kubo, C.; Koga, Y. Postnatal microbial colonization programs the hypothalamic-pituitary-adrenal system for stress response in mice. J. Physiol. 2004, 558, 263–275. [Google Scholar] [CrossRef]
- Ryu, J.K.; McLarnon, J.G. A leaky blood-brain barrier, fibrinogen infiltration and microglial reactivity in inflamed Alzheimer’s disease brain. J. Cell. Mol. Med. 2009, 13, 2911–2925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkins, B.T.; Davis, T.P. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol. Rev. 2005, 57, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Obermeier, B.; Daneman, R.; Ransohoff, R.M. Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 2013, 19, 1584–1596. [Google Scholar] [CrossRef] [Green Version]
- Musa, N.H.; Mani, V.; Lim, S.M.; Vidyadaran, S.; Abdul Majeed, A.B.; Ramasamy, K. Lactobacilli-fermented cow’s milk attenuated lipopolysaccharide-induced neuroinflammation and memory impairment in vitro and in vivo. J. Dairy Res. 2017, 84, 488–495. [Google Scholar] [CrossRef]
- Plaza-Diaz, J.; Ruiz-Ojeda, F.J.; Vilchez-Padial, L.M.; Gil, A. Evidence of the Anti-Inflammatory Effects of Probiotics and Synbiotics in Intestinal Chronic Diseases. Nutrients 2017, 9, 555. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
When the hippocampus is stimulated by stress, it secretes ACTH to stimulate the adrenal cortex, from which glucocorticoids and glutamates are overproduced. If a high level of glucocorticoids is maintained due to persistent stress, resistance occurs in GR, resulting in impaired glucocorticoids acceptance, which leads to impairment in anti-inflammatory action. In addition, it over-produces glutamate, an excitatory amino acid, which over-generates NO, resulting in NO-mediated neuroinflammation, and induces apoptosis in neural cells by upregulating the pro-apoptotic transcription factors. Glucocorticoids themselves also contribute to apoptosis by inducing intracellular calcium excess. The black arrow indicates that it contributes to the following response. The red arrow indicates increased neuroinflammation. ACTH: adrenocorticotropin hormone, GR: glucocorticoids receptor, HPA: hypothalamic-pituitary-adrenal, NO: nitric oxide.
Figure 1.
When the hippocampus is stimulated by stress, it secretes ACTH to stimulate the adrenal cortex, from which glucocorticoids and glutamates are overproduced. If a high level of glucocorticoids is maintained due to persistent stress, resistance occurs in GR, resulting in impaired glucocorticoids acceptance, which leads to impairment in anti-inflammatory action. In addition, it over-produces glutamate, an excitatory amino acid, which over-generates NO, resulting in NO-mediated neuroinflammation, and induces apoptosis in neural cells by upregulating the pro-apoptotic transcription factors. Glucocorticoids themselves also contribute to apoptosis by inducing intracellular calcium excess. The black arrow indicates that it contributes to the following response. The red arrow indicates increased neuroinflammation. ACTH: adrenocorticotropin hormone, GR: glucocorticoids receptor, HPA: hypothalamic-pituitary-adrenal, NO: nitric oxide.
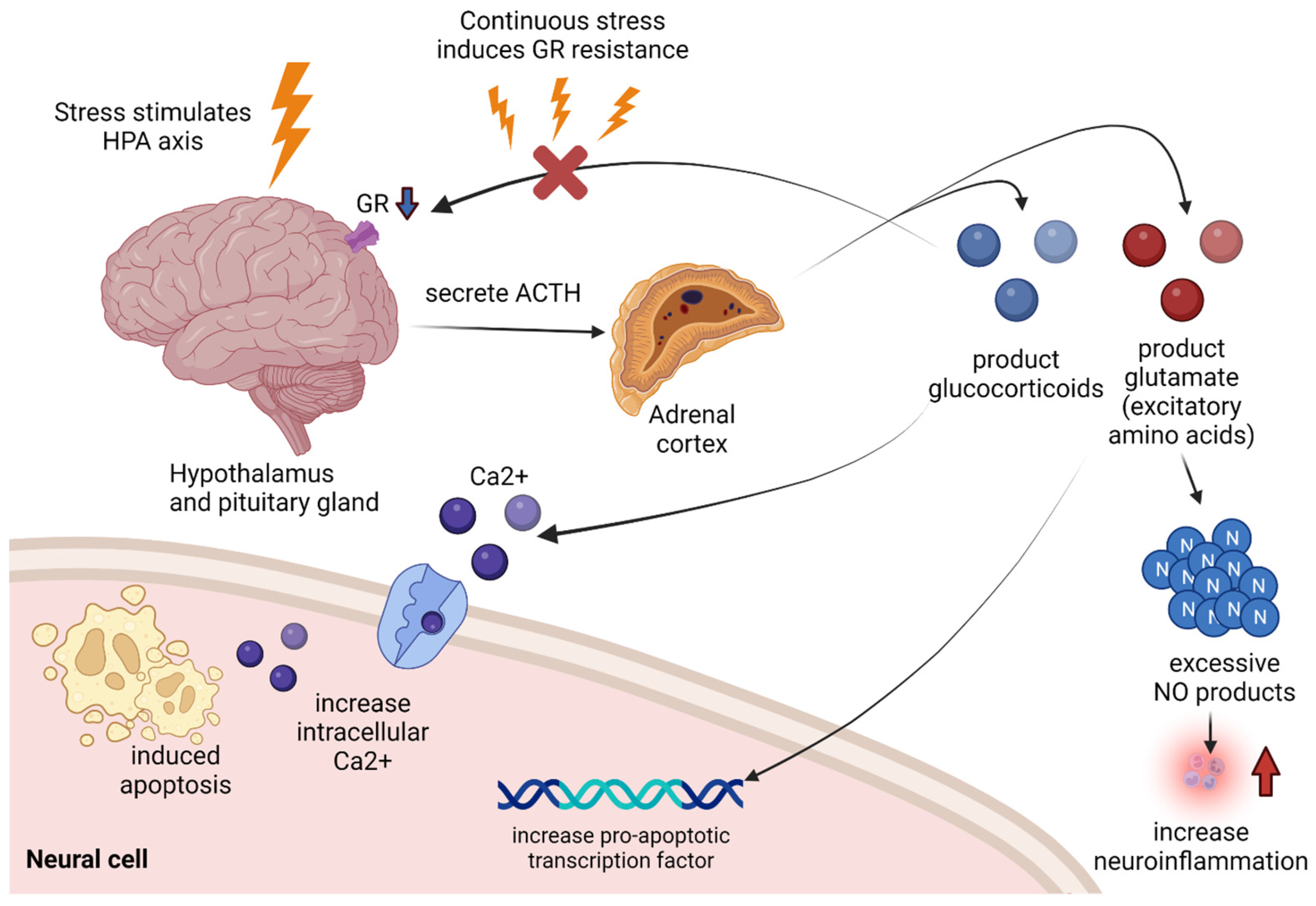
Figure 2.
When stress stimulates sympathetic nerves, excessive Ang II is induced. It boosts ROS production and produces inflammatory cytokines. Pro-inflammatory cytokines, such as NF-κB, contribute to the production of COX-2 and PGE2. COX-2 is an important reactant contributing to the inflammatory response and can also contribute to neuroinflammation by inducing PGE2 activity by PGE2 RAS. PGE2 improves the infiltration of leukocyte. Ang II also contributes to ATCH and glucocorticoid upregulation, which may affect other mechanisms described above. The black arrow indicates that it contributes to the following response. The red arrow indicates an increase in ROS. ACTH: adrenocorticotropin hormone, Ang II: angiotensin II, COX-2: cyclooxygenase-2, PGE2: prostaglandin E2, ROS: reactive oxygen species.
Figure 2.
When stress stimulates sympathetic nerves, excessive Ang II is induced. It boosts ROS production and produces inflammatory cytokines. Pro-inflammatory cytokines, such as NF-κB, contribute to the production of COX-2 and PGE2. COX-2 is an important reactant contributing to the inflammatory response and can also contribute to neuroinflammation by inducing PGE2 activity by PGE2 RAS. PGE2 improves the infiltration of leukocyte. Ang II also contributes to ATCH and glucocorticoid upregulation, which may affect other mechanisms described above. The black arrow indicates that it contributes to the following response. The red arrow indicates an increase in ROS. ACTH: adrenocorticotropin hormone, Ang II: angiotensin II, COX-2: cyclooxygenase-2, PGE2: prostaglandin E2, ROS: reactive oxygen species.


{kind=link}
{kind=link}
Table 1.
Various pharmacological treatments for the treatment of neuroinflammation drugs.
| Types of Treatments | Mechanism | Typical Drugs |
|---|---|---|
| ACE inhibitor and ARB | Inhibits converting enzymes that activate Ang II, or converts Ang II into Ang(1–7), which exhibits an opposite effect | Benazepril, captopril, enalapril, lisinopril, perindopril, ramipril, trandolapril |
| Antidepressant | Stress suppression, or up-regulation of Treg cells that limit cytokines | Amitriptyline (Elavil), fluoxetine, imipramine, paroxetine |
| COX-2 inhibitor | Direct inhibition of COX-2, which is important for inflammatory reactions, improves neuroinflammatory pathways, and improves depression | celecoxib, etoricoxib, rofecoxib |
| NMDA receptor antagonist | Reduces oxidative and NO damage by inhibiting excessive glutamate in cells and improves neuroinflammation | dizocilpine |
| PPAR agonist | Prevents various post-stress neuroinflammatory pathways by inhibiting NF-κB, TNF-α, COX-2 and iNOS when injecting agents. | ciglitazone, rosiglitazone, troglitazone, pioglitazone |
ACE: angiotensin-converting enzyme, ARB: angiotensin receptor blocker, COX-2: cyclooxygenase-2, iNOS: induced nitric oxide synthase, NO: nitric oxide, TNF-α: tumor necrosis factor-α.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lee, D.-H.; Lee, J.-Y.; Hong, D.-Y.; Lee, E.-C.; Park, S.-W.; Lee, Y.-K.; Oh, J.-S. Pharmacological Treatment for Neuroinflammation in Stress-Related Disorder. Biomedicines 2022, 10, 2518. https://doi.org/10.3390/biomedicines10102518
AMA Style
Lee D-H, Lee J-Y, Hong D-Y, Lee E-C, Park S-W, Lee Y-K, Oh J-S. Pharmacological Treatment for Neuroinflammation in Stress-Related Disorder. Biomedicines. 2022; 10(10):2518. https://doi.org/10.3390/biomedicines10102518
Chicago/Turabian StyleLee, Dong-Hun, Ji-Young Lee, Dong-Yong Hong, Eun-Chae Lee, Sang-Won Park, Yun-Kyung Lee, and Jae-Sang Oh. 2022. "Pharmacological Treatment for Neuroinflammation in Stress-Related Disorder" Biomedicines 10, no. 10: 2518. https://doi.org/10.3390/biomedicines10102518
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.