
Mitochondrial Calcium-Triggered Oxidative Stress and Developmental Defects in Dopaminergic Neurons Differentiated from Deciduous Teeth-Derived Dental Pulp Stem Cells with MFF Insufficiency

 , , ,
, , ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. SHED Isolation, Culture, and Differentiation into Dopaminergic Neurons
2.2. RNA Interference to Knock down MFF Expression
2.3. Folic Acid, Ruthenium Red, and Xestospongin C Treatment
2.4. Immunocytochemistry
2.5. Analyses of Neuronal Morphology, Mitochondrial Amount, Distribution, and Length in Dopaminergic Neurons
2.6. Western Blotting
2.7. Immunoprecipitation
2.8. Quantitative Reverse Transcription Polymerase Chain Reaction
2.9. Analyses of Intracellular, Mitochondrial, and Endoplasmic Reticulum Ca2+ Levels
2.10. Analyses of NADH and NAD+ Levels
2.11. Analysis of Dopamine Levels
2.12. Measurement of Mitochondrial Reactive Oxygen Species
2.13. Measurement of Mitochondrial Membrane Potential
2.14. Analysis of Intracellular Adenosine Triphosphate Levels
2.15. Statistical Analyses
3. Results
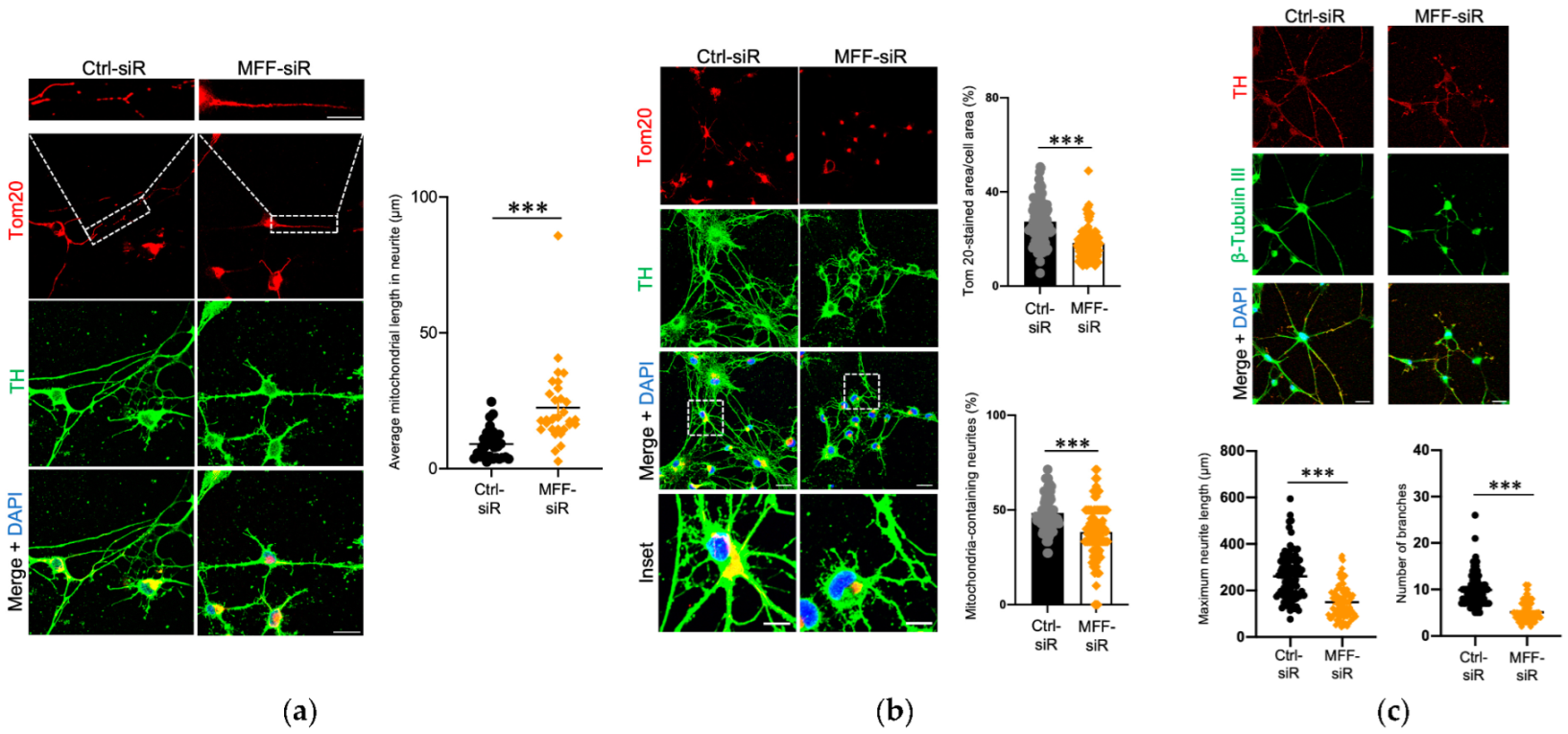
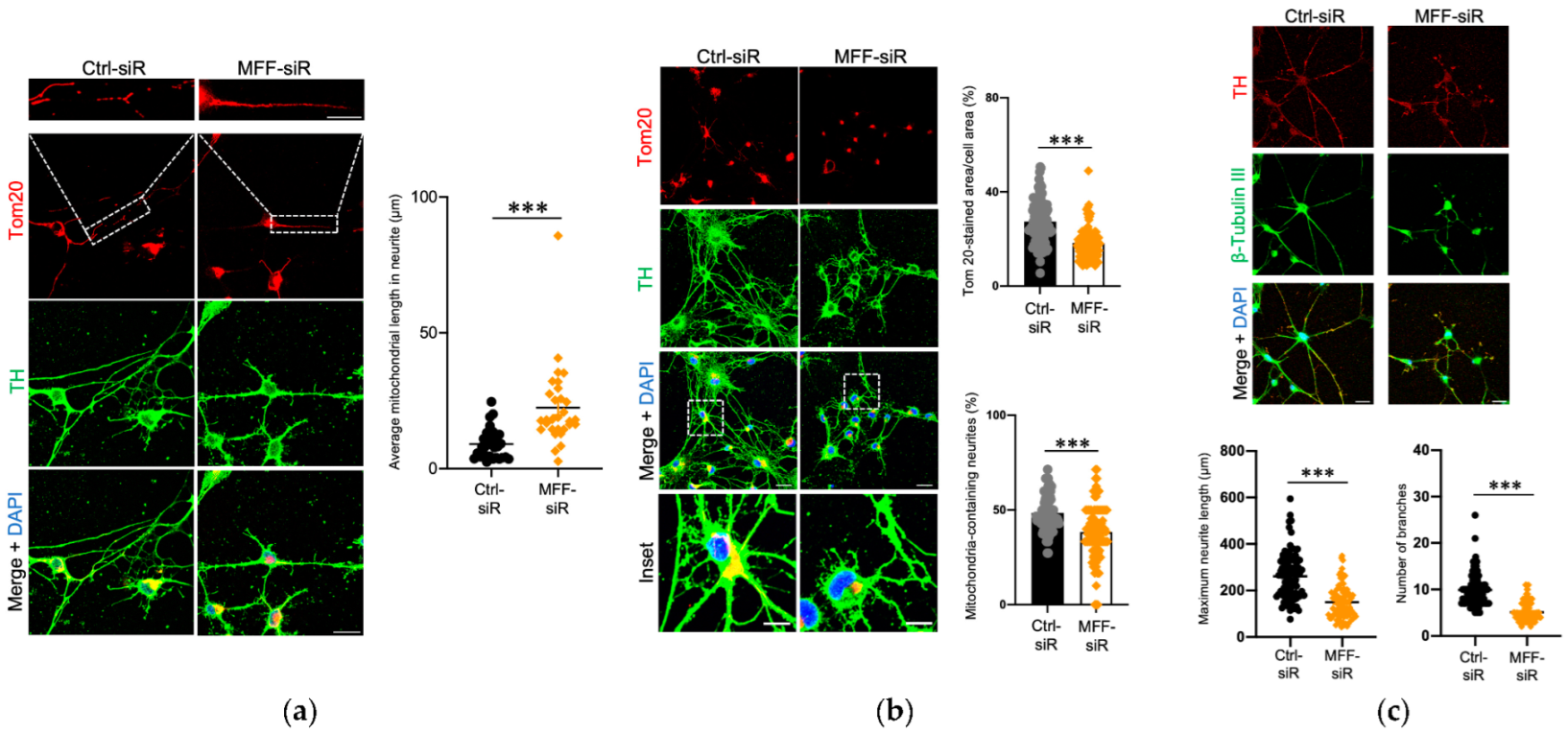
3.1. Impaired Mitochondrial Morphology and Neurite Development in Dopaminergic Neurons with MFF Insufficiency
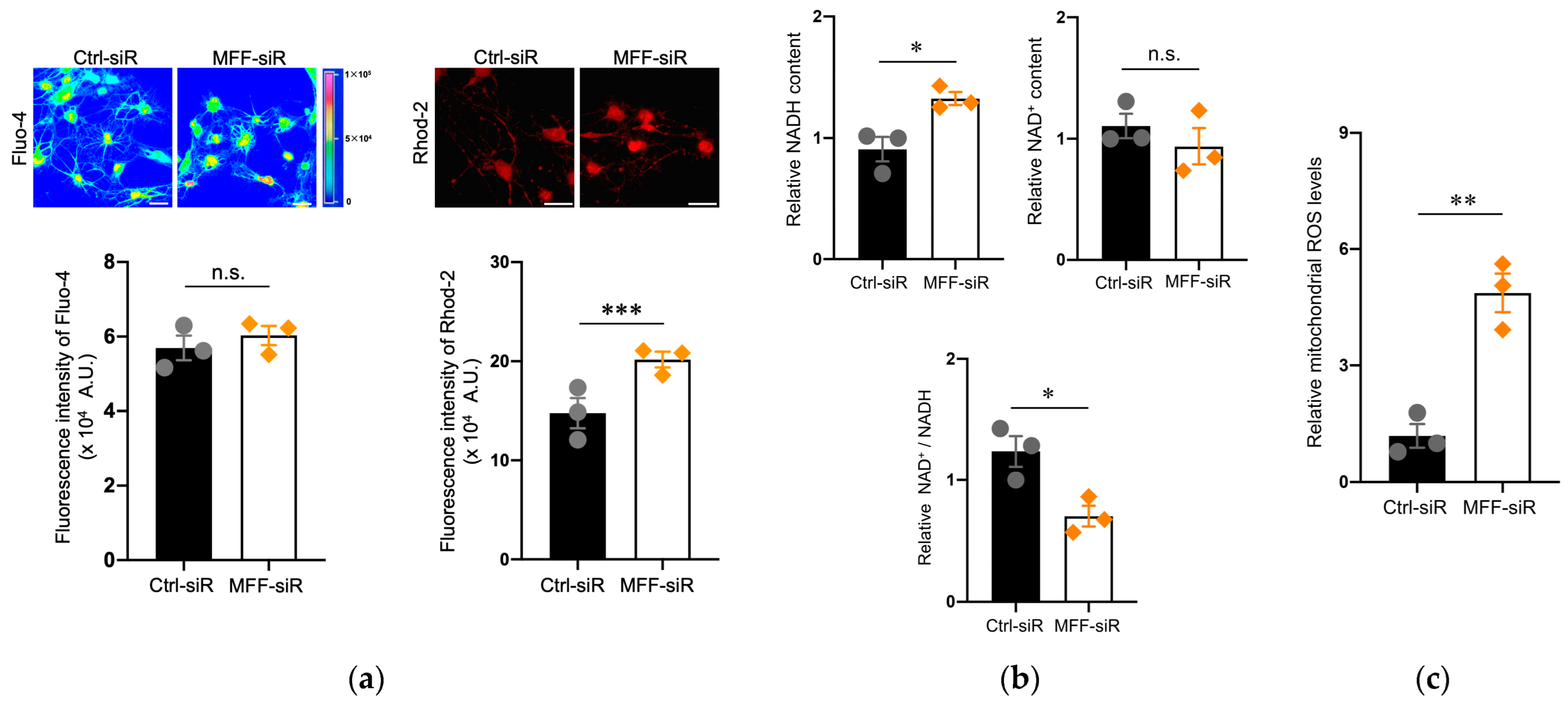
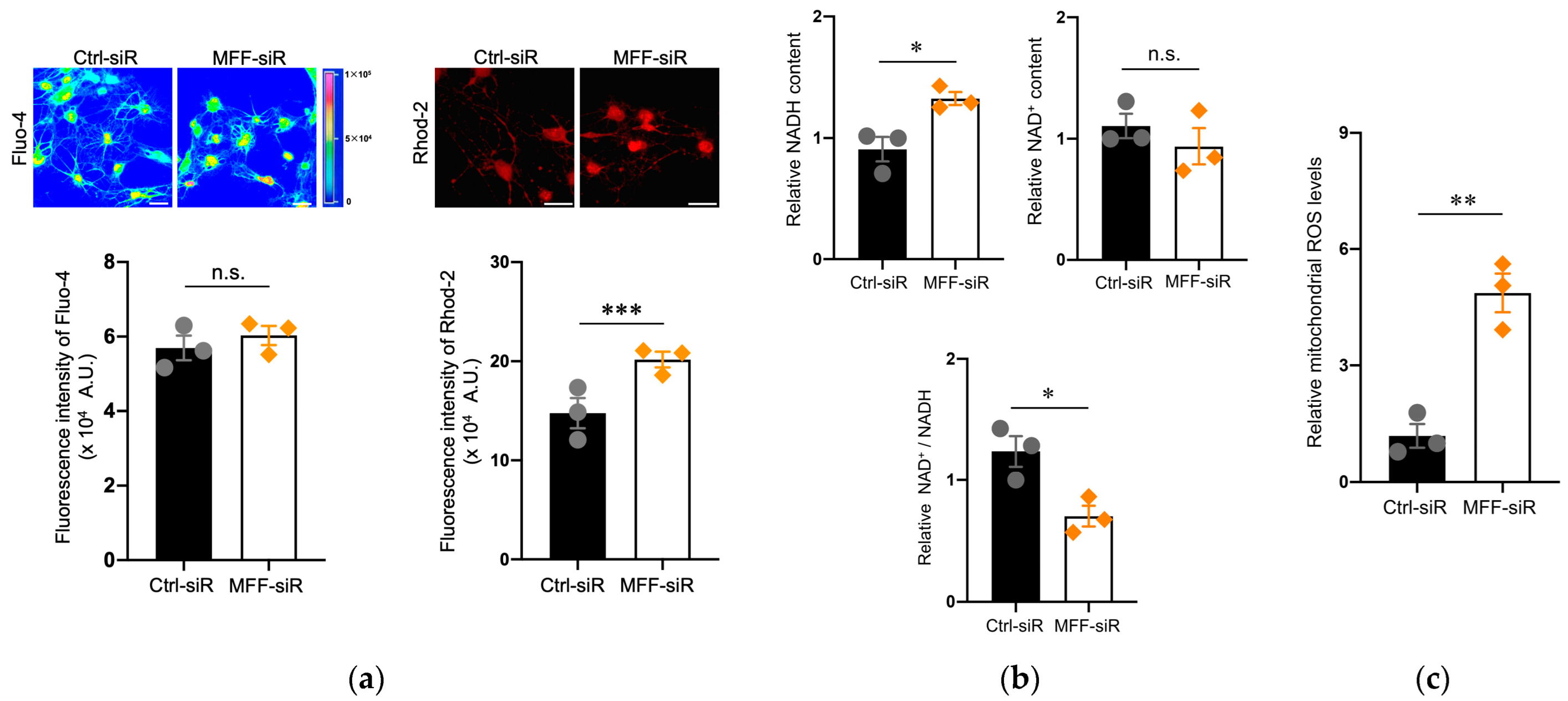
3.2. Mitochondrial Ca2+ and ROS Accumulation in Dopaminergic Neurons with MFF Insufficiency
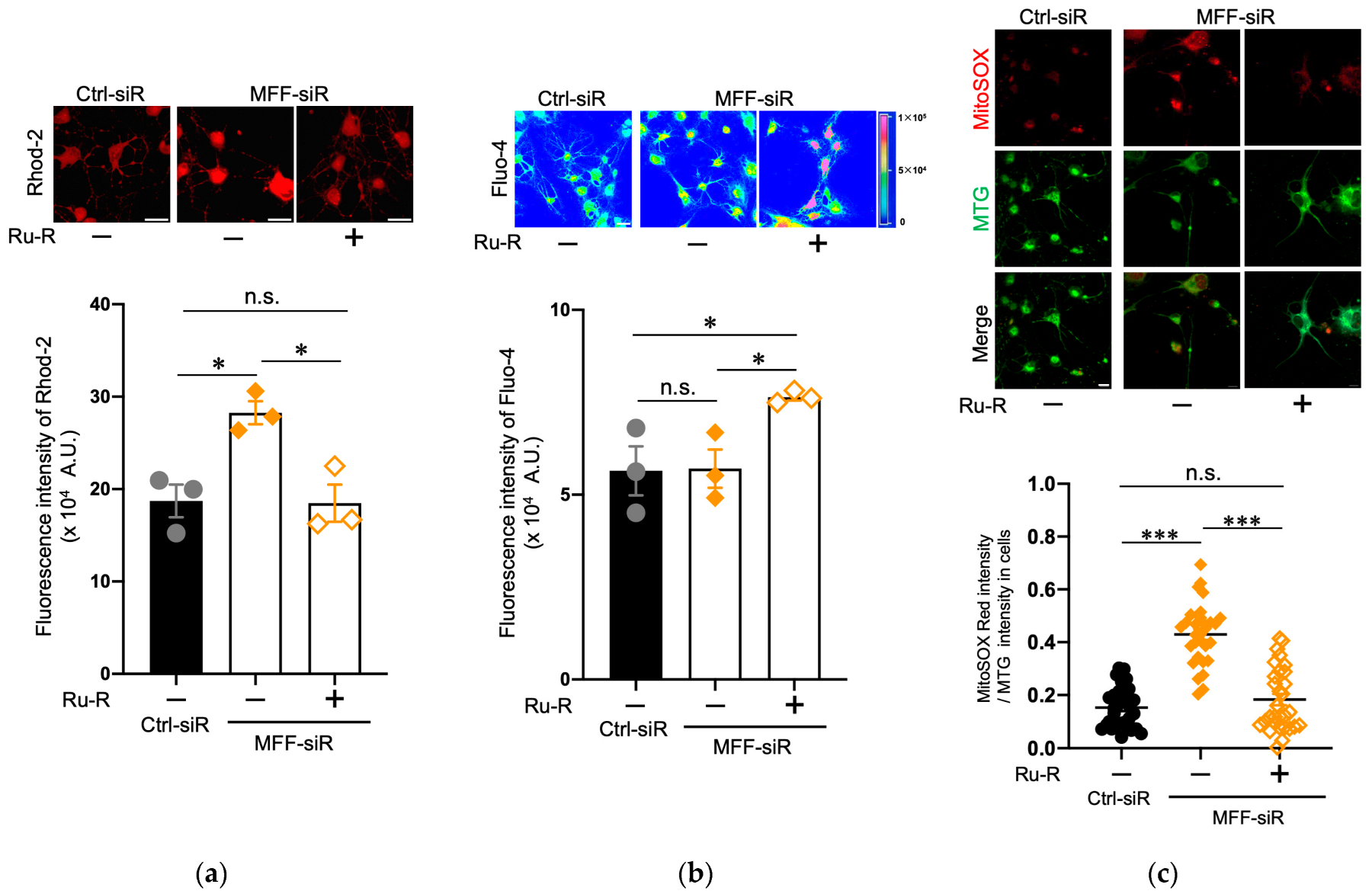
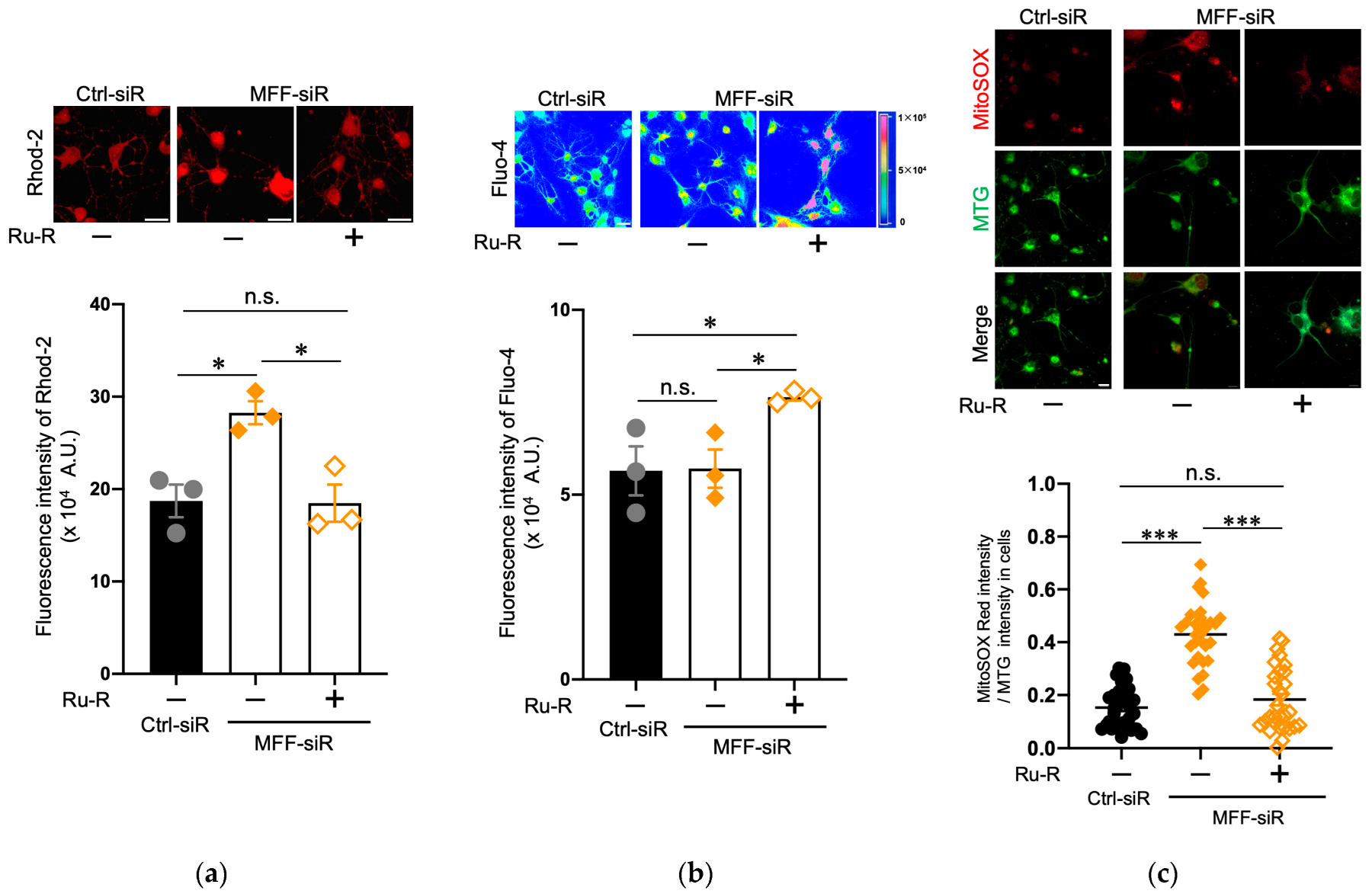
3.3. Suppression of Mitochondrial Ca2+ and ROS Levels by Blocking the Mitochondrial Calcium Uniporter Channel in Dopaminergic Neurons with MFF Insufficiency
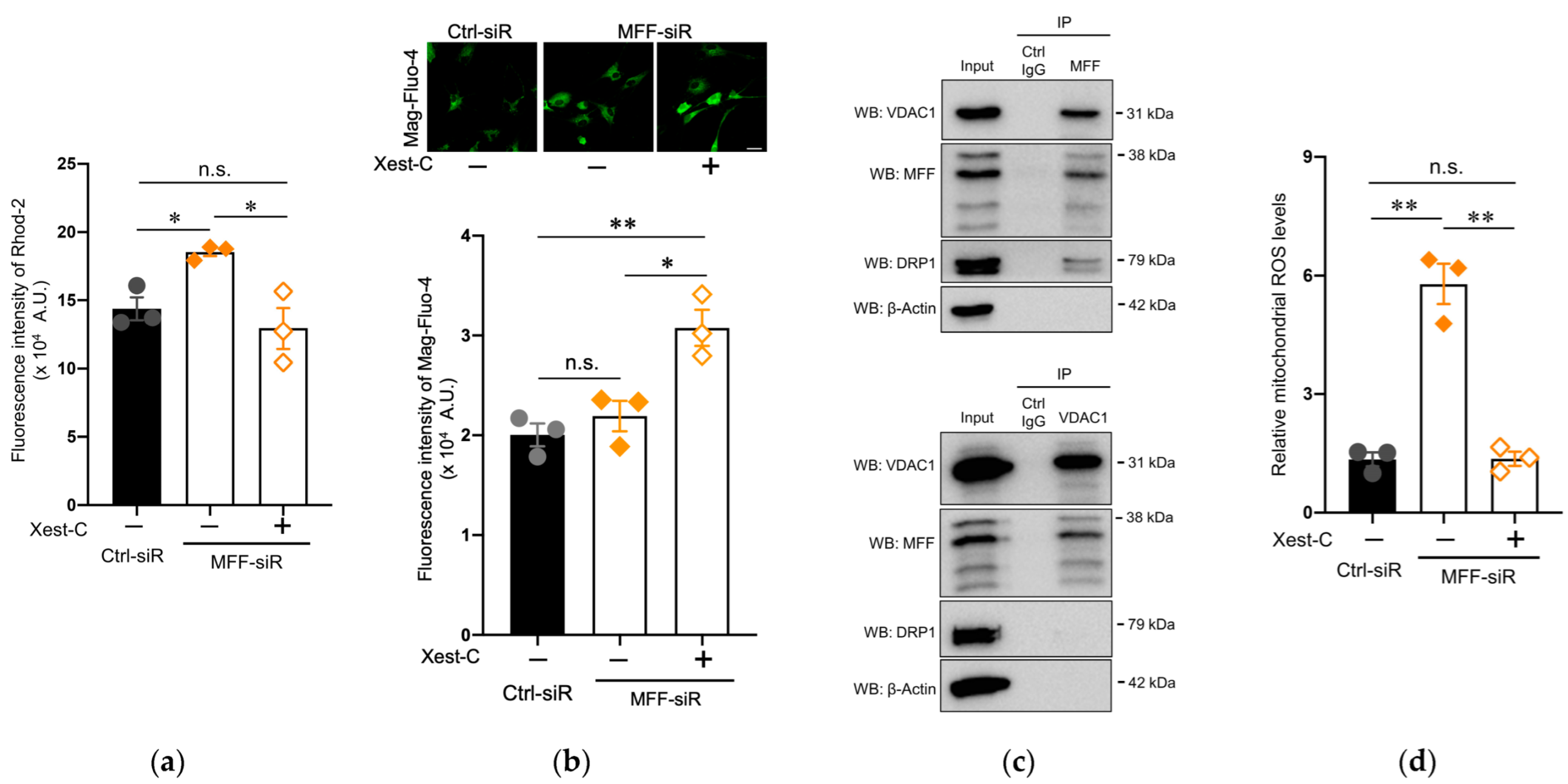
3.4. Suppression of Mitochondrial Ca2+ and ROS Levels by Blocking IP3R in Dopaminergic Neurons with MFF Insufficiency
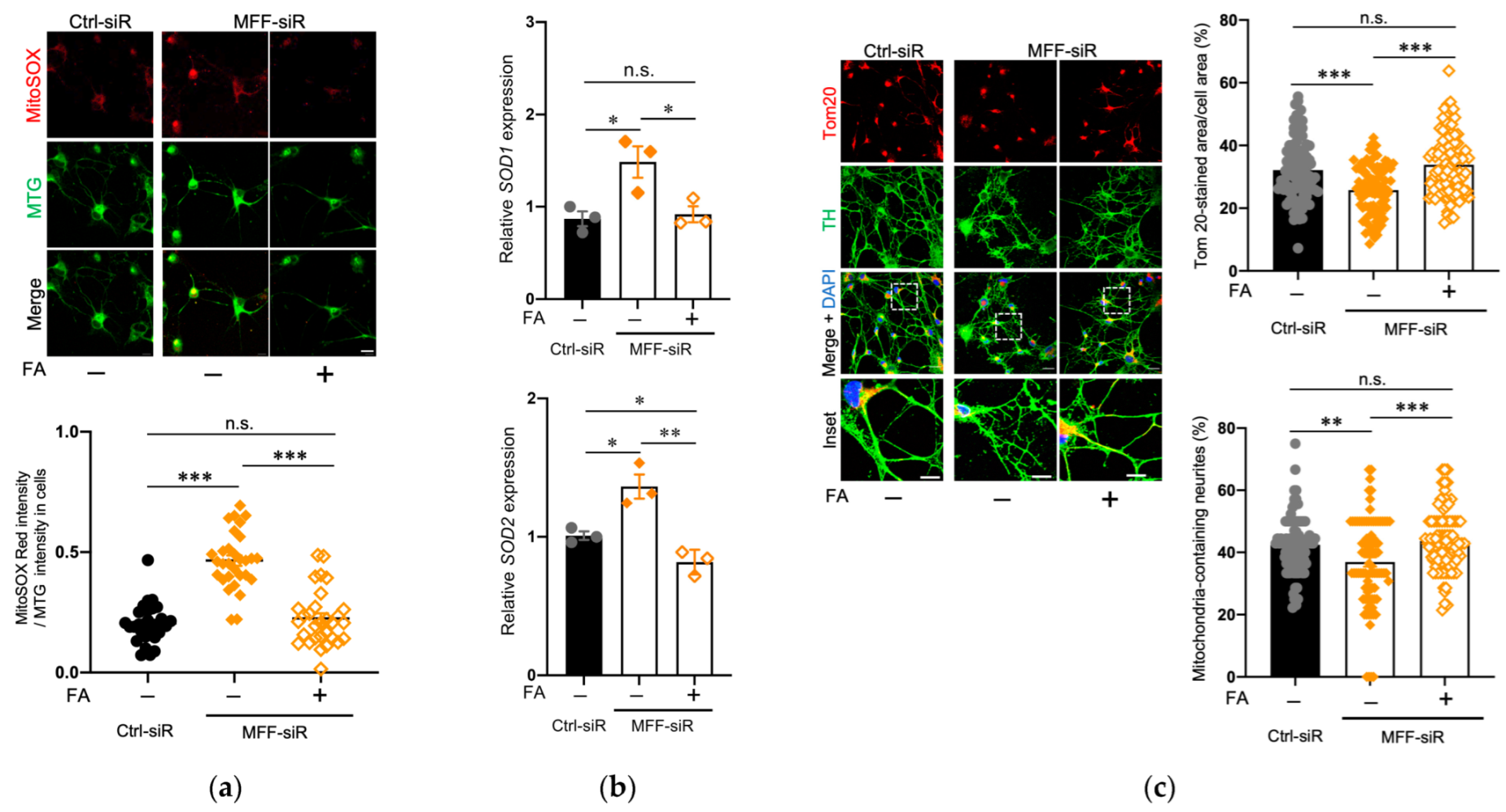
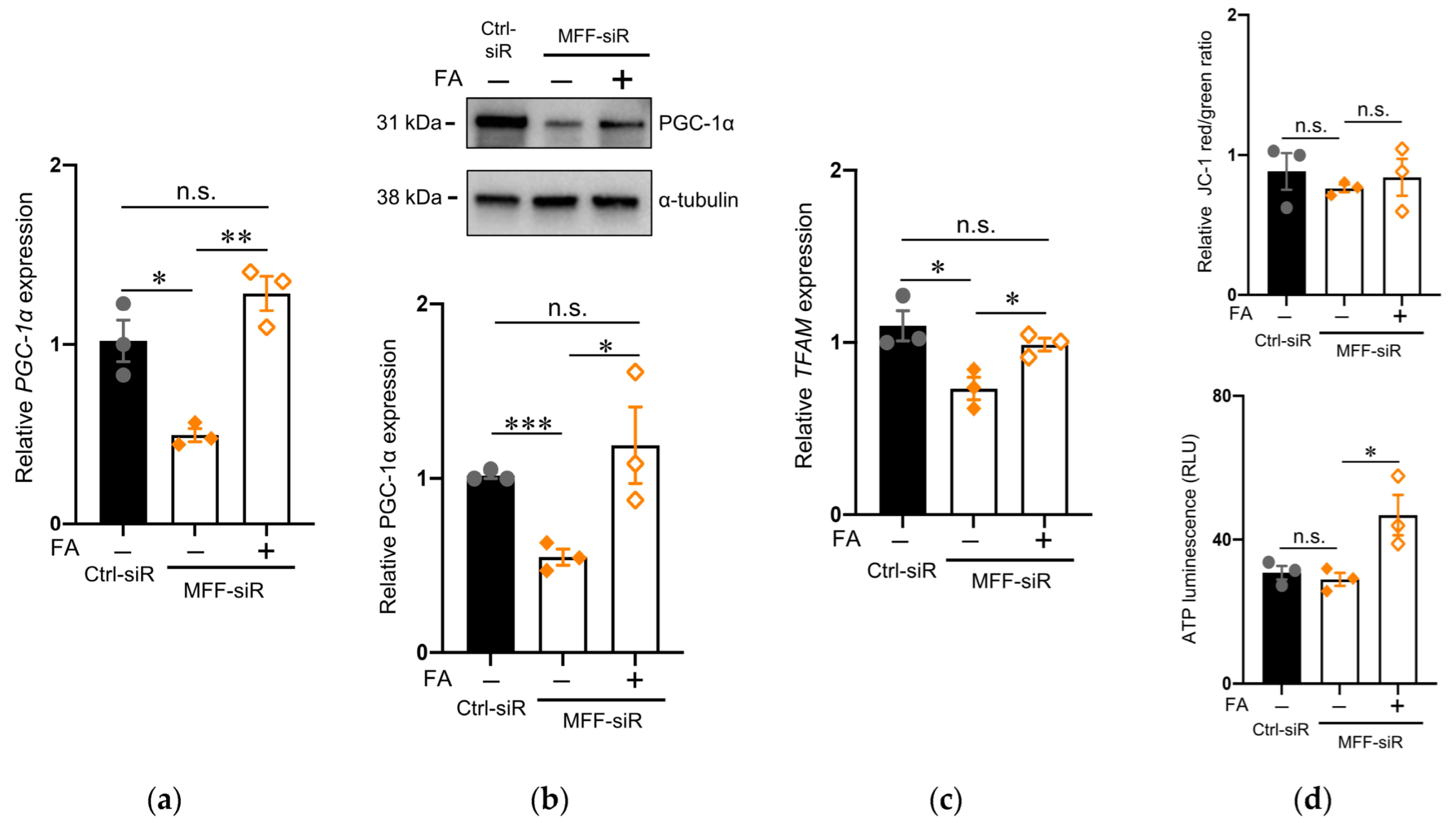
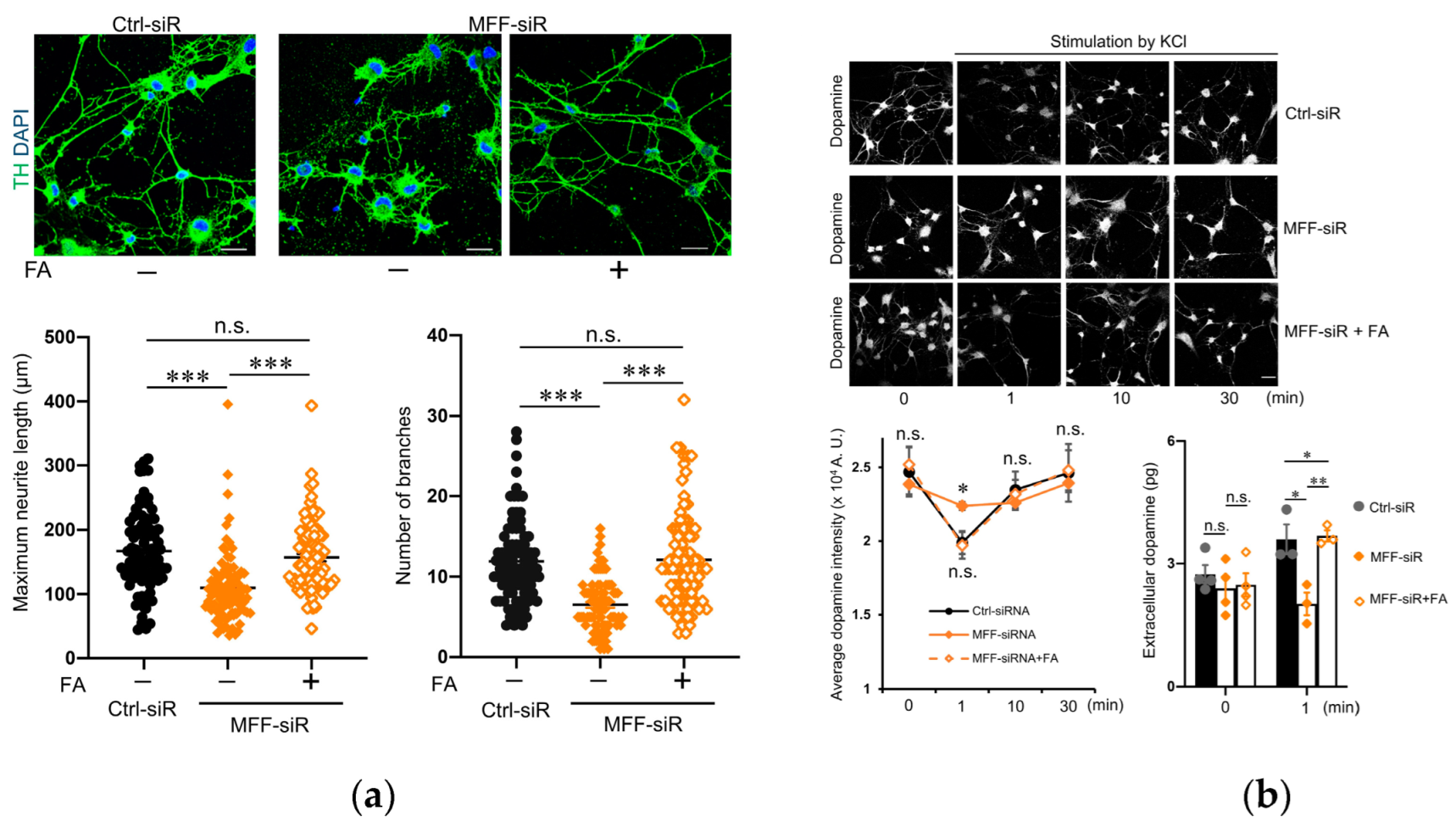
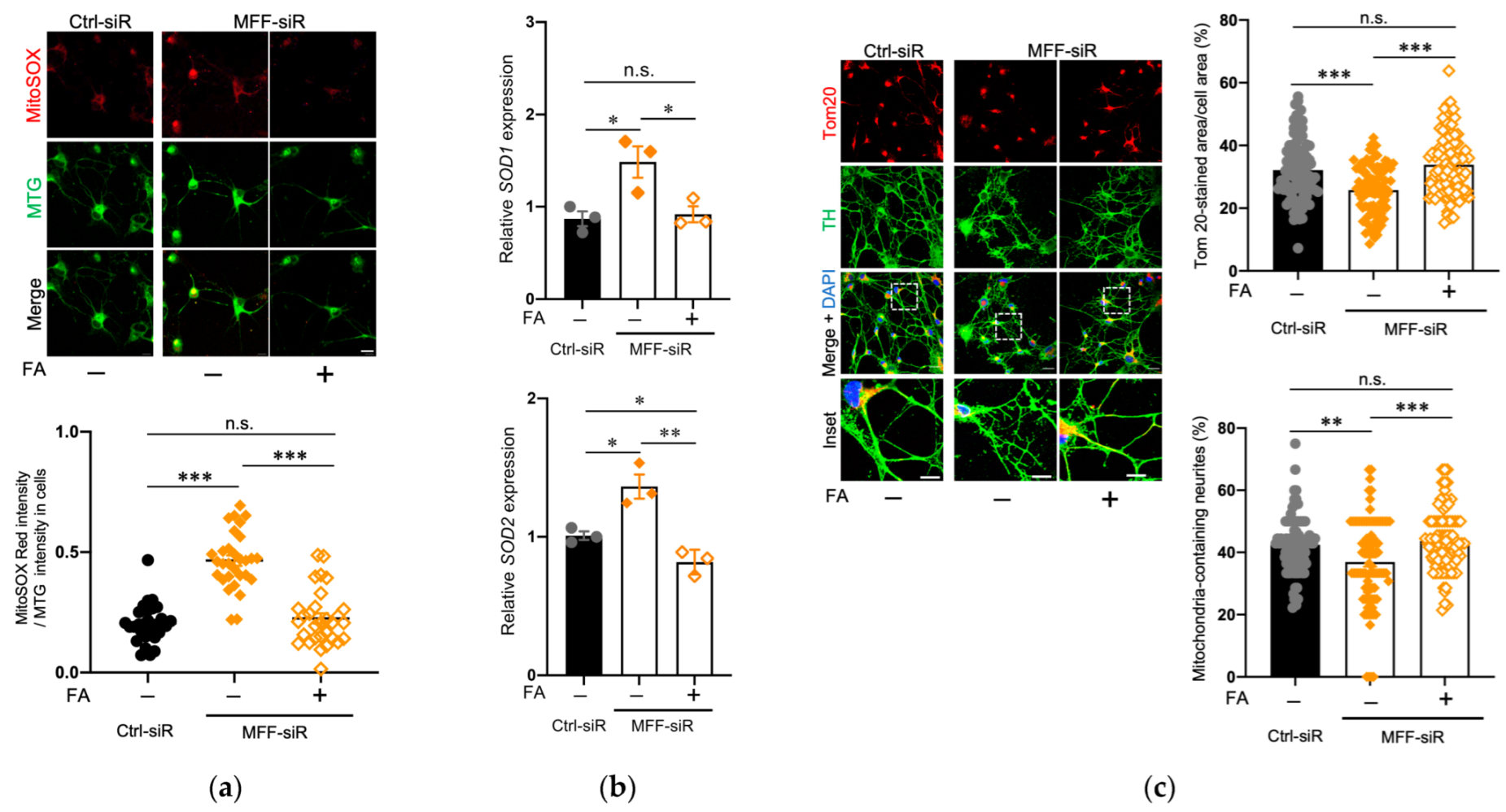
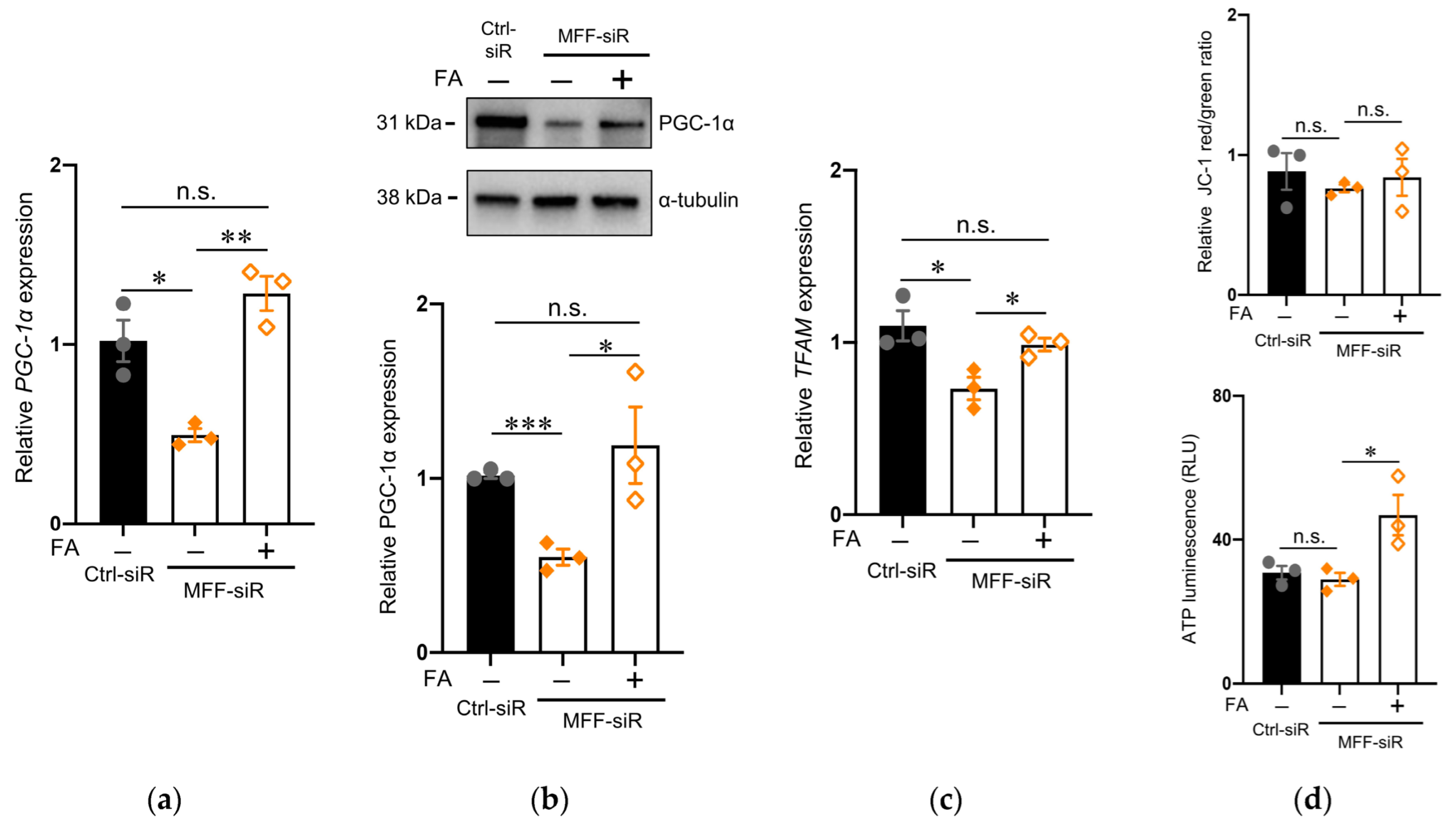
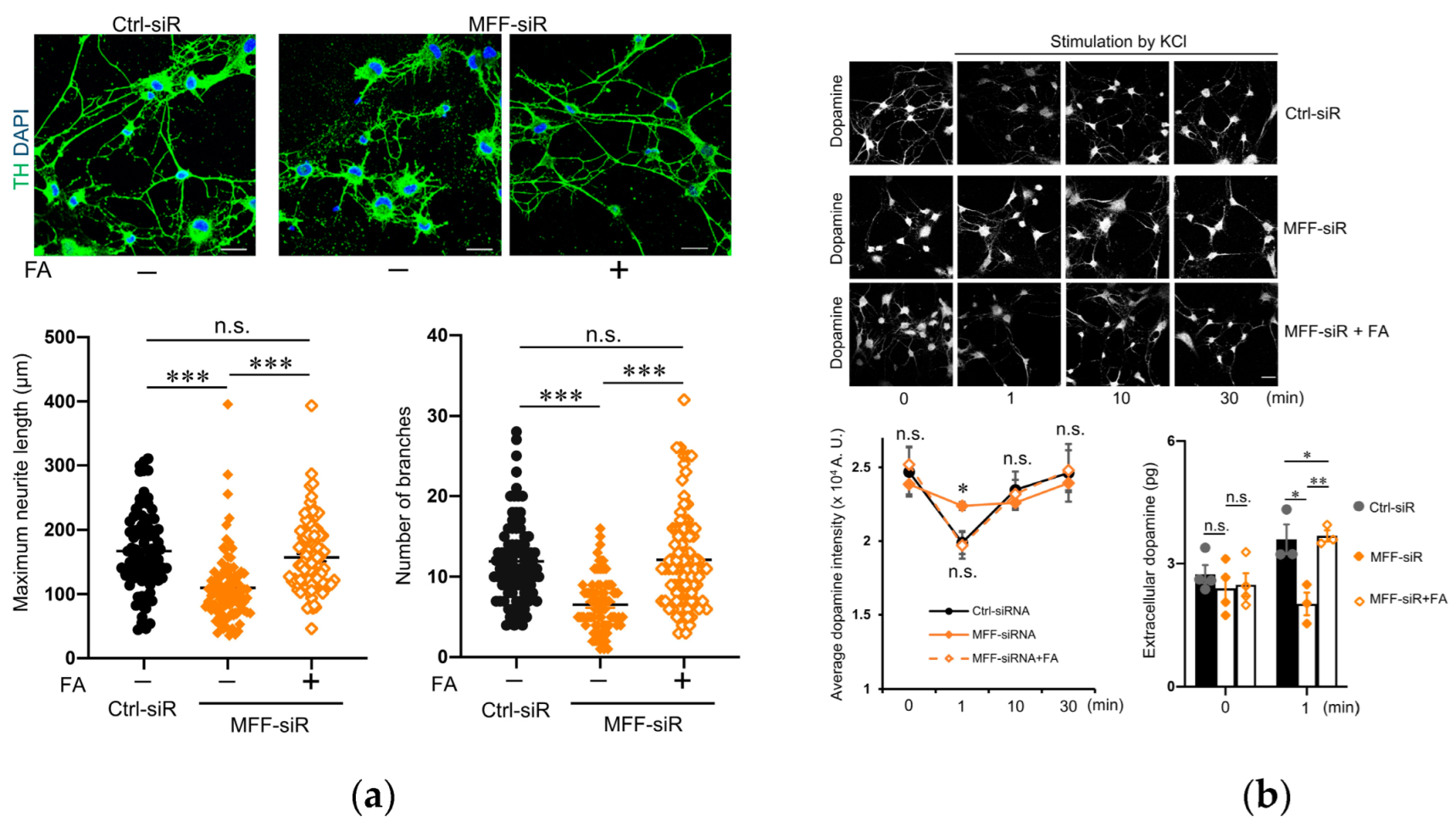
3.5. Effect of Folic Acid on Developmental Defects of Dopaminergic Neurons with MFF Insufficiency
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 12, 872884. [Google Scholar] [CrossRef]
- Adebayo, M.; Singh, S.; Singh, A.P.; Dasgupta, S. Mitochondrial fusion and fission: The fine-tune balance for cellular homeostasis. FASEB J. 2021, 6, e21620. [Google Scholar] [CrossRef]
- Brookes, R.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef]
- Archer, S.L. Mitochondrial dynamics—Mitochondrial fission and fusion in human diseases. N. Engl. J. Med. 2013, 369, 2236–2251. [Google Scholar] [CrossRef] [Green Version]
- Yapa, N.M.B.; Lisnyak, V.; Reljic, B.; Ryan, M.T. Mitochondrial dynamics in health and disease. FEBS Lett. 2021, 595, 1184–1204. [Google Scholar] [CrossRef]
- Waterham, H.R.; Koster, J.; van Roermund, C.W.T.; Mooyer, P.A.W.; Wanders, R.J.A.; Leonard, J.V. A lethal defect of mitochondrial and peroxisomal fission. N. Engl. J. Med. 2007, 356, 1736–1741. [Google Scholar] [CrossRef]
- Sheffer, R.; Douiev, L.; Edvardson, S.; Shaag, A.; Tamimi, K.; Soiferman, D.; Meiner, V.; Saada, A. Postnatal microcephaly and pain insensitivity due to a de novo heterozygous DNM1L mutation causing impaired mitochondrial fission and function. Am. J. Med. Genet. 2016, 170, 1603–1607. [Google Scholar] [CrossRef]
- Vanstone, J.R.; Smith, A.M.; McBride, S.; Naas, T.; Holcik, M.; Antoun, G.; Harper, M.-E.; Michaud, J.; Sell, E.; Chakraborty, P.; et al. DNM1L-related mitochondrial fission defect presenting as refractory epilepsy. Eur. J. Hum. Genet. 2016, 24, 1084–1088. [Google Scholar] [CrossRef] [Green Version]
- Fahrner, J.A.; Liu, R.; Perry, M.S.; Klein, J.; Chan, D.C. A novel de novo dominant negative mutation in DNM1L impairs mitochondrial fission and presents as childhood epileptic encephalopathy. Am. J. Med. Genet. A 2016, 170, 2002–2011. [Google Scholar] [CrossRef] [Green Version]
- Chao, Y.-H.; Robak, L.A.; Xia, F.; Koenig, M.K.; Adesina, A.; Bacino, C.A.; Scaglia, F.; Bellen, H.J.; Wangler, M.F. Missense variants in the middle domain of DNM1L in cases of infantile encephalopathy alter peroxisomes and mitochondria when assayed in Drosophila. Hum. Mol. Genet. 2016, 25, 1846–1856. [Google Scholar] [CrossRef] [Green Version]
- Yoon, G.; Malam, Z.; Paton, T.; Marshall, C.R.; Hyatt, E.; Ivakine, Z.; Scherer, S.W.; Lee, K.-S.; Hawkins, C.; Cohn, R.D. Lethal disorder of mitochondrial fission caused by mutations in DNM1L. J. Pediat. 2016, 171, 313–316. [Google Scholar] [CrossRef]
- Nasca, A.; Legati, A.; Baruffini, E.; Nolli, C.; Moroni, I.; Ardissone, A.; Goffrini, P.; Ghezzi, D. Biallelic mutations in DNM1L are associated with a slowly progressive infantile encephalopathy. Hum. Mutat. 2016, 37, 898–903. [Google Scholar] [CrossRef] [Green Version]
- Vandeleur, D.; Chen, C.V.; Huang, E.J.; Connolly, A.J.; Sanchez, H.; Moon-Grady, A.J. Novel and lethal case of cardiac involvement in DNM1L mitochondrial encephalopathy. Am. J. Med. Genet. A 2019, 179, 2486–2489. [Google Scholar] [CrossRef]
- Ishihara, N.; Nomura, M.; Jofuku, A.; Kato, H.; Suzuki, S.O.; Masuda, K.; Otera, H.; Nakanishi, Y.; Nonaka, I.; Goto, Y.; et al. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat. Cell Biol. 2009, 11, 958–966. [Google Scholar] [CrossRef]
- Gandre-Babbe, S.; van der Bliek, A.M. The novel tail-anchored membrane protein MFF controls mitochondrial and peroxisomal fission in mammalian cells. Mol. Biol. Cell 2008, 19, 2402–2412. [Google Scholar] [CrossRef] [Green Version]
- Wasiak, S.; Zunino, R.; McBride, H.M. Bax/Bak promote sumoylation of DRP1 and its stable association with mitochondria during apoptotic cell death. J. Cell Biol. 2007, 177, 439–450. [Google Scholar] [CrossRef] [Green Version]
- Karbowski, M.; Neutzner, A.; Youle, R.J. The mitochondrial E3 ubiquitin ligase MARCH5 is required for Drp1 dependent mitochondrial division. J. Cell Biol. 2007, 178, 71–84. [Google Scholar] [CrossRef]
- Otera, H.; Wang, C.; Cleland, M.M.; Setoguchi, K.; Yokota, S.; Youle, R.J.; Mihara, K. MFF is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 2010, 191, 1141–1158. [Google Scholar] [CrossRef] [Green Version]
- Shamseldin, H.E.; Alshammari, M.; Al-Sheddi, T.; Salih, M.A.; Alkhalidi, H.; Kentab, A.; Repetto, G.M.; Hashem, M.; Alkuraya, F.S. Genomic analysis of mitochondrial diseases in a consanguineous population reveals novel candidate disease genes. J. Med. Genet. 2012, 49, 234–241. [Google Scholar] [CrossRef]
- Koch, J.; Feichtinger, R.G.; Freisinger, P.; Pies, M.; Schrodl, F.; Iuso, A.; Sperl, W.; Mayr, J.A.; Prokisch, H.; Haack, T.B. Disturbed mitochondrial and peroxisomal dynamics due to loss of MFF causes Leigh-like encephalopathy, optic atrophy and peripheral neuropathy. J. Med. Genet. 2016, 53, 270–278. [Google Scholar] [CrossRef]
- Nasca, A.; Nardecchia, F.; Commone, A.; Semeraro, M.; Legati, A.; Garavaglia, B.; Ghezzi, D.; Leuzzi, V. Clinical and biochemical features in a patient with mitochondrial fission factor gene alteration. Front. Genet. 2018, 9, 625. [Google Scholar] [CrossRef] [Green Version]
- Panda, I.; Ahmad, I.; Sagar, S.; Zahra, S.; Shamim, U.; Sharma, S.; Faruq, M. Encephalopathy due to defective mitochondrial and peroxisomal fission 3 caused by a novel MFF gene mutation in a young child. Clin. Genet. 2020, 97, 933–937. [Google Scholar] [CrossRef]
- Lewis, T.L., Jr.; Kwon, S.K.; Lee, A.; Shaw, R.; Polleux, F. MFF-dependent mitochondrial fission regulates presynaptic release and axon branching by limiting axonal mitochondria size. Nat. Commun. 2018, 9, 5008. [Google Scholar] [CrossRef] [Green Version]
- Feissner, R.F.; Skalska, J.; Gaum, W.E.; Sheu, S.S. Crosstalk signaling between mitochondrial Ca2+ and ROS. Front. Biosci. 2009, 14, 1197–1218. [Google Scholar] [CrossRef] [Green Version]
- Baev, A.Y.; Vinokurov, A.Y.; Novikova, I.N.; Dremin, V.V.; Potapova, E.V.; Abramov, A.Y. Interaction of Mitochondrial Calcium and ROS in Neurodegeneration. Cells 2022, 11, 706. [Google Scholar] [CrossRef]
- Gronthos, S.; Mankani, M.; Brahim, J.; Robey, P.G.; Shi, S. Postnatal human dental pulp stem cells (DPSCs) in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2000, 97, 13625–13630. [Google Scholar] [CrossRef] [Green Version]
- Miura, M.; Gronthos, S.; Zhao, M.; Lu, B.; Fisher, L.W.; Robey, P.G.; Shi, S. SHED: Stem cells from human exfoliated deciduous teeth. Proc. Natl. Acad. Sci. USA 2003, 100, 5807–5812. [Google Scholar] [CrossRef] [Green Version]
- Fujii, H.; Matsubara, K.; Sakai, K.; Ito, M.; Ohno, K.; Ueda, M.; Yamamoto, A. Dopaminergic differentiation of stem cells from human deciduous teeth and their therapeutic benefits for Parkinsonian rats. Brain Res. 2015, 1613, 59–72. [Google Scholar] [CrossRef]
- Kanafi, M.; Majumdar, D.; Bhonde, R.; Gupta, P.; Datta, I. Midbrain cues dictate differentiation of human dental pulp stem cells towards functional dopaminergic neurons. J. Cell Physiol. 2014, 229, 1369–1377. [Google Scholar] [CrossRef]
- Majumdar, D.; Kanafi, M.; Bhonde, R.; Gupta, P.; Datta, I. Differential neuronal plasticity of dental pulp stem cells from exfoliated deciduous and permanent teeth towards dopaminergic neurons. J. Cell Physiol. 2016, 231, 2048–2063. [Google Scholar] [CrossRef]
- Masuda, K.; Han, X.; Kato, H.; Sato, H.; Zhang, Y.; Sun, X.; Hirofuji, Y.; Yamaza, H.; Yamada, A.; Fukumoto, S. Dental pulp-derived mesenchymal stem cells for modeling genetic disorders. Int. J. Mol. Sci. 2021, 22, 2269. [Google Scholar] [CrossRef]
- Kato, H.; Pham, T.M.T.; Yamaza, H.; Masuda, K.; Hirofuji, Y.; Han, X.; Sato, H.; Taguchi, T.; Nonaka, K. Mitochondria regulate the differentiation of stem cells from human exfoliated deciduous teeth. Cell Struct. Funct. 2017, 42, 105–116. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Kato, H.; Sato, H.; Han, X.; Hirofuji, Y.; Kato, A.; Sakai, Y.; Ohga, S.; Fukumoto, S.; Masuda, K. Dopamine-related oxidative stress and mitochondrial dysfunction in dopaminergic neurons differentiated from deciduous teeth-derived stem cells of children with Down syndrome. FASEB BioAdv. 2022, 4, 454–467. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Sun, X.; Kato, H.; Sato, H.; Torio, M.; Han, X.; Zhang, Y.; Hirofuji, Y.; Kato, A.; Sakai, Y.; Ohga, S.; et al. Impaired neurite development and mitochondrial dysfunction associated with calcium accumulation in dopaminergic neurons differentiated from the dental pulp stem cells of a patient with metatropic dysplasia. Biochem. Biophys. Rep. 2021, 26, 100968. [Google Scholar] [CrossRef]
- Kageyama, Y.; Zhang, Z.; Roda, R.; Fukaya, M.; Wakabayashi, J.; Wakabayashi, N.; Kensler, T.W.; Reddy, P.H.; Iijima, M.; Sesaki, H. Mitochondrial division ensures the survival of postmitotic neurons by suppressing oxidative damage. J. Cell Biol. 2012, 197, 535–551. [Google Scholar] [CrossRef] [Green Version]
- Rossi, A.; Pizzo, P.; Filadi, R. Calcium, mitochondria and cell metabolism: A functional triangle in bioenergetics. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1068–1078. [Google Scholar] [CrossRef]
- Garbincius, J.F.; Elrod, J.W. Mitochondrial calcium exchange in physiology and disease. Physiol. Rev. 2022, 102, 893–992. [Google Scholar] [CrossRef]
- Márta, K.; Hasan, P.; Rodríguez-Prados, M.; Paillard, M.; Hajnóczky, G. Pharmacological inhibition of the mitochondrial Ca2+ uniporter: Relevance for pathophysiology and human therapy. J. Mol. Cell Cardiol. 2021, 151, 135–144. [Google Scholar] [CrossRef]
- Rizzuto, R.; Brini, M.; Murgia, M.; Pozzan, T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science 1993, 262, 744–747. [Google Scholar] [CrossRef]
- Gafni, J.; Munsch, J.A.; Lam, T.H.; Catlin, M.C.; Costa, L.G.; Molinski, T.F.; Pessah, I.N. Xestospongins: Potent membrane permeable blockers of the inositol 1,4,5-trisphosphate receptor. Neuron 1997, 19, 723–733. [Google Scholar] [CrossRef] [Green Version]
- Joshi, R.; Adhikari, S.; Patro, B.S.; Chattopadhyay, S.; Mukherjee, T. Free radical scavenging behavior of folic acid: Evidence for possible antioxidant activity. Free Radic. Biol. Med. 2001, 30, 1390–1399. [Google Scholar] [CrossRef]
- Mentch, S.J.; Locasale, J.W. One-carbon metabolism and epigenetics: Understanding the specificity. Ann. N. Y. Acad. Sci. 2016, 1363, 91–98. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Kato, H.; Sato, H.; Yamaza, H.; Hirofuji, Y.; Han, X.; Masuda, K.; Nonaka, K. Folic acid-mediated mitochondrial activation for protection against oxidative stress in human dental pulp stem cells derived from deciduous teeth. Biochem. Biophys. Res. Commun. 2019, 508, 850–856. [Google Scholar] [CrossRef]
- Lee, S.; Min, K.T. The Interface between ER and Mitochondria: Molecular Compositions and Functions. Mol. Cells 2018, 41, 1000–1007. [Google Scholar]
- Szabadkai, G.; Bianchi, K.; Varnai, P.; de Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, T.; Yamada, A.; Watanabe, M.; Yoshimura, Y.; Yamazaki, N.; Yoshimura, Y.; Yamauchi, T.; Kataoka, M.; Nagata, T.; Terada, H.; et al. VDAC1, having a shorter N-terminus than VDAC2 but showing the same migration in an SDS-polyacrylamide gel, is the predominant form expressed in mitochondria of various tissues. J. Proteome Res. 2006, 5, 3336–3344. [Google Scholar] [CrossRef]
- Seo, J.H.; Chae, Y.C.; Kossenkov, A.V.; Lee, Y.G.; Tang, H.Y.; Agarwal, E.; Gabrilovich, D.I.; Languino, L.R.; Speicher, D.W.; Shastrula, P.K.; et al. MFF Regulation of Mitochondrial Cell Death Is a Therapeutic Target in Cancer. Cancer Res. 2019, 79, 6215–6226. [Google Scholar] [CrossRef] [Green Version]
- Hansford, R.G. Physiological role of mitochondrial Ca2+ transport. J. Bioenerg. Biomembr. 1994, 26, 495–508. [Google Scholar] [CrossRef]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552 Pt 2, 335–344. [Google Scholar] [CrossRef]
- Palty, R.; Silverman, W.F.; Hershfinkel, M.; Caporale, T.; Sensi, S.L.; Parnis, J.; Nolte, C.; Fishman, D.; Shoshan-Barmatz, V.; Herrmann, S.; et al. NCLX is an essential component of mitochondrial Na+/Ca2+; exchange. Proc. Natl. Acad. Sci. USA 2010, 107, 436–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardi, P.; Azzone, G.F. Regulation of Ca2+ efflux in rat liver mitochondria. Role of membrane potential. Eur. J. Biochem. 1983, 134, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Thirupathi, A.; de Souza, C.T. Multi-regulatory network of ROS: The interconnection of ROS, PGC-1 alpha, and AMPK-SIRT1 during exercise. J. Physiol. Biochem. 2017, 73, 487–494. [Google Scholar] [CrossRef]
- Bouchez, C.; Devin, A. Mitochondrial Biogenesis and Mitochondrial Reactive Oxygen Species (ROS): A Complex Relationship Regulated by the cAMP/PKA Signaling Pathway. Cells 2019, 8, 287. [Google Scholar] [CrossRef] [Green Version]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jäger, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef] [Green Version]
- Garnier, A.; Fortin, D.; Deloménie, C.; Momken, I.; Veksler, V.; Ventura-Clapier, R. Depressed mitochondrial transcription factors and oxidative capacity in rat failing cardiac and skeletal muscles. J. Physiol. 2003, 551, 491–501. [Google Scholar] [CrossRef]
- Javadov, S.; Purdham, D.M.; Zeidan, A.; Karmazyn, M. NHE-1 inhibition improves cardiac mitochondrial function through regulation of mitochondrial biogenesis during postinfarction remodeling. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H1722–H1730. [Google Scholar] [CrossRef]
- Wang, B.; Sun, J.; Ma, Y.; Wu, G.; Tian, Y.; Shi, Y.; Le, G. Resveratrol preserves mitochondrial function, stimulates mitochondrial biogenesis, and attenuates oxidative stress in regulatory T cells of mice fed a high-fat diet. J. Food Sci. 2014, 79, H1823–H1831. [Google Scholar] [CrossRef]
- Baldelli, S.; Aquilano, K.; Ciriolo, M.R. PGC-1α buffers ROS-mediated removal of mitochondria during myogenesis. Cell Death Dis. 2014, 5, e1515. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.C.; Li, L.C.; Chen, J.B.; Chang, H.W. Indoxyl sulfate-induced oxidative stress, mitochondrial dysfunction, and impaired biogenesis are partly protected by vitamin C and N-acetylcysteine. Sci. World J. 2015, 2015, 620826. [Google Scholar] [CrossRef] [Green Version]
- Takeichi, Y.; Miyazawa, T.; Sakamoto, S.; Hanada, Y.; Wang, L.; Gotoh, K.; Uchida, K.; Katsuhara, S.; Sakamoto, R.; Ishihara, T.; et al. Non-alcoholic fatty liver disease in mice with hepatocyte-specific deletion of mitochondrial fission factor. Diabetologia 2021, 64, 2092–2107. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Accession Number | Sequence (5′-3′) | |
|---|---|---|---|
| 18S | X03205.1 | Forward | CGGCTACCACATCCAAGGAA |
| Reverse | GCTGGAATTACCGCGGCT | ||
| SOD1 | NM_000454.5 | Forward | GGTGGGCCAAAGGATGAAGAG |
| Reverse | CCACAAGCCAAACGACTTCC | ||
| SOD2 | NM_000636.4 | Forward | AAACCTCAGCCCTAACGGTG |
| Reverse | GCCTGTTGTTCCTTGCAGTG | ||
| PGC-1α | NM_001330751.2 | Forward | GGCAGAAGGCAATTGAAGAG |
| Reverse | TCAAAACGGTCCCTCAGTTC | ||
| TFAM | NM_001270782.2 | Forward | GATGCTTATAGGGCGGAGTGG |
| Reverse | GCTGAACGAGGTCTTTTTGGT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, X.; Dong, S.; Kato, H.; Kong, J.; Ito, Y.; Hirofuji, Y.; Sato, H.; Kato, T.A.; Sakai, Y.; Ohga, S.; et al. Mitochondrial Calcium-Triggered Oxidative Stress and Developmental Defects in Dopaminergic Neurons Differentiated from Deciduous Teeth-Derived Dental Pulp Stem Cells with MFF Insufficiency. Antioxidants 2022, 11, 1361. https://doi.org/10.3390/antiox11071361
Sun X, Dong S, Kato H, Kong J, Ito Y, Hirofuji Y, Sato H, Kato TA, Sakai Y, Ohga S, et al. Mitochondrial Calcium-Triggered Oxidative Stress and Developmental Defects in Dopaminergic Neurons Differentiated from Deciduous Teeth-Derived Dental Pulp Stem Cells with MFF Insufficiency. Antioxidants. 2022; 11(7):1361. https://doi.org/10.3390/antiox11071361
Chicago/Turabian StyleSun, Xiao, Shuangshan Dong, Hiroki Kato, Jun Kong, Yosuke Ito, Yuta Hirofuji, Hiroshi Sato, Takahiro A. Kato, Yasunari Sakai, Shouichi Ohga, and et al. 2022. "Mitochondrial Calcium-Triggered Oxidative Stress and Developmental Defects in Dopaminergic Neurons Differentiated from Deciduous Teeth-Derived Dental Pulp Stem Cells with MFF Insufficiency" Antioxidants 11, no. 7: 1361. https://doi.org/10.3390/antiox11071361
APA StyleSun, X., Dong, S., Kato, H., Kong, J., Ito, Y., Hirofuji, Y., Sato, H., Kato, T. A., Sakai, Y., Ohga, S., Fukumoto, S., & Masuda, K. (2022). Mitochondrial Calcium-Triggered Oxidative Stress and Developmental Defects in Dopaminergic Neurons Differentiated from Deciduous Teeth-Derived Dental Pulp Stem Cells with MFF Insufficiency. Antioxidants, 11(7), 1361. https://doi.org/10.3390/antiox11071361