Abstract
The respiratory epithelium can be affected by many diseases that could be treated using aerosol gene therapy. Among these, cystic fibrosis (CF) is a lethal inherited disease characterized by airways complications, which determine the life expectancy and the effectiveness of aerosolized treatments. Beside evaluations performed under in vivo settings, cell culture models mimicking in vivo pathophysiological conditions can provide complementary insights into the potential of gene transfer strategies. Such models must consider multiple parameters, following the rationale that proper gene transfer evaluations depend on whether they are performed under experimental conditions close to pathophysiological settings. In addition, the mucus layer, which covers the epithelial cells, constitutes a physical barrier for gene delivery, especially in diseases such as CF. Artificial mucus models featuring physical and biological properties similar to CF mucus allow determining the ability of gene transfer systems to effectively reach the underlying epithelium. In this review, we describe mucus and cellular models relevant for CF aerosol gene therapy, with a particular emphasis on mucus rheology. We strongly believe that combining multiple pathophysiological features in single complex cell culture models could help bridge the gaps between in vitro and in vivo settings, as well as viral and non-viral gene delivery strategies.
1. Introduction
1.1. Cystic Fibrosis and Current Protein Treatment
Cystic fibrosis (CF) is one of the most common inherited genetic disorders, affecting over 70,000 people worldwide [1]. CF is induced by numerous mutations listed in a single gene encoding the cystic fibrosis transmembrane conductance regulator (CFTR) [2]. The CFTR protein is a channel that participates in the exit of chloride ions from the cytoplasm. This channel also plays a key role in the regulation of another channel called ENaC (epithelial sodium channel), involved in the reabsorption of sodium. Altogether, those channels control the correct hydration of mucus on airway epithelial cells. Alteration or dysfunction of the CFTR channel leads thus to abnormal ion transport and mucus dehydration (Figure 1A). Such dehydrated sticky mucus is responsible for the diminution of mucociliary clearance (MCC), leading to a mucosal accumulation. This mucus stagnation provides a propitious environment for bacterial colonization and infections, inducing related chronic inflammation. In the CF context, polynuclear neutrophils accumulation in the mucus is responsible for the production and the excretion of lysosomal enzymes which directly contribute to tissue remodeling [3,4]. The remodeling of secretory cells induces an overproduction of mucin, the main constituent of mucus, leading to severe consequences for the respiratory exchanges [5].
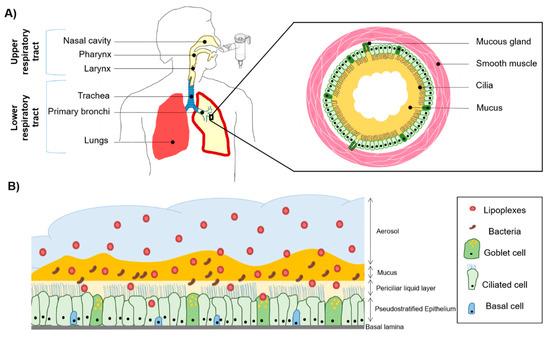
Figure 1.
Schematic representation of the respiratory system and the use of an aerosol to target the pulmonary epithelium. (A) The upper and lower respiratory tract with an emphasis on a bronchial section constituted by mucous glands and smooth muscles. In the case of cystic fibrosis (CF), sticky mucus is overproduced and creates a bronchial plug. (B) Pseudostratified airway epithelium composed of goblet, ciliated, and basal cells. The mucosa is covered with airways surface liquid (ASL) which contains two layers, a periciliary liquid layer directly in contact with the cilia and mucus layer. CF mucus is colonized with opportunistic bacteria such as Staphylococcus aureus and Pseudomonas aeruginosa. These bacteria form a biofilm participating in the viscous and dense aspect of the CF mucus. When nanoparticles (NPs) such as lipoplexes are aerosolized, they have to cross the mucus barrier before reaching the epithelial cells to deliver therapeutic compounds such as acids nucleic construction.
Today, pulmonary complications are the primary cause of CF-related morbidity and mortality. Similar manifestations exist in the gastrointestinal and genital tracts but are, at present, dealt with by symptomatic treatments such as supplementary pancreatic enzymes [6]. During the last decade, new drugs have been developed regarding the CFTR mutations. Ivacaftor (Kalydeco®), a CFTR potentiator channel, is indicated for the G551D mutation. This potentiator can be associated with correctors such as lumacaftor (Orkambi®) or tezacaftor (Symdeko®) for the homozygote F508del mutation. More recently, a tritherapy, namely, elexacaftor-tezacaftor-ivacaftor (Trikafta®), showed an FEV1 (forced expiratory volume) higher than 10% benefit for patients with a single F508del allele [7]. Unfortunately, these molecules are mutation-dependent and are sometimes responsible for several side effects (such as hepatitis, abdominal pain) [8]. Even though pharmacology using CFTR potentiators is a real hope for most CF patients, rare mutations cannot currently be treated with those kinds of molecules.
1.2. Gene Therapy in CF Disease
Gene therapy may be a relevant approach to treat CF patients whatever their genotype. This technique was primarily used during the first trials of gene therapy in 1989–1990, which were coordinated by the NIH Clinical Center in Bethesda (Maryland) for adenosine deaminase (ADA) deficiency [9]. Since then, this technology kept on developing and became more and more efficient. The principle of gene transfer is based on the introduction of nucleic acids sequences. The plasmid, pDNA, containing a transgene encoding for the gene of interest, can be inserted into targeted cells through different systems. Viral vectors were the first to be used and have already proven efficient. In 2017, gene therapy was proved to be efficient in treating patients with hemophilia A and B using viral AAV vectors, providing a breakthrough to cure these monogenetic diseases [10,11]. The last gene therapy approved by the FDA and the EMA is Zolgensma (Novartis) for the treatment of spinal muscular atrophy [12]. However, viral vectors present some disadvantages. For instance, their immunogenicity limits the possibility of re-administration and they are difficult to produce and to purify. On the other hand, non-viral vectors are based on the use of chemical compounds, mostly lipids or cationic polymers, which directly cross the cytoplasmic membrane of the cells [13,14]. Compared to the viral strategy, the non-viral approach can easily be re-administrated thanks to the absence of an immunogenic reaction and to their good tolerance [15]. This re-administration is mandatory because of the constant renewal of epithelium pulmonary cells leading to the decrease in and eventually the disappearance of transgene expression. Moreover, non-viral vectors have the ability to compact longer nucleic acid sequences, which is useful for transgenes such as CFTR. However, the in vivo efficiency of non-viral vectors is still too low to get closer to the normal phenotype significantly, even if a change in the clinical state of the patient can be observed [15]. These strategies still have to be improved.
Given its characteristics, CF is a particularly complex and challenging model to approach pulmonary diseases and is a helpful model for other pathologies affecting the lungs and the respiratory tract. Successfully developing a gene therapy treatment for CF can potentially open the door to the treatment of other lung disorders. To directly target the lung, inhaled drugs can be administrated under different forms: pressurized metered-dose inhalers, nebulizers, soft mist inhalers, nasal sprays, and dry powders for inhalation (Figure 1A) [16,17]. The choice of devices has some consequences and influences the lung deposition [18]. This type of administration is a non-invasive method performed locally as drugs can easily reach the large absorptive surface area of the epithelium. Moreover, this technique has the benefit of avoiding the first-pass metabolism, intestinal efflux transporters, and degradation in the digestive system [19,20]. Deposition of an aerosol is dependent on the physical properties of generated micro-droplets. The droplet size determines the delivery location in pulmonary airways: the smaller the size, the deeper the deposition. The anatomy of the upper and lower airways and the breathing frequency are also key parameters that influence aerosol deposition [21,22]. Nebulization represents the process of transforming a liquid into an aerosol which can reach the respiratory tract (Figure 1B). This technique has the advantage of not requiring galenic formulation to a dry powder, which could damage the quality of the product. Qiu et al. showed that the lyophilization technique did not impact the in vitro transfection efficiency but, when tested on mice models using tracheal administration, nebulization of a reconstituted formulation was more efficient than the dry powder [23].
1.3. Current Challenges and Models
However, currently, over 25 clinical trials of gene delivery based on viral or non-viral gene vectors have failed to demonstrate significant clinical benefits, mainly due to inefficient gene transfer into the target cells [24]. Additionally, some viral CF gene therapy trials elicited host immune response. Consequently, clinical assays have been discontinuous, making subsequent treatments ineffective. Moreover, lifetime repeated treatment is required when using non-integrative systems for CF patients because of the transient nature of episomal transgene expression in addition to the natural life expectancy of the airways epithelial cells [25]. In 2015, the UK CF Gene Therapy Consortium completed a CF gene therapy clinical trial and observed that the use of a non-viral gene vector (GL67A, Genzyme corp.) had a significant, but still modest, benefit on the respiratory function (+3.7% of FEV1) compared to placebo controls [15]. They showed that a single aerosol per month, for one year, of a solution composed of nanoparticles (NPs) containing 13 mg of plasmid was very well tolerated. However, a more efficient gene delivery vector is needed to really make gene therapy a realistic option for the treatment of CF patients [15].
Extracellular barriers, indeed, have to be taken into account to adapt and finely tune their formulations (Figure 1B). Due to the presence of mucus as a protective barrier for xenobiotics, the passage through the cell is considerably reduced. The great majority of non-viral vectors as well as viral ones are stuck in the thick mucus of CF patients [26]. Inhaled gene vectors can be trapped within the mucus layer via non-covalent bonds (electrostatic and hydrophobic interactions) or simply cannot enter the network structure [26]. Many clinical studies have failed to demonstrate a gene therapy efficiency in the CF lung because the mucus was partially or totally neglected in in vivo and in vitro models. For instance, GL67A was selected using healthy sheep, and therefore without taking the mucus into account [27]. Moreover, animal models currently used for gene transfection studies are not totally satisfactory for CF model systems. For instance, CF mice and rats do not present spontaneous lung infections as well as mucus production [28]. W. K. O’Neal et al. successfully developed a transgenic mice model with overexpression of the ENaC β-subunit [29]. This model has the advantage of spontaneously exhibiting a CF-like lung disease more similar to human pathology with airways inflammation, bacterial infection, mucus dehydration, and obstruction [30,31]. Due to their anatomy, however, larger animal models represent better models to test inhaled gene therapy because of closer physiological and pathological similarities to human lungs. CFTR-knockout ferrets can be used as a potential model due to a lung structure close to that of humans. They exhibit altered chloride transport, leading to mucus hypersecretion and lung infections [32,33]. CF pigs also represent a potential model, as they present pulmonary manifestations similar to those found in human CF lung (infection, inflammation, airway remodeling, and mucus hypersecretion). They have, however, a short life expectancy (some days to few weeks) [34,35]. Nonetheless, every animal experimentation has to respect the 3R regulation (Reduce, Replace, and Refine). Difficulties in accessing animal models coupled with 3R restriction lead to the need for a more accurate cellular model. If the models used in vitro or in vivo are not sufficiently complex and representative of clinical conditions, the results obtained will not be reproducible during clinical trials. Therefore, it appears to be essential to have a reliable in vitro model close enough to the pathology studied to test and develop new vectorization strategies for nucleic acids with the ultimate aim of gene therapy. This review will focus on the use of mucus models and cellular cultures to obtain results closer to the in vivo pulmonary tracts and to avoid, as far as possible, failures in clinical development. Such models are fundamental to allow the development of non-viral gene carrier formulations able to overcome cellular and extracellular barriers faced in the respiratory tract.
2. Mucus Model for Gene Therapy
2.1. Mucus Composition and Biological Fuctions
2.1.1. Airway Mucus
As previously mentioned, epithelial pulmonary cells are covered with airways surface liquid (ASL). The thickness of ASL has been estimated to be between 10 and 20 µm [36]. Two layers form this surface liquid: the periciliary layer (PCL) and the mucus layer. The PCL is the deepest film that covers the epithelial cilia and facilitates the beating. This layer is less viscous than the upper coat forming the mucus layer. Mucus is a hydrogel that can be found on the surface of wet epithelia such as the lungs but also the stomach, eyes, and vagina. It has a key role in protecting epithelial cells. Mucus is a dynamic semipermeable barrier that enables the exchange of nutrients, water, gases, odorants, and hormones, but retains pathogens and particles through its network structure [37]. Mucus is constitutively excreted by secretory cells in the airway and removed by MCC. Mucus combined with the mucociliary escalator of the upper airways allows the trapped particles to be moved back up to the airway and mainly swallowed into the esophagus [38]. The clearance of inhaled matter generally occurs in 15 min to 2 h and is dependent on several factors (anatomy, size and deposition of particles, tobacco exposure, and pulmonary disease) [39]. Along the lower airways, in the alveolar region, a surfactant film is present and participates in the oxygenation function. Pulmonary surfactant is a surface-active complex composed of lipids and proteins. This fluid prevents alveolar collapse at low lung volume. It also preserves bronchiolar patency during respiration while protecting against injuries, inhaled particles, and micro-organisms [40].
2.1.2. Mucins
In a healthy context, mucus is constantly maintained in a state of hydration thanks to the coordination of CFTR and ENaC channels. Mucus is mainly composed of 95% water, but one of the main constituents playing a key role in this physical barrier is the structural glycoprotein mucin (size 0.5–50 MDa) [41]. Mucin acts as a net that traps molecules which cannot cross the pores, whose size lies between 10 and 500 nm [41,42]. Two major classes of mucins exist: mucins that are secreted and mucins attached to the cytoplasmic membrane (MUC1, MUC3A, MUC3B, MUC4, MUC12, MUC13, MUC15, MUC16, MUC17, and MUC20) [43]. Membrane-tethered mucins possess transmembrane and cytoplasmic domains. Some of them also present an epidermal growth factor-like domain [44]. Among the secreted mucins, five oligomeric gel-forming mucins (MUC2, MUC5AC, MUC5B, MUC6, and MUC19) and two non-polymeric mucins (MUC7 and MUC8) form the structure of the protein backbone [43]. Secreted mucins are stored within secretory vesicles and, constitutively or in response to a stress, are released to protect the airway epithelium. The airway mucus is mainly composed of MUC5AC released from goblet cells and MUC5B produced by the mucous cells in submucosal glands [43]. MUC2, also present in the respiratory mucus, is found at the bronchus level at a low concentration [45]. Mucin secretion is mediated by different factors such as pathogenic agents and inflammatory mediators by increasing mucin gene expression [5,46]. Secreted or not, mucins share some common structures. The mucin central region is composed of a high content of serine, threonine, and proline residues (PTS domain), leading to a dense glycosylation (Figure 2) [47]. The glycosylated portion represents 70 to 80% of the total mass of mucin and is composed of carbohydrates such as N-acetylglucosamine, N-acetylgalactosamine, fucose, galactose, N-acetylneuraminic acid (sialic acid), and traces of mannose and sulfate [37,48]. Altogether, they form oligosaccharides linked by O-glycosylation that are responsible for the bottle-brush structure and for the global negative charge of mucin [47]. However, PTS domains are interrupted by cysteine residues playing a key role in the mucin network [49]. Moreover, amino-terminal and carboxy-terminal regions are rich in cysteine and create intradomain disulfide bonds (Figure 2). N- and C-terminals of the protein backbone also have some similarities with the sequence of von Willebrand factors C and D and are involved in polymerization of the mucin network [50]. In addition to disulfide bonds, hydrophobic and electrostatic interactions also contribute to the formation of a 3D network responsible for the gel-type characteristics of mucins [51].
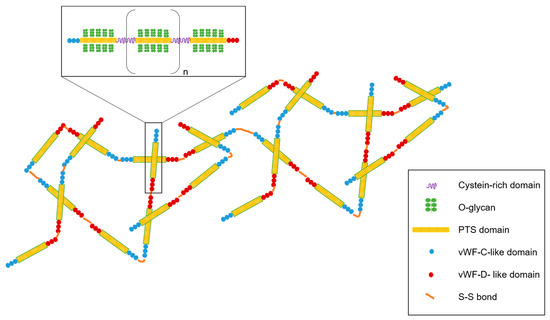
Figure 2.
General structure of mucins, the main constituent of mucus. Mucin contains the PTS (proline, threonine, and serine) domain and supporting O-glycan. The PTS domain is interspaced with cysteine-rich domains. Mucin glycopolymer ends with the von Willebrand factor (vWF) domain in N- and C-terminal regions. Mucin polymerized via disulfide bonds and mucin polymers interact with each other via hydrophobic and electrostatic interactions.
2.2. Mucus Physical Properties
2.2.1. Mucus Rheology
Rheology is the science of deformation and flow of matter. Rheological techniques are often used to investigate the relationships between the behavior of complex fluids and their multi-scale structure [52]. Since pioneer works in the 1960s, the rheological properties of mucus, which is a natural complex fluid, have attracted much attention [53]. Using classical macrorheology, the effect of mucus viscoelastic behavior on mucus transport properties was highlighted [54,55]. From a therapeutic aspect, the better understanding of the role played by rheology in mucus transport allowed improving mucus drainage [56,57]. More recently, the interest in using microrheology in order to investigate local viscoelastic properties of mucus was underlined [58]. Indeed, microrheology is more interesting to investigate the viscoelasticity that will encounter microparticles or nanoparticles. This technique can appreciate the heterogeneity inside the mucus samples and represents a necessary complement to macrorheology by characterizing local mechanical properties in biological fluids [58]. At last, there are numerous recent studies concerning the search for rheological properties as characteristic markers of respiratory diseases [59]. From that point of view, characteristics of the rheological response in a non-linear regime (large deformation or flow regime) seem to be better biomarkers than the viscoelastic response in the linear regime [60].
2.2.2. Factors Influencing Mucus Rheology
Mucus is mainly composed of water and mucins. However, other molecules such as immunoglobulins, lipids, DNA, electrolytes, inorganic salts, enzymes, cells, cellular debris, and mucopolysaccharides take part in the mucus composition [61]. Taken together, the normal mucus gel barrier constituents create a heterogeneous environment and directly influence the Newtonian viscosity, which is 1000 to 10,000 times higher than that of water [41]. In diseases such as CF and chronic obstructive pulmonary disease (COPD), MUC5B is found to be predominant, whereas in healthy patients, MUC5AC is the main mucin [62]. These unbalanced situations lead to different rheological properties. Mucus exhibits viscoelastic behaviors which correlate with the hydration ratio and mucus composition. Moreover, vitamin A (acid retinoic) has also been shown to increase mucin gene expression [63,64]. Furthermore, in the CF and COPD context, some changes in glycosylation may occur, leading to an increase in sulfatation of mucins and contributing to phenotype degradation [65]. In these diseases, mucins are overproduced and the transport properties of nanoparticles onto the epithelial cells are very limited, and, instead of protection, mucus provides a propitious environment for bacteria development. The emblematic pathogen infection in the CF patient is Pseudomonas aeruginosa (PA). PA is a Gram-negative opportunistic bacterium with flagella, which is capable of biofilm formation in the airway respiratory tract. When PA aggregates, it loses its mobility and adopts anaerobic respiration, avoiding macrophage destruction [66,67]. Moreover, this bacterium can be hooked to the epithelial via adhesion or to the mucus layer thanks to its flagella that have the ability to bind to MUC1 [68]. Bacterial infections lead to chronic inflammation and pulmonary damage. Infection and the inflammatory environment have also been shown to change mucin glycosylation. In chronic respiratory diseases, mucin production increases due to inflammation which recruits and activates T cells and macrophages, but also neutrophils and eosinophils [5]. For example, Delmotte et al. pointed out the fact that the cytokine tumor necrosis factor (TNFα) has the faculty to alternate sialylation in tracheal cells [69]. Some studies have demonstrated that neutrophil elastase and interleukin-9 increased the expression of MUC5AC in epithelial pulmonary cells [44,70]. Pro-inflammatory cytokines such TNFα, IL-6, and IL-8 directly influence the rheological properties of mucus. Moreover, in the CF airway, a high level of DNA and actin filaments is observed due to the necrotic neutrophils present in this pathology. They contribute to the dense network structure of the mucus. For instance, DNA makes up about 0.02% of mucus mass and is originated from debris of epithelial cells [71]. However, in CF patients, there is an accumulation of DNA, which increases both the elasticity and viscosity of the mucus [72]. This DNA results from the destruction of neutrophils and its mass ratio in mucus can rise up to 1.5% [73]. A commonly used mucolytic for CF sputa is rhDNAse (Pulmozyme®) that directly targets and hydrolyzes the extracellular DNA. Lipids are also present in the mucus, with a mass ratio up to 1–2% [71]. In healthy mucus, pH is around 7; however, in CF, because of the bacterial development, pH is around 5 [74]. This variation also influences the physical properties of the mucus barrier and its viscosity and impedes the nanoparticle diffusion. Lipids are also mainly found in hydrophobic domains of mucin and play a crucial role in the mucus viscosity [75]. For instance, phosphatidylethanolamine, sphingomyelin, and lysophosphatidyl-choline increase the viscosity, whereas phosphatidylglycerol decreases the CF mucus viscosity [76,77]. Finally, mineral salts present in mucus influence the ionic strength and consequently the structural and rheological properties of mucus.
2.3. Mucus Sources
To determine the capacity of gene vectors to overcome the mucus barrier, mucus models used in vitro should possess physical and biological properties similar to those of native mucus (Table 1). Purified mucin is commercially available, but in 2010, Crater and Carrier showed that gel reconstituted from purified gastric mucin (PGM, Sigma-Aldrich) did not represent an ideal model [78]. They concluded that this model was not accurate to study particle diffusion into the mucus. Moreover, using rheological techniques, Kočevar-Nared et al. compared crude gastric mucin and natural gastric mucus and pointed out the fact that they had different rheological behaviors, due to purified mucins which did not form a proper network [51]. The extraction protocol for mucin isolation perturbs the interaction involved in the supramolecular organization. When disulfide bonds are disrupted by chemical reduction, the gel collapses. Mucin purification alters native interactions but also interactions between the other mucosal compounds. This disruptive condition leads to a poor rheological model for studying the physical behavior of the airway mucus. More recently, Meldrum et al. showed that the mucin gel assembly is formed by a synergistic interaction between mucin, but also non-mucin proteins, and Ca2+ [79]. Artificial models have also been used to study nanovector interaction. D. Sriramulu and coworkers developed an artificial sputum medium (ASM) to mimic the sputum of CF patients [80]. To study microparticles of mannitol containing ciprofloxacin, Yang et al. used this ASM model [81]. These microparticles were proved to be able to penetrate artificial mucus, but this ability remains to be demonstrated on human mucus. Freshly collected mucus should ideally be used for such studies. The mucus origin and the species are also important. Ideally, human mucus would be used. The physical and biological properties of gastric mucus are different from vaginal mucus as well as airways mucus. For instance, under a non-pathological condition, the pH of gastric and vaginal mucus is acidic, whereas pulmonary mucus is neutral [47]. Human airway samples can be collected by an endotracheal tube, brushed or directly collected from patients’ expectoration. To really assess the mucus properties and determine if nanoparticles are capable of crossing over, the storage of samples has to be specific and carefully controlled. When the mucus is kept at 37 °C up to 60 min, proteases will degrade mucins. Frozen mucus (between −20 and −80 °C), due to the reduction in protease activities, retains its bulk viscoelastic properties. However, when sputum is kept at 4 °C for more than 48 h, the diffusion of NPs decreases [26].

Table 1.
Example of different sources of mucus and their oscillatory shear viscoelastic properties. ω, angular frequency (rad/s); f, frequency (Hz); G’, storage modulus (Pa); G’’, loss modulus (Pa); η*, complex viscosity.
2.4. Crossing the Mucus Barrier
Gene vectors that have been tested for the CF treatment in clinical trials have not been proved to efficiently penetrate the mucus barrier, even for viral vectors [90]. Non-viral vectors, mostly cationic, are also unable to pass through the mucus network mesh due to their size and their electrostatic interactions. Cationic gene vectors aggregate in the presence of albumin that is abundant in the airway secretions [91]. Aggregation can also be observed with the DNA and mucin glycoprotein present in the mucus, especially in CF patients. Moreover, the size of the nanoparticles is primordial for the penetration into the mucus even in healthy donors [92]. They have to be small enough to get through the mesh of the network structure. Airway mucus pores are about 500 nm large; however, for CF mucus, the pore size is smaller, approximatively 150 nm, meaning that NPs also have to be smaller [93]. Murgia et al. also showed that the penetration of nanoparticles into porcine respiratory mucus after aerosol deposition is size-limited [82]. After aerosol deposition on mucus, NPs of 200 nm and larger could not penetrate the mucus. However, a small size is not enough to ensure the rapid transport of NPs in mucus. Nanoparticles (NPs) surface chemistry represents an important factor influencing the capacity to overcome the mucus barrier. There are different strategies to break through the mucus barrier. Uncharged NPs are needed to avoid electrostatic interactions with the negatively charged mucins. For example, the use of poly-ethylene-glycol (PEG) covalently attached on the NPs surface has been shown to reduce the electrostatic interactions between the positively charged nanoparticles and the negatively charged mucus [94]. Suk et al., in 2008, showed that non-adhesive NPs (PEG-PS) of 100 nm were more hindered in CF sputum than 200 nm PEG-PS, but less hindered than 500 nm PEG-PS [42]. The authors explained the results by the insufficient PEG coating of 100 nm PEG-PS, thus underlying the importance of the chemistry surface. PEG coating techniques also need to be considered. NPs densely coated with linear PEG have been shown to efficiently pass the mucus barrier. Xu et al. demonstrated that PEG with a low density (mushroom conformation) did not sufficiently cross mucus and interacted with mucin. On the other hand, PEG with a high density (dense brush conformation) largely diffuses in mucus [95]. Mansfield et al. tried to replace PEG with poly(2-oxazoline) (POZ) and reported that silica NPs with POZ enhanced diffusion into mucin [96]. Another option to coat NPs is the use of triblock copolymers of poly(propylene oxide) and poly(ethylene oxide) (PEO-PPO-PEO) [97]. In this case, the triblock copolymers, namely, Pluronics, were physically adsorbed on the surface. They showed that Pluronic F127, which is the most commonly used Pluronic, improved the transport of polymeric particles. It was also shown that the hydrophobic part, PPO, can be associated with the hydrophobic NPs surface, whereas PEO avoids muco-adhesion. Instead of fixed Pluronic F127 in the NP surface, Ensign et al. pretreated cervico-vaginal mucus with it [98]. They reported that F127 improved the distribution, retention, and efficiency of nanoparticles. The use of the HPMA (N-(2-hydroxypropyl) methacrylamide) copolymer was also explored as an alternative to PEG. In parallel, Shan et al. designed insulin-loaded NPs coated with HPMA that allowed penetration of the mucus and released insulin inside epithelial cells [99]. Alternative strategies focus on the mucus mesh perturbation by using mucolytic agents. Two main mucolytic approaches exist: the use of disulfide-breaking molecules and the use of proteolytic enzymes. In each case, the mucus structure is disrupted, enabling the penetration of nanoparticles. Dithiothreithol (DTT) is commonly known to cut disulfide bridges but is also cytotoxic. As a consequence, it cannot be used in clinical applications but remains a valuable control in vitro. Another agent that can be used to reduce disulfide interactions is N-acetyl cysteine (NAC), which has one sulfhydryl group that allows decreasing the mucus viscosity. Suk et al. proved that the pretreatment of CF sputum with NAC enhanced rapid transport of muco-nanoparticles [100]. Based on PEGylated nanoparticles, they showed that the average size of the sputum mesh increased from 145 ± 50 to 230 ± 50 nm when pretreated with NAC. Developing nanoparticles releasing mucolytic agents therefore represents a good strategy to allow nanoparticles to find their way inside the mucus structure. Proteolytic enzymes, such as trypsin, papain, and bromelain, were immobilized on nanoparticle polymers to study their ability to digest the glycoprotein network and disulfide bridges [101]. The authors used this particle on intestinal mucus and noticed that such nanoparticles could penetrate the mucus.
3. Cellular Models for Gene Transfer Evaluation
3.1. Respiratory Tract
The use of relevant in vitro models is essential to mimic the in vivo situation and properly evaluate the efficiency of innovative treatments (Table 2). In fact, cellular models can be a valuable alternative to in vivo efficiency studies, as they can be highly reproducible. The airway pulmonary tract constitutes a major organ due to its function and represents the location where gas exchanges occur in mammals. This system can be roughly subdivided into two parts: the upper and the lower respiratory tracts (Figure 1A). The upper part is composed of nasal cavities, the pharynx, and the larynx, while the lower tract includes trachea, bronchi, bronchioles, alveolar ducts, and alveolar sacs [102]. The proximal part, from nasal cavities to the second or the third segmentations of bronchioles, is a cartilaginous airway, while the distal airway is non-cartilaginous, corresponding to the terminal and respiratory bronchioles [103]. Different types of cells, constituting the pulmonary pseudostratified epithelial, can be found in the pulmonary tract. All these cells play a direct or indirect role in the oxygenation function. Between 6 and 12 L of air are inhaled every minute and constantly expose the lungs to microorganisms, allergens, and particles (sized nm to µm). To prevent such aggression, the lungs have to possess different barriers to contain any kind of particle. The integrity of the epithelium combined with the ASL presence is essential to prevent potential pathogens from penetrating the bloodstream [104].
Table 2.
Examples of commonly used immortalized airway epithelial cells and primary cell models.
3.2. Airway Epithelium
The airway epithelium is mainly composed of ciliated and secretory cells contributing to the protection and humidification of the pulmonary mucosa. The epithelium of the respiratory mucosa is a physical barrier maintained by junctions between cells. These junctions are composed of occluding tight junctions (TJ), anchoring adherent junctions, desmosomes, and Gap junctions (Figure 3) [105]. TJ formation appears during the polarization of epithelial cells. This process is led by the sorting and the distribution of certain protein and lipid membranes which organize the cell in three surface domains (apical, basal, lateral) [106]. TJ complexes are made by occluding, claudins, and junction adhesion molecules (JAM). A TJ complex consists of a TJ part and an adherence junction mainly composed of E-cadherin [107,108]. The selection of cell type to model the airway epithelium requires, in fact, the formation of functional TJ by cells in culture when cultured in air–liquid interphase (ALI) conditions (see ALI and TEER measurement paragraph) [109]. These properties can be estimated through the measurement of the transepithelial electrical resistance value (TEER expressed in Ω·cm2). This technique allows the determination of the integrity of TJ in culture of epithelial cells through the use of electrodes [110]. For instance, tracheal and bronchial epithelial cells culture can be used as a model due to the cells’ ability to differentiate and to provide a permeability barrier. The pulmonary epithelium is also a polarized pseudostratified epithelium which is composed of three major kinds of cells with distinct properties. Among them, ciliated cells represent over 50% of all epithelial cells [111]. In the upper airway, the predominant cells are ciliated columnar cells interspersed with goblet cells, while cuboidal ciliated cells interspersed with club cells are present in the lower airway [5]. In the epithelial airway, there is approximately one secretory cell for five ciliated cells; this ratio increases in diseases such COPD, asthma, or CF [5,104].
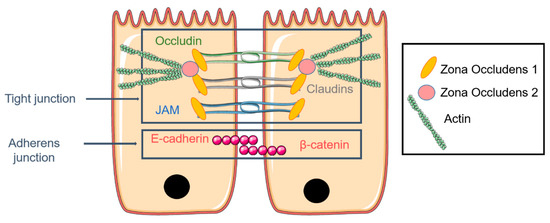
Figure 3.
The pulmonary epithelium is maintained by adherent junctions and the tight junction (TJ) complex composed of occludin, claudins, and junction adhesion molecules (JAM). Zona occludens (ZO) is directly attached to actin and connects the TJ to the cytoskeleton, notably actin F.
3.2.1. Ciliated Cells
Ciliated cells are elongated columnar cells that contain abundant mitochondria in the apical region to ensure the production of ATP, mandatory for the ciliary motion [103]. About 200 to 300 cilia recover the apical pole of each ciliated cell [104]. Airway cilia are 6.5 to 7 µm long, with a diameter of 0.1 µm, and are structured by microtubules [112,113]. The main role played by these cells is to propel the mucus layer produced by the secretory cells. Coordinated beatings take place to sweep the mucus and everything trapped in it from the luminal space towards the pharynx where it is most often swallowed. The ciliary beating frequency (CBF) is approximatively 12–14 Hz for cells cultivated in vitro. The CBF is dependent on different kinds of stimuli [114]. For instance, it has been shown that Ca2+ plays a key role in the CBF regulation. When [Ca2+] is increased in mammalian cells, CBF is increased, and vice versa [115,116]. The composition of the ASL directly influences the mucociliary clearance (MCC), especially for diseases such as CF where dehydrated mucus sticks to cilia and prevents them from expelling the mucus by reducing their motion.
3.2.2. Secretory Cells
The principal secretory cells present on the airway epithelia are the goblet cells which are intercalated between ciliated cells. Goblet cells are highly polarized cells. Nucleus and organelles are observed at the basal pole, whereas membrane-bound secretory granules of mucin are present at the apical pole, giving the cell its characteristic shape [103]. Secretion is mediated by a secretory apparatus engaged in mucin granules exocytosis. This phenome is ATP-dependent and involves the P2Y2 receptor (G protein-coupled receptors) [93]. When ATP is attached on this apical membrane receptor, phospholipase C (PLC) is activated, which triggers the second messengers: diacylglycerol (DAG) and inositol triphosphate (IP3). DAG activates Munc13 that liberates Syntaxin and therefore the formation of a four-helix SNARE (soluble N-ethylmaleimide–sensitive factor attachment protein receptor). When complexed with SNAP-23 (synaptosomal-associated protein 23) and VAMP (vesicle-associated membrane protein), the membrane of the secretory vesicle is allowed to be closer to the plasma membrane. As for IP3, attachment to its receptor enables the release of intracellular calcium from the endoplasmic reticulum. This calcium increase induces the activation of synaptotagmin, leading to the final confirmation of the SNARE complex [117]. All these events lead to the fusion of both vesicle and plasmatic membranes and to the release of mucins [118]. Submucosal glands constitutively secrete mucins, but its production can be increased through adrenergic and cholinergic stimulation [119]. In the pathologic context, the number or size of the cells is disturbed and hyperplasia in goblet cell occurs in COPD and asthma, while hypertrophy concerns CF patients [120]. Other kinds of secretory cells can be found in the small airways, namely, serous and club cells, which also participate in the humidification of epithelial cells. Club cells also have the ability to differentiate into ciliated cells or goblet cells [103].
3.2.3. Other Cell Types
Another kind of cell called basal cells are progenitor cells, linked to the basement membrane through hemi-desmosomes; they interact with columnar cells as well as neuronal cells. In addition to their ability to repopulate the epithelium cells of the conductive airway, they can also regulate inflammation response, water movement, or oxidant defense [121]. Apart from these tissue-specific epithelial cells, there are a variety of non-epithelial cells, such as immune cells (lymphocytes, leukocytes, and mast cells), and squamous alveolar type I pneumocytes covering 96% of the alveolar region, which are responsible for the gas exchange, or cuboidal alveolar type II pneumocytes involved in the synthesis and secretion of antimicrobial and surface-active components [122,123,124]. More recently, a new kind of cell, ionocytes, has been identified; they have been described as a major source of CFTR activity [125,126]. Although these cells represent only 1% to 2% of the total cells of the pulmonary epithelium, they accumulate more than 50% of cftr transcripts and could represent an important target for CF gene therapy.
3.3. Immortalized Cells
Immortalized cells have undergone genetic modifications leading to an indefinite replication. They can be either native from a spontaneous tumor or can be transformed by oncogenic or viral agents. Among airways epithelial cells, these kinds of cells are of particular interest because they proved to be useful in screening new drugs and are available in bulk quantities. Immortalized epithelial cells from airways were established by the introduction of exogenous DNA in order to provide better characteristics of the relationship between transmembrane conductance regulation and CF pathology [127]. Different strategies were tested to immortalize cell lines, the most used and particularly efficient one is the introduction of DNA encoding the SV40 T-antigen. The development of this cell line allows high-throughput screening to identify new drugs as well as genetic and pharmacological therapies. However, these systems present some disadvantages, especially for in situ extrapolation [128]. For instance, the clone is from a mixture of primary epithelial cells with different phenotypes; therefore, an isolated clone cannot represent all the characteristics of the original tissue. Furthermore, when the cells are kept in culture, they can express different properties than their original features. The transformation itself also influences the phenotype expression. As an example, they can lose their ability to form TJ, ciliary beating, mucus secretion, and their properties to transport ions, depending on the cell lines [129].
3.3.1. BEAS-2B, NuLi-1, NCI-H292, NCI-H358
BEAS-2B is a normal human epithelial cell transformed by an adenovirus 12 and SV40 hybrid [130]. This cell line is commonly used for studying airway epithelial structure and function and the interaction between xenobiotics and the cells themselves [131]. NuLi-1 is an airway epithelia cell which is useful for studying passive and active drug transportation through the epithelial barrier [132]. NuLi-1 cells have the ability to form TJ with a TEER value around 685 ± 31 Ω and can be used for active and passive drug transport studies [132]. NCI-H292 is a cell line obtained from a 32-year-old woman with mucoepidermoid pulmonary carcinoma. They do not form tight junctions but express desmosomes and adherens junctions. They also present a low level of CFTR, while they express mucin and can be used for mucin expression studies [133]. NCI-H358 cells were isolated from a man with a bronchioalveolar carcinoma and have the ability to express lung surfactant associated protein A (SP-A) [134].
3.3.2. 16HBE14o- and CFBE41o-
The 16HBE14o- cell line was developed by Cozens et al. It exposes a transepithelial resistance at 250 Ω·cm2 after 6 days of optimized culture depending on initial seeding densities and surface coating [135,136]. This cell line expressed more P-glycoproteins, lung resistance-related proteins (LRP), and caveolin-1 [137]. 16HBEo- was initially developed for the study of CFTR but can also be used for transport studies. Under ALI conditions, they do not polarize and do not display a normal epithelial phenotype (Figure 4A). Another cell line, the immortalized CF tracheo-bronchial epithelial cell CFBE41o-, is homozygous for ∆F508-CFTR and was first used by D. Gruenert and his team [138]. Ehrhardt et al. reported that CFBE41o- had a TEER peak at 1156 Ω·cm2 after 16 days of liquid-covered culture, while TEER is around 200 Ω·cm2 for cells cultured under ALI conditions [139]. This cell line is particularly interesting to study gene transfection or drugs screening in the CF context, due to its homozygous state.

Figure 4.
(A) Scanning electron microscopy of 16HBE41o- and Calu-3 cultivated in the liquid–liquid interphase (LLI) or air–liquid interphase (ALI) for 3 weeks after seeding. When cultivated under ALI conditions, unlike 16HBE41o-, Calu-3 cells produce mucus and form microvilli. For all pictures, the scale bar corresponds to 20.0 µm. (B) Transition from LLI culture to ALI allowing cell differentiation and in some case, depending on the cell line, mucus production.
3.3.3. A549
The A549 cell line is a human lung adenocarcinoma from a Caucasian male with lung cancer [140]. This cell line is frequently used as an in vitro model of pulmonary cuboidal epithelium or alveolar cells and screening of biopharmaceuticals molecules. For example, this cell line can be used for transfection assays, drug testing, and infection studies. However, A549 does not form TJ when cultured in ALI conditions. This feature leads to the formation of a very permeable monolayer with low transepithelial electrical resistances. In some cases and in optimized co-culture conditions, TEER values of this cell line can be increased [141]. Moreover, A549 possesses several multilamellar cytoplasmic inclusion bodies that are reminiscent of alveolar cells and can also be used as an alveolar model [142].
3.3.4. Calu-3
Calu-3 cells are cells from a 25-year-old Caucasian male with lung adenocarcinoma. When cultured in ALI, they formed a pseudostratified columnar epithelium similar to the normal columnar bronchial epithelium [143]. In such culture conditions, Calu-3 presents far more secretory vesicles after 3 weeks and mucus secretion can be observed. This secretion is also due to the fact that Calu-3 can differentiate into goblet cells. From our own observations, scanning electron microscopy reveals the presence of abundant microvilli in the apical surface (Figure 4A). Moreover, Calu-3 cells form a confluent monolayer and express TJ protein occludin, adherens junction protein E-cadherin, and epithelial-specific cytokeratin 7 (CK7) [144]. Calu-3, in ALI conditions, presents a morphology very close to the in vivo airway epithelium in terms of ultrastructure, secretory component, and electrical resistance [145,146]. The TEER values of Calu-3 in ALI culture show a variable TEER, ranging approximately between 400 and 800 Ω cm2, but confirming the formation of a tight epithelium [109,147]. A previous study has shown that Calu-3 could be transfected by polyethylenimine (l-PEI) and therefore could be used as a potential model for gene delivery [148]. Calu-3 is close to the in vivo situation, as it secrets mucus and generates microvilli. For instance, Calu-3 can be used as a good reproducible aerosol model for gene delivery. Hein et al. developed the new Pharmaceutical Aerosol Deposition Device on Cell Cultures (PADDOCC) and used the Calu-3 cell line to mimic the inhalation of dry powder on an epithelial monolayer [149]. Furthermore, Jeong et al. very recently proposed a system using the Calu-3 epithelium, able to reproduce the biology of the respiratory tract when it is exposed to chemicals [150].
3.4. Primary Cell Culture
Immortalized cell lines have low culture cost and provide large amounts of reproducible cells, yet despite their advantages, they frequently lose their characteristics during in vitro culture and senesce after a certain number of divisions. In addition, they are not a perfect representation of the original cells in primary culture; this is especially true for cancerous cells which are morphologically different [137]. Primary cell cultures, on the other hand, represent a closer model to the in vivo situation than immortalized cells, due to the fact that they are directly isolated from animal or human tissue and used as they are. However, cell isolation is hardly reproducible and collected cells are heterogeneous, meaning that each isolation is somewhat unique. There is also the non-negligible risk of bacteria and fungi contamination, as well as a limited life expectancy compared to immortalized cell lines.
3.4.1. Mammalians Cells
Primary epithelial cells can be derived from the upper and the lower airways. These cells can be collected from different species such as mouse, pig, bovine, or sheep. Lam et al. characterized a porcine bronchial epithelial cell culture model [151]. They took bronchi from the slaughterhouse and, after a dedicated protocol of digestion and cell culture, obtained a valuable differentiated airway epithelium. Moreover, Boyle et al. used trachea picked from sheep and cultivated on an air–liquid interphase. They observed the formation of ciliated epithelial cells from day 12, as well as mucus production [152]. TEER values were measured to have a peak at 1049 Ω·cm2 and stabilized around 200 Ω·cm2. Mouse trachea can also be used for airway epithelial cell isolation. Lam et al. thoroughly described the procedure to isolate mouse respiratory epithelial cells [153]. This model has demonstrated a TEER value between 1000 to 2000 Ω·cm2 and has been, thereafter, exposed to mainstream cigarette smoke. The formed epithelium exhibited several subtypes of cells: ciliated cells, basal cells, and non-ciliated cells highlighted by transmission electron microscopy observations. Cozen et al. characterized bovine bronchial epithelial cells (BBECs) when cultivated in ALI and proved that this model exhibited TJ, cilia, and mucus production [154].
3.4.2. Human Cells
Human primary cells are the most relevant model because they are closest to the in vivo situation but remain difficult to access. The trachea and lung can be collected during organ transplantation, biopsies, excised human nasal polyps, or autopsies, reducing their accessibility considerably. Non-traumatic methods can also be used, such as cellular scraping or brushings, but the donor’s agreement is required. A primary cell culture is composed of different kinds of cells, including ciliated and goblet cells, which all contribute to mimic the in vivo airway. NHBE (Normal Human Bronchial Epithelial) is an in vitro model for human bronchial epithelial cells that has the property of forming functional barriers. When cultured in ALI conditions, NHBE presents peak TEER values at 800 Ω·cm2 after 8 days of cultivation [155]. The small airway epithelial cells (SAECs) model is a normal human lung tissue from the bronchiole area. It exhibits basal cell properties with expression of ICAM-1 (Intercellular Adhesion Molecule 1) and club cell specific protein (CCSP) [156]. This model has already been used for infection and co-infection studies but also for the study of nanoparticles and cigarette smoke exposure [156,157]. In CF, a pharmaceutical company, Vertex, has successfully used human primary cells isolated from the bronchi of a patient with G551D and F508del CFTR mutations to determine the efficiency of Ivacaftor and Lumacaftor drugs [158]. In parallel, Epithelix has developed a commercial model, MucilAirTM, which is a reconstituted epithelium using human primary cells issued from a single donor or a pool of donors. Cells could come from different anatomical sites (nasal, trachea, or bronchial) and could belong to healthy donors or patients. Several pathologies are available, such as COPD, asthma, or CF. It has been shown that these cells present a cilia-beating in ALI conditions with, amongst others, the formation of TJ, the production of mucus, and active ions transport. For instance, this model was used by Ishikawa et al. to study the direct aerosol exposure of cigarette smoke [159]. Moreover, Firth et al. used induced pluripotent stem cells (iPSC) from homozygote F508del patients as a model to test the CRISPR/Cas9 system in order to correct the mutation [160]. Vaidynathan et al. also used this technique on primary cells from patients to correct the mutation and succeeded in regenerating a well-differentiated epithelium on a porcine small intestinal submucosal membrane [161]. Conversely, the use of the CRISPR/Cas9 technique can generate new cellular models to study, for example, the influence of mutation on cell phenotype.
3.5. Methods of Culture
3.5.1. Air–Liquid Interphase and TEER Measurement
Air–liquid interphase is a technique which can be used to mimic the respiratory tract epithelium (Figure 4B). This kind of culture is obtained by seeding airway epithelial cells (primary or not) in a Transwell system. After a proliferation phase, during which TEER is monitored, the media in the apical surface in the Transwell are removed to allow the cells differentiation into a pseudostratified epithelium. All nutrients, required to maintain the cells, are present in the apical chamber containing the culture media. Typically, in order to obtain a well-differentiated epithelial monolayer, cells have to be kept in ALI conditions for at least 3 weeks [143]. Primary cells, as well as immortalized cells, can be cultivated in such conditions. For instance, Calu-3 cells have the ability to differentiate under ALI conditions, forming cilia and producing mucus. NHBE also demonstrates such ability. This culture mode also presents the advantage of making a more complex model by adding mucus (artificial or from patient) on the apical pole of the cells that does not produce mucus spontaneously. To monitor the integrity of epithelium forms, TEER measurements can be used as previously mentioned (Figure 5A). They measure the electrical resistance across the cellular monolayer. This technique is very sensitive and informs researchers about the permeability of the epithelium [110]. The measurement method is based on Ohm’s law and reflects the ionic conductance. For the airway cells, it has been established that human tracheal and bronchial epithelial cells from healthy donors present a TEER value between 700 and 1200 Ω·cm2 when cultured in ALI conditions [162]. Pezzulo et al. also showed that the transcriptional profile of human primary ALI culture cells is closely similar to the native in vivo epithelium, demonstrating that ALI culture is more representative than immerged culture [162]. However, different TEER values have been reported for the same kind of cells, depending on the measurement protocol (Figure 5B). For instance, Foster et al. measured TEER values between 300 and 600 Ω·cm2 for Calu-3 cells, while Mathias et al. reported a TEER value for the same cell line at 1126 ± 222 Ω·cm2 [163,164]. It is important to note that in this study, Calu-3 was cultivated on a modified surface in the presence of collagen. In fact, there are numerous parameters to consider in TEER measurements: temperature, culture medium composition, cell culture passage, and shear stress [110].
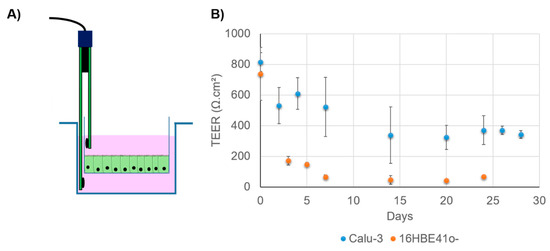
Figure 5.
(A) Transepithelial electrical resistance (TEER) measurement using a Transwell and an electrode. (B) Evolution of TEER in Calu-3 and 16HBE41o- when cultivated in ALI conditions. Cells are seeded in a Transwell (150,000 cells per insert) and cultivated in LLI conditions for one week. Then, the apical media are removed. TEER is monitored for 25 to 30 days. Data are averages of at least three Transwells and are expressed in Ω·cm2. Calu-3 exhibits a higher TEER value, demonstrating its ability to form TJ under this condition.
3.5.2. Culture Media
Culture media play a key role in the phenotype of ALI-cultured cells. For instance, primary cells, such as mouse trachea epithelial cells, when supplemented with IL-13, have the ability to produce mucus [165]. This interleukin up-regulates the production of mucus by inducing the production of MUC5AC [166,167]. Furthermore, cells (primary or not) can differentiate when cultured on collagen and supplemented with retinoic acid (RA), meaning that growth factors are important factors to be considered [168,169]. In the respiratory system, RA is the natural metabolite of retinol (vitamin A), controlling the development and the maintenance of the mucociliary epithelium [170]. Koo et al. showed that the incubation of RA on airway epithelial cells under squamous differentiation restored a mucous phenotype, leading to a mucin secretion 72 h after treatment [171]. Culture media can also influence ciliary motility. Compounds such as phenylmercuric borate and thiomersal have the ability to irreversibly decrease the CBF frequency. Conversely, chlorobutanol can inhibit motility with a reversible action. Another compound, benzalkonuim chloride, induces a slow, but irreversible, ciliotoxic effect. It also has the capacity to slow down CBF and disorganize the mucus structure through electrostatic interactions with anionic substances [172]. Mucociliary clearance constituted one of the extracellular barriers for gene transfer. Sinn et al. used formulations containing a viral vector with viscoelastic gels in order to slow down mucociliary clearance. This strategy was shown to considerably improve vector transduction [173].
3.5.3. Polyculture
The pulmonary tract is composed of a multitude of airway epithelial cells and constitutes a complex tissue. To mimic the in vivo context as far as possible, polycultures have been considered. Altogether, cells create a particular airway microenvironment contributing to the pulmonary function and its protection. When mimicking, in vitro, the respiratory epithelium, polycultures represent a valuable approach and can bring results close to in vivo conditions. For instance, Rothen-Rutishauser et al. developed three lung cell lines associating A549 cultured with airway macrophages and dendritic cells to study particles interactions [141]. The use of a 3D model instead of a 2D model allows a mostly in vivo-like morphology, allowing a greater resemblance to the physiological conditions. Three-dimensional single or co-culture models also constitute an interesting system for in vitro studies. To obtain such models, the extracellular matrix is necessary to support cells. For that purpose, synthetic or natural hydrogels can be used and provide a valuable extracellular matrix [174,175]. Other kinds of support can be used as a scaffold such as porous sheets to mimic basal lamina. As an example, Harrington et al. used electrospun fibers of PET in order to create a 3D model of the airway epithelium [176]. Meenach et al. developed a lung composed of multicellular spheroids by using type I rat tail collagen with A549 and H358 cell lines. Two techniques were used in this study, liquid-covered culture and air–liquid interphase. These spheroids were then evaluated for aerosolization of dry powder to model lung cancer therapy [177]. Moreover, with the development of microfluidic technologies, a new approach of culture cells has been described, namely, the organ-on-chip approach. This in vitro approach allows high-resolution, real-time imaging analysis of living cells (structure and function) in a tissue or at the organ level [178]. The lung-on-chip model could become a relevant model for pulmonary drug delivery and especially for nanotoxicology studies because it can mimic the effect of lung–particle integration [142]. In fact, cells still form a tight epithelium barrier under this culture condition. Finally, an alternative approach is to use directly living tissue from the lung, trachea, or bronchus for constituting a relevant in vitro model. Different kinds of explant origins can be used such as human or porcine [179,180]. In 1994, Seeger et al. developed a model system using isolated and perfused rabbit lung [181]. This ex vivo model was afterward used as a pulmonary drug delivery for aerosolizable nanoparticles [182].
4. Conclusions and Outlooks
In vitro cell culture models of human airways are crucial complementary tools to properly study nucleic acid delivery under normal or pathophysiological conditions. As reviewed herein, gene delivery systems have to go through successive steps during their journey into the respiratory tract, which includes crossing the mucus barrier, reaching the underlying epithelium, and releasing their cargo in the cytoplasm of target cells. In this respect, CF may be considered as a challenging model disease since these steps are further complicated with noticeable hurdles, including mucus dehydration and accumulation, chronic inflammation, and microbial colonization. Importantly, improvements obtained for CF aerosol-mediated gene therapy could benefit other similar respiratory disorders such as asthma, chronic obstructive pulmonary disease (COPD), or α-1 antitrypsin (AAT) deficiency. Asthma is a chronic inflammation affecting the lower respiratory tract and characterized by difficulties to breathe due to reversible airflow obstruction and bronchospasms. COPD is a slowly progressive chronic disease mainly affecting bronchial tubes and alveoli and also characterized by a chronic airway obstruction that is not completely reversible with drugs [183]. Another genetic disease, α-1 antitrypsin deficiency, is caused by a missense mutation in the AAT-encoding gene involved in inflammatory processes. Finally, aerosol gene therapy may also be applied for treating lung cancers and metastasis located in the airways. Though different therapeutic nucleic acids may be used for each of the abovementioned conditions (e.g., miRNA, siRNA, or suicide gene), delivery systems and strategies will be needed in every case, which could be more properly designed thanks to optimized in vitro airway cell culture systems.
Author Contributions
Writing—original draft preparation, R.G.; writing—review and editing, V.L., P.R., T.H., S.R., T.L.G., T.A., T.M., and R.G. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by “Vaincre la mucoviscidose” (Paris, France), ANR CE18, and “Association de transfusion sanguine et de biogénétique Gaétan Saleün” (Brest, France). RG is grateful for a PhD fellowship from the Brest Métropole and Association Gaétan Saleün.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
The authors are grateful to the “Plateforme d’Imagerie et de Mesure en Microscopie, PIMM” (Brest, France) for the scanning electron microscopy of cells. The authors are also grateful to Julian Ravel for English reviewing.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kim, N.; Duncan, G.A.; Hanes, J.; Suk, J.S. Barriers to inhaled gene therapy of obstructive lung diseases: A review. J. Control. Release 2016, 240, 465. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, B.P.; Freedman, S.D. Cystic fibrosis. Lancet 2009, 373, 1891–1904. [Google Scholar] [CrossRef]
- Ratjen, F.; Rietschel, E.; Griese, M.; Ballmann, M.; Kleinau, I.; Doring, G.; Reinhardt, D.; Paul, K. Fractional analysis of bronchoalveolar lavage fluid cytology in cystic fibrosis patients with normal lung function. Bronchoalveolar lavage for the evaluation of anti-inflammatory treatment (BEAT) study group. Eur. Respir. J. 2000, 15, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, N.; Chen, C.I.U.; Loppow, D.; Schink, T.; Kleinau, I.; Jörres, R.A.; Wahn, U.; Magnussen, H.; Paul, K.P. Cellular profiles of induced sputum in children with stable cystic fibrosis: Comparison with BAL. Eur. Respir. J. 2003, 22, 497–502. [Google Scholar] [CrossRef]
- Jackson, A.D. Airway goblet-cell mucus secretion. Trends Pharmacol. Sci. 2001, 22, 39–45. [Google Scholar] [CrossRef]
- Elborn, J.S. Cystic fibrosis. Lancet 2016, 388, 2519–2531. [Google Scholar] [CrossRef]
- Middleton, P.G.; Mall, M.A.; Dřevínek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef]
- Burgel, P.-R.; Munck, A.; Durieu, I.; Chiron, R.; Mely, L.; Prevotat, A.; Murris-Espin, M.; Porzio, M.; Abely, M.; Reix, P.; et al. Real-Life Safety and Effectiveness of Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2019. [Google Scholar] [CrossRef]
- Collins, F.S.; Gottlieb, S. The Next Phase of Human Gene-Therapy Oversight. N. Engl. J. Med. 2018, 379, 1393–1395. [Google Scholar] [CrossRef]
- Rangarajan, S.; Walsh, L.; Lester, W.; Perry, D.; Madan, B.; Laffan, M.; Yu, H.; Vettermann, C.; Pierce, G.F.; Wong, W.Y.; et al. AAV5-Factor VIII Gene Transfer in Severe Hemophilia A. N. Engl. J. Med. 2017, 377, 2519–2530. [Google Scholar] [CrossRef]
- George, L.A.; Sullivan, S.K.; Giermasz, A.; Rasko, J.E.J.; Samelson-Jones, B.J.; Ducore, J.; Cuker, A.; Sullivan, L.M.; Majumdar, S.; Teitel, J.; et al. Hemophilia B Gene Therapy with a High-Specific-Activity Factor IX Variant. N. Engl. J. Med. 2017, 377, 2215–2227. [Google Scholar] [CrossRef]
- Al-Zaidy, S.A.; Kolb, S.J.; Lowes, L.; Alfano, L.N.; Shell, R.; Church, K.R.; Nagendran, S.; Sproule, D.M.; Feltner, D.E.; Wells, C.; et al. AVXS-101 (Onasemnogene Abeparvovec) for SMA1: Comparative Study with a Prospective Natural History Cohort. J. Neuromuscul. Dis. 2019, 6, 307–317. [Google Scholar] [CrossRef]
- Belmadi, N.; Midoux, P.; Loyer, P.; Passirani, C.; Pichon, C.; Le Gall, T.; Jaffres, P.-A.; Lehn, P.; Montier, T. Synthetic vectors for gene delivery: An overview of their evolution depending on routes of administration. Biotechnol. J. 2015, 10, 1370–1389. [Google Scholar] [CrossRef]
- Ia, P.; Sc, H.; Dr, G. Non-viral vectors in cystic fibrosis gene therapy: Recent developments and future prospects. Expert Opin. Biol. Ther. 2009, 9, 991–1003. [Google Scholar]
- Alton, E.W.; Armstrong, D.K.; Ashby, D.; Bayfield, K.J.; Bilton, D.; Bloomfield, E.V.; Boyd, A.C.; Brand, J.; Buchan, R.; Calcedo, R.; et al. A Randomised, Double-Blind, Placebo-Controlled Trial of Repeated Nebulisation of Non-Viral Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Gene Therapy in Patients with Cystic Fibrosis. Efficacy and Mechanism Evaluation; NIHR Journals Library: Southampton, UK, 2016. [Google Scholar]
- d’Angelo, I.; Conte, C.; La Rotonda, M.I.; Miro, A.; Quaglia, F.; Ungaro, F. Improving the efficacy of inhaled drugs in cystic fibrosis: Challenges and emerging drug delivery strategies. Adv. Drug Deliv. Rev. 2014, 75, 92–111. [Google Scholar] [CrossRef]
- Ibrahim, M.; Verma, R.; Garcia-Contreras, L. Inhalation drug delivery devices: Technology update. Med. Devices 2015, 8, 131–139. [Google Scholar] [CrossRef]
- Cheng, Y.S. Mechanisms of Pharmaceutical Aerosol Deposition in the Respiratory Tract. AAPS PharmSciTech 2014, 15, 630–640. [Google Scholar] [CrossRef] [PubMed]
- Audus, K.L.; Bartel, R.L.; Hidalgo, I.J.; Borchardt, R.T. The Use of Cultured Epithelial and Endothelial Cells for Drug Transport and Metabolism Studies. Pharm. Res. 1990, 7, 435–451. [Google Scholar] [CrossRef] [PubMed]
- Ehrhardt, C.; Laue, M.; Kim, K.-J. In Vitro Models of the Alveolar Epithelial Barrier. In Drug Absorption Studies. Biotechnology: Pharmaceutical Aspects; Ehrhardt, C., Kim, K.J., Eds.; Springer: Boston, MA, USA, 2008; Volume VII, pp. 258–282. ISBN 978-0-387-74901-3. [Google Scholar]
- Byron, P.R.; Patton, J.S. Drug delivery via the respiratory tract. J. Aerosol Med. 1994, 7, 49–75. [Google Scholar] [CrossRef] [PubMed]
- Resnier, P.; Mottais, A.; Sibiril, Y.; Le Gall, T.; Montier, T. Challenges and Successes Using Nanomedicines for Aerosol Delivery to the Airways. Curr. Gene Ther. 2016, 16, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Man, R.C.H.; Liao, Q.; Kung, K.L.K.; Chow, M.Y.T.; Lam, J.K.W. Effective mRNA pulmonary delivery by dry powder formulation of PEGylated synthetic KL4 peptide. J. Control. Release 2019, 314, 102–115. [Google Scholar] [CrossRef] [PubMed]
- Griesenbach, U.; Alton, E.W.F.W. Gene transfer to the lung: Lessons learned from more than 2 decades of CF gene therapy. Adv. Drug Deliv. Rev. 2009, 61, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Conese, M.; Boyd, A.C.; Di Gioia, S.; Auriche, C.; Ascenzioni, F. Genomic context vectors and artificial chromosomes for cystic fibrosis gene therapy. Curr. Gene Ther. 2007, 7, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Duncan, G.A.; Jung, J.; Hanes, J.; Suk, J.S. The Mucus Barrier to Inhaled Gene Therapy. Mol. Ther. 2016, 24, 2043–2053. [Google Scholar] [CrossRef]
- Alton, E.W.F.W.; Baker, A.; Baker, E.; Boyd, A.C.; Cheng, S.H.; Coles, R.L.; Collie, D.D.S.; Davidson, H.; Davies, J.C.; Gill, D.R.; et al. The safety profile of a cationic lipid-mediated cystic fibrosis gene transfer agent following repeated monthly aerosol administration to sheep. Biomaterials 2013, 34, 10267–10277. [Google Scholar] [CrossRef]
- Rosen, B.H.; Chanson, M.; Gawenis, L.R.; Liu, J.; Sofoluwe, A.; Zoso, A.; Engelhardt, J.F. Animal and model systems for studying cystic fibrosis. J. Cyst. Fibros. 2018, 17, S28–S34. [Google Scholar] [CrossRef]
- Livraghi-Butrico, A.; Grubb, B.R.; Kelly, E.J.; Wilkinson, K.J.; Yang, H.; Geiser, M.; Randell, S.H.; Boucher, R.C.; O’Neal, W.K. Genetically determined heterogeneity of lung disease in a mouse model of airway mucus obstruction. Physiol. Genom. 2012, 44, 470–484. [Google Scholar] [CrossRef]
- Livraghi-Butrico, A.; Kelly, E.J.; Klem, E.R.; Dang, H.; Wolfgang, M.C.; Boucher, R.C.; Randell, S.H.; O’Neal, W.K. Mucus clearance, MyD88-dependent and MyD88-independent immunity modulate lung susceptibility to spontaneous bacterial infection and inflammation. Mucosal Immunol. 2012, 5, 397–408. [Google Scholar] [CrossRef]
- Livraghi-Butrico, A.; Kelly, E.J.; Wilkinson, K.J.; Rogers, T.D.; Gilmore, R.C.; Harkema, J.R.; Randell, S.H.; Boucher, R.C.; O’Neal, W.K.; Grubb, B.R. Loss of Cftr function exacerbates the phenotype of Na+ hyperabsorption in murine airways. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 304, L469–L480. [Google Scholar] [CrossRef]
- Fisher, J.T.; Liu, X.; Yan, Z.; Luo, M.; Zhang, Y.; Zhou, W.; Lee, B.J.; Song, Y.; Guo, C.; Wang, Y.; et al. Comparative Processing and Function of Human and Ferret Cystic Fibrosis Transmembrane Conductance Regulator. J. Biol. Chem. 2012, 287, 21673–21685. [Google Scholar] [CrossRef]
- Li, Z.; Engelhardt, J.F. Progress toward generating a ferret model of cystic fibrosis by somatic cell nuclear transfer. Reprod. Biol. Endocrinol. RBE 2003, 1, 83. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rogers, C.S.; Stoltz, D.A.; Meyerholz, D.K.; Ostedgaard, L.S.; Rokhlina, T.; Taft, P.J.; Rogan, M.P.; Pezzulo, A.A.; Karp, P.H.; Itani, O.A.; et al. Disruption of the CFTR Gene Produces a Model of Cystic Fibrosis in Newborn Pigs. Science 2008, 321, 1837–1841. [Google Scholar] [CrossRef] [PubMed]
- Stoltz, D.A.; Meyerholz, D.K.; Pezzulo, A.A.; Ramachandran, S.; Rogan, M.P.; Davis, G.J.; Hanfland, R.A.; Wohlford-Lenane, C.; Dohrn, C.L.; Bartlett, J.A.; et al. Cystic Fibrosis Pigs Develop Lung Disease and Exhibit Defective Bacterial Eradication at Birth. Sci. Transl. Med. 2010, 2, 29ra31. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Namkung, W.; Nielson, D.W.; Lee, J.-W.; Finkbeiner, W.E.; Verkman, A.S. Airway surface liquid depth measured in ex vivo fragments of pig and human trachea: Dependence on Na+ and Cl− channel function. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L1131–L1140. [Google Scholar] [CrossRef] [PubMed]
- Cone, R.A. Barrier properties of mucus. Adv. Drug Deliv. Rev. 2009, 61, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Kilburn, K.H. A hypothesis for pulmonary clearance and its implications. Am. Rev. Respir. Dis. 1968, 98, 449–463. [Google Scholar] [CrossRef]
- Donaldson, S.H.; Corcoran, T.E.; Laube, B.L.; Bennett, W.D. Mucociliary Clearance as an Outcome Measure for Cystic Fibrosis Clinical Research. Proc. Am. Thorac. Soc. 2007, 4, 399–405. [Google Scholar] [CrossRef]
- Griese, M. Pulmonary surfactant in health and human lung diseases: State of the art. Eur. Respir. J. 1999, 13, 1455–1476. [Google Scholar] [CrossRef]
- Lai, S.K.; O’Hanlon, D.E.; Harrold, S.; Man, S.T.; Wang, Y.-Y.; Cone, R.; Hanes, J. Rapid transport of large polymeric nanoparticles in fresh undiluted human mucus. Proc. Natl. Acad. Sci. USA 2007, 104, 1482–1487. [Google Scholar] [CrossRef]
- Suk, J.S.; Lai, S.K.; Wang, Y.-Y.; Ensign, L.M.; Zeitlin, P.L.; Boyle, M.P.; Hanes, J. The penetration of fresh undiluted sputum expectorated by cystic fibrosis patients by non-adhesive polymer nanoparticles. Biomaterials 2009, 30, 2591–2597. [Google Scholar] [CrossRef]
- Thornton, D.J.; Rousseau, K.; McGuckin, M.A. Structure and function of the polymeric mucins in airways mucus. Annu. Rev. Physiol. 2008, 70, 459–486. [Google Scholar] [CrossRef] [PubMed]
- Rose, M.C.; Nickola, T.J.; Voynow, J.A. Airway mucus obstruction: Mucin glycoproteins, MUC gene regulation and goblet cell hyperplasia. Am. J. Respir. Cell Mol. Biol. 2001, 25, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Vinall, L.E.; Fowler, J.C.; Jones, A.L.; Kirkbride, H.J.; de Bolós, C.; Laine, A.; Porchet, N.; Gum, J.R.; Kim, Y.S.; Moss, F.M.; et al. Polymorphism of Human Mucin Genes in Chest Disease. Am. J. Respir. Cell Mol. Biol. 2000, 23, 678–686. [Google Scholar] [CrossRef] [PubMed]
- Rose, M.C.; Piazza, F.M.; Chen, Y.A.; Alimam, M.Z.; Bautista, M.V.; Letwin, N.; Rajput, B. Model systems for investigating mucin gene expression in airway diseases. J. Aerosol Med. 2000, 13, 245–261. [Google Scholar] [CrossRef]
- Demouveaux, B.; Gouyer, V.; Gottrand, F.; Narita, T.; Desseyn, J.-L. Gel-forming mucin interactome drives mucus viscoelasticity. Adv. Colloid Interface Sci. 2018, 252, 69–82. [Google Scholar] [CrossRef]
- Bansil, R.; Turner, B.S. Mucin structure, aggregation, physiological functions and biomedical applications. Curr. Opin. Colloid Interface Sci. 2006, 11, 164–170. [Google Scholar] [CrossRef]
- Verdugo, P. Supramolecular Dynamics of Mucus. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef]
- Turner, B.S.; Bhaskar, K.R.; Hadzopoulou-Cladaras, M.; LaMont, J.T. Cysteine-rich regions of pig gastric mucin contain von Willebrand factor and cystine knot domains at the carboxyl terminal. Biochim. Biophys. Acta BBA Gene Struct. Expr. 1999, 1447, 77–92. [Google Scholar] [CrossRef]
- Kočevar-Nared, J.; Kristl, J.; Šmid-Korbar, J. Comparative rheological investigation of crude gastric mucin and natural gastric mucus. Biomaterials 1997, 18, 677–681. [Google Scholar] [CrossRef]
- Larson, R. The Structure and Rheology of Complex Fluids; Oxford University Press: Oxford, UK, 1999. [Google Scholar]
- Hwang, S.H.; Litt, M.; Forsman, W.C. Rheological properties of mucus. Rheol. Acta 1969, 8, 438–448. [Google Scholar] [CrossRef]
- Puchelle, E.; Zahm, J.M.; Quemada, D. Rheological properties controlling mucociliary frequency and respiratory mucus transport. Biorheology 1987, 24, 557–563. [Google Scholar] [CrossRef] [PubMed]
- King, M.; Zahm, J.M.; Pierrot, D.; Vaquez-Girod, S.; Puchelle, E. The role of mucus gel viscosity, spinnability, and adhesive properties in clearance by simulated cough. Biorheology 1989, 26, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Puchelle, E.; Zahm, J.M.; de Bentzmann, S.; Grosskopf, C.; Shak, S.; Mougel, D.; Polu, J.M. Effects of rhDNase on purulent airway secretions in chronic bronchitis. Eur. Respir. J. 1996, 9, 765–769. [Google Scholar] [CrossRef] [PubMed]
- App, E.M.; Kieselmann, R.; Reinhardt, D.; Lindemann, H.; Dasgupta, B.; King, M.; Brand, P. Sputum rheology changes in cystic fibrosis lung disease following two different types of physiotherapy: Flutter vs autogenic drainage. Chest 1998, 114, 171–177. [Google Scholar] [CrossRef]
- Lai, S.K.; Wang, Y.-Y.; Wirtz, D.; Hanes, J. Micro- and macrorheology of mucus. Adv. Drug Deliv. Rev. 2009, 61, 86–100. [Google Scholar] [CrossRef]
- Serisier, D.J.; Carroll, M.P.; Shute, J.K.; Young, S.A. Macrorheology of cystic fibrosis, chronic obstructive pulmonary disease & normal sputum. Respir. Res. 2009, 10, 63. [Google Scholar] [CrossRef]
- Patarin, J.; Ghiringhelli, É.; Darsy, G.; Obamba, M.; Bochu, P.; Camara, B.; Quétant, S.; Cracowski, J.-L.; Cracowski, C.; Robert de Saint Vincent, M. Rheological analysis of sputum from patients with chronic bronchial diseases. Sci. Rep. 2020, 10, 15685. [Google Scholar] [CrossRef]
- Smart, J.D. The basics and underlying mechanisms of mucoadhesion. Adv. Drug Deliv. Rev. 2005, 57, 1556–1568. [Google Scholar] [CrossRef]
- Kirkham, S.; Sheehan, J.K.; Knight, D.; Richardson, P.S.; Thornton, D.J. Heterogeneity of airways mucus: Variations in the amounts and glycoforms of the major oligomeric mucins MUC5AC and MUC5B. Biochem. J. 2002, 361, 537–546. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, Y.H.; Di, Y.-P.; Wu, R. Characterization of Human Mucin 5B Gene Expression in Airway Epithelium and the Genomic Clone of the Amino-Terminal and 5′ -Flanking Region. Am. J. Respir. Cell Mol. Biol. 2001, 25, 542–553. [Google Scholar] [CrossRef]
- Aggarwal, S.; Kim, S.-W.; Cheon, K.; Tabassam, F.H.; Yoon, J.-H.; Koo, J.S. Nonclassical Action of Retinoic Acid on the Activation of the cAMP Response Element-binding Protein in Normal Human Bronchial Epithelial Cells. Mol. Biol. Cell 2005, 17, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Lamblin, G.; Degroote, S.; Perini, J.M.; Delmotte, P.; Scharfman, A.; Davril, M.; Lo-Guidice, J.M.; Houdret, N.; Dumur, V.; Klein, A.; et al. Human airway mucin glycosylation: A combinatory of carbohydrate determinants which vary in cystic fibrosis. Glycoconj. J. 2001, 18, 661–684. [Google Scholar] [CrossRef] [PubMed]
- Luzar, M.A.; Thomassen, M.J.; Montie, T.C. Flagella and motility alterations in Pseudomonas aeruginosa strains from patients with cystic fibrosis: Relationship to patient clinical condition. Infect. Immun. 1985, 50, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Lovewell, R.R.; Hayes, S.M.; O’Toole, G.A.; Berwin, B. Pseudomonas aeruginosa flagellar motility activates the phagocyte PI3K/Akt pathway to induce phagocytic engulfment. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, L698–L707. [Google Scholar] [CrossRef]
- Lillehoj, E.P.; Kim, B.T.; Kim, K.C. Identification of Pseudomonas aeruginosa flagellin as an adhesin for Muc1 mucin. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 282, L751–L756. [Google Scholar] [CrossRef]
- Delmotte, P.; Degroote, S.; Merten, M.D.; Van Seuningen, I.; Bernigaud, A.; Figarella, C.; Roussel, P.; Périni, J.M. Influence of TNFalpha on the sialylation of mucins produced by a transformed cell line MM-39 derived from human tracheal gland cells. Glycoconj. J. 2001, 18, 487–497. [Google Scholar] [CrossRef]
- Louahed, J.; Toda, M.; Jen, J.; Hamid, Q.; Renauld, J.C.; Levitt, R.C.; Nicolaides, N.C. Interleukin-9 upregulates mucus expression in the airways. Am. J. Respir. Cell Mol. Biol. 2000, 22, 649–656. [Google Scholar] [CrossRef]
- Matthews, L.W.; Spector, S.; Lemm, J.; Potter, J.L. Studies on pulmonary secretions. I. The over-all chemical composition of pulmonary secretions from patients with cystic fibrosis, bronchiectasis, and laryngectomy. Am. Rev. Respir. Dis. 1963, 88, 199–204. [Google Scholar] [CrossRef]
- Lethem, M.I.; James, S.L.; Marriott, C. The Role of Mucous Glycoproteins in the Rheologic Properties of Cystic Fibrosis Sputum. Am. Rev. Respir. Dis. 1990, 142, 1053–1058. [Google Scholar] [CrossRef]
- Mrsny, R.; Daugherty, A.; Short, S.; Widmer, R.; Siegel, M.; Keller, G.-A. Distribution of DNA and Alginate in Purulent Cystic Fibrosis Sputum: Implications to Pulmonary Targeting Strategies. J. Drug Target. 1996, 4, 233–243. [Google Scholar] [CrossRef]
- Luk, C.K.; Dulfano, M.J. Effect of pH, viscosity and ionic-strength changes on ciliary beating frequency of human bronchial explants. Clin. Sci. 1983, 64, 449–451. [Google Scholar] [CrossRef] [PubMed]
- Nadziejko, C.E.; Slomiany, B.L.; Slomiany, A. Most of the Lipid in Purulent Sputum is Bound to Mucus Glycoprotein. Exp. Lung Res. 1993, 19, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Galabert, C.; Jacquot, J.; Zahm, J.M.; Puchelle, E. Relationships between the lipid content and the rheological properties of airway secretions in cystic fibrosis. Clin. Chim. Acta 1987, 164, 139–149. [Google Scholar] [CrossRef]
- Girod, S.; Galabert, C.; Lecuire, A.; Zahm, J.; Puchelle, E. Phospholipid composition and surface-active properties of tracheobronchial secretions from patients with cystic fibosis and chronic obstructive pulmonary disease. Pediatric Pulmonol. 1992, 13, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Crater, J.S.; Carrier, R.L. Barrier Properties of Gastrointestinal Mucus to Nanoparticle Transport. Macromol. Biosci. 2010, 10, 1473–1483. [Google Scholar] [CrossRef]
- Meldrum, O.W.; Yakubov, G.E.; Bonilla, M.R.; Deshmukh, O.; McGuckin, M.A.; Gidley, M.J. Mucin gel assembly is controlled by a collective action of non-mucin proteins, disulfide bridges, Ca2+-mediated links, and hydrogen bonding. Sci. Rep. 2018, 8, 5802. [Google Scholar] [CrossRef] [PubMed]
- Sriramulu, D.D.; Lünsdorf, H.; Lam, J.S.; Römling, U. Microcolony formation: A novel biofilm model of Pseudomonas aeruginosa for the cystic fibrosis lung. J. Med. Microbiol. 2005, 54, 667–676. [Google Scholar] [CrossRef]
- Yang, Y.; Tsifansky, M.D.; Shin, S.; Lin, Q.; Yeo, Y. Mannitol-Guided delivery of ciprofloxacin in artificial cystic fibrosis mucus model. Biotechnol. Bioeng. 2011, 108, 1441–1449. [Google Scholar] [CrossRef]
- Murgia, X.; Pawelzyk, P.; Schaefer, U.F.; Wagner, C.; Willenbacher, N.; Lehr, C.-M. Size-Limited Penetration of Nanoparticles into Porcine Respiratory Mucus after Aerosol Deposition. Biomacromolecules 2016, 17, 1536–1542. [Google Scholar] [CrossRef]
- Dawson, M.; Wirtz, D.; Hanes, J. Enhanced Viscoelasticity of Human Cystic Fibrotic Sputum Correlates with Increasing Microheterogeneity in Particle Transport. J. Biol. Chem. 2003, 278, 50393–50401. [Google Scholar] [CrossRef]
- Fernandez-Petty, C.M.; Hughes, G.W.; Bowers, H.L.; Watson, J.D.; Rosen, B.H.; Townsend, S.M.; Santos, C.; Ridley, C.E.; Chu, K.K.; Birket, S.E.; et al. A glycopolymer improves vascoelasticity and mucociliary transport of abnormal cystic fibrosis mucus. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Nettle, C.J.; Jenkins, L.; Curtis, D.; Badiei, N.; Lewis, K.; Williams, P.R.; Daniels, D.R. Linear rheology as a potential monitoring tool for sputum in patients with Chronic Obstructive Pulmonary Disease (COPD). Biorheology 2018, 54, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Nordgård, C.T.; Draget, K.I. Oligosaccharides As Modulators of Rheology in Complex Mucous Systems. Biomacromolecules 2011, 12, 3084–3090. [Google Scholar] [CrossRef] [PubMed]
- Tomaiuolo, G.; Rusciano, G.; Caserta, S.; Carciati, A.; Carnovale, V.; Abete, P.; Sasso, A.; Guido, S. A New Method to Improve the Clinical Evaluation of Cystic Fibrosis Patients by Mucus Viscoelastic Properties. PLoS ONE 2014, 9, e82297. [Google Scholar] [CrossRef]
- Sanders, N.N.; De Smedt, S.C.; Van Rompaey, E.; Simoens, P.; De Baets, F.; Demeester, J. Cystic fibrosis sputum: A barrier to the transport of nanospheres. Am. J. Respir. Crit. Care Med. 2000, 162, 1905–1911. [Google Scholar] [CrossRef]
- Vukosavljevic, B.; Murgia, X.; Schwarzkopf, K.; Schaefer, U.F.; Lehr, C.-M.; Windbergs, M. Tracing molecular and structural changes upon mucolysis with N-acetyl cysteine in human airway mucus. Int. J. Pharm. 2017, 533, 373–376. [Google Scholar] [CrossRef]
- Hida, K.; Lai, S.K.; Suk, J.S.; Won, S.Y.; Boyle, M.P.; Hanes, J. Common Gene Therapy Viral Vectors Do Not Efficiently Penetrate Sputum from Cystic Fibrosis Patients. PLoS ONE 2011, 6. [Google Scholar] [CrossRef]
- Noël-Georis, I.; Bernard, A.; Falmagne, P.; Wattiez, R. Database of bronchoalveolar lavage fluid proteins. J. Chromatogr. B 2002, 771, 221–236. [Google Scholar] [CrossRef]
- Schuster, B.S.; Suk, J.S.; Woodworth, G.F.; Hanes, J. Nanoparticle diffusion in respiratory mucus from humans without lung disease. Biomaterials 2013, 34, 3439–3446. [Google Scholar] [CrossRef]
- Fahy, J.V.; Dickey, B.F. Airway Mucus Function and Dysfunction. N. Engl. J. Med. 2010, 363, 2233. [Google Scholar] [CrossRef]
- Huckaby, J.T.; Lai, S.K. PEGylation for enhancing nanoparticle diffusion in mucus. Adv. Drug Deliv. Rev. 2018, 124, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Ensign, L.M.; Boylan, N.J.; Schön, A.; Gong, X.; Yang, J.-C.; Lamb, N.W.; Cai, S.; Yu, T.; Freire, E.; et al. Impact of Surface Polyethylene Glycol (PEG) Density on Biodegradable Nanoparticle Transport in Mucus ex Vivo and Distribution in Vivo. ACS Nano 2015, 9, 9217–9227. [Google Scholar] [CrossRef] [PubMed]
- Mansfield, E.D.H.; Sillence, K.; Hole, P.; Williams, A.C.; Khutoryanskiy, V.V. POZylation: A new approach to enhance nanoparticle diffusion through mucosal barriers. Nanoscale 2015, 7, 13671–13679. [Google Scholar] [CrossRef]
- Yang, M.; Lai, S.K.; Wang, Y.-Y.; Zhong, W.; Happe, C.; Zhang, M.; Fu, J.; Hanes, J. Biodegradable Nanoparticles Composed Entirely of Safe Materials that Rapidly Penetrate Human Mucus. Angew. Chem. Int. Ed. Engl. 2011, 50, 2597–2600. [Google Scholar] [CrossRef] [PubMed]
- Ensign, L.M.; Lai, S.K.; Wang, Y.-Y.; Yang, M.; Mert, O.; Hanes, J.; Cone, R. Pretreatment of human cervicovaginal mucus with pluronic F127 enhances nanoparticle penetration without compromising mucus barrier properties to herpes simplex virus. Biomacromolecules 2014, 15, 4403–4409. [Google Scholar] [CrossRef] [PubMed]
- Shan, W.; Zhu, X.; Liu, M.; Li, L.; Zhong, J.; Sun, W.; Zhang, Z.; Huang, Y. Overcoming the Diffusion Barrier of Mucus and Absorption Barrier of Epithelium by Self-Assembled Nanoparticles for Oral Delivery of Insulin. ACS Nano 2015, 9, 2345–2356. [Google Scholar] [CrossRef]
- Suk, J.S.; Lai, S.K.; Boylan, N.J.; Dawson, M.R.; Boyle, M.P.; Hanes, J. Rapid transport of muco-inert nanoparticles in cystic fibrosis sputum treated with N-acetyl cysteine. Nanomedicine 2011, 6, 365–375. [Google Scholar] [CrossRef]
- Müller, C.; Leithner, K.; Hauptstein, S.; Hintzen, F.; Salvenmoser, W.; Bernkop-Schnürch, A. Preparation and characterization of mucus-penetrating papain/poly(acrylic acid) nanoparticles for oral drug delivery applications. J. Nanoparticle Res. 2012, 15, 1353. [Google Scholar] [CrossRef]
- Itoh, H.; Nishino, M.; Hatabu, H. Architecture of the Lung: Morphology and Function. J. Thorac. Imaging 2004, 19, 221–227. [Google Scholar] [CrossRef]
- Bustamante-Marin, X.M.; Ostrowski, L.E. Cilia and Mucociliary Clearance. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef]
- Ganesan, S.; Comstock, A.T.; Sajjan, U.S. Barrier function of airway tract epithelium. Tissue Barriers 2013, 1. [Google Scholar] [CrossRef] [PubMed]
- De Rose, V.; Molloy, K.; Gohy, S.; Pilette, C.; Greene, C.M. Airway Epithelium Dysfunction in Cystic Fibrosis and COPD. Mediat. Inflamm. 2018. [Google Scholar] [CrossRef] [PubMed]
- Papazian, D.; Würtzen, P.A.; Hansen, S.W.K. Polarized Airway Epithelial Models for Immunological Co-Culture Studies. Int. Arch. Allergy Immunol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Furuse, M.; Hirase, T.; Itoh, M.; Nagafuchi, A.; Yonemura, S.; Tsukita, S.; Tsukita, S. Occludin: A novel integral membrane protein localizing at tight junctions. J. Cell Biol. 1993, 123, 1777–1788. [Google Scholar] [CrossRef]
- Martìn-Padura, I.; Lostaglio, S.; Schneemann, M.; Williams, L.; Romano, M.; Fruscella, P.; Panzeri, C.; Stoppacciaro, A.; Ruco, L.; Villa, A.; et al. Junctional Adhesion Molecule, a Novel Member of the Immunoglobulin Superfamily That Distributes at Intercellular Junctions and Modulates Monocyte Transmigration. J. Cell Biol. 1998, 142, 117–127. [Google Scholar] [CrossRef]
- Forbes, B.; Ehrhardt, C. Human respiratory epithelial cell culture for drug delivery applications. Eur. J. Pharm. Biopharm. 2005, 60, 193–205. [Google Scholar] [CrossRef]
- Srinivasan, B.; Kolli, A.R.; Esch, M.B.; Abaci, H.E.; Shuler, M.L.; Hickman, J.J. TEER measurement techniques for in vitro barrier model systems. J. Lab. Autom. 2015, 20, 107–126. [Google Scholar] [CrossRef]
- Spina, D. Epithelium Smooth Muscle Regulation and Interactions. Am. J. Respir. Crit. Care Med. 1998, 158 (Suppl. S2), S141–S145. [Google Scholar] [CrossRef]
- Wanner, A.; Salathé, M.; O’Riordan, T.G. Mucociliary clearance in the airways. Am. J. Respir. Crit. Care Med. 1996, 154, 1868–1902. [Google Scholar] [CrossRef]
- Satir, P.; Christensen, S.T. Overview of Structure and Function of Mammalian Cilia. Annu. Rev. Physiol. 2007, 69, 377–400. [Google Scholar] [CrossRef]
- Salathe, M. Regulation of Mammalian Ciliary Beating. Annu. Rev. Physiol. 2007, 69, 401–422. [Google Scholar] [CrossRef] [PubMed]
- Villalón, M.; Hinds, T.R.; Verdugo, P. Stimulus-response coupling in mammalian ciliated cells. Demonstration of two mechanisms of control for cytosolic [Ca2+]. Biophys. J. 1989, 56, 1255–1258. [Google Scholar] [CrossRef]
- Salathe, M.; Bookman, R.J. Coupling of [Ca2+] and ciliary beating in cultured tracheal epithelial cells. J. Cell Sci. 1995, 108 Pt 2, 431–440. [Google Scholar]
- Tuvim, M.J.; Mospan, A.R.; Burns, K.A.; Chua, M.; Mohler, P.J.; Melicoff, E.; Adachi, R.; Ammar-Aouchiche, Z.; Davis, C.W.; Dickey, B.F. Synaptotagmin 2 Couples Mucin Granule Exocytosis to Ca2+ Signaling from Endoplasmic Reticulum. J. Biol. Chem. 2009, 284, 9781–9787. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.W.; Dickey, B.F. Regulated airway goblet cell mucin secretion. Annu. Rev. Physiol. 2008, 70, 487–512. [Google Scholar] [CrossRef]
- Wine, J.J.; Joo, N.S. Submucosal Glands and Airway Defense. Proc. Am. Thorac. Soc. 2004, 1, 47–53. [Google Scholar] [CrossRef]
- Ma, J.; Rubin, B.K.; Voynow, J.A. Mucins, Mucus, and Goblet Cells. Chest 2018, 154, 169–176. [Google Scholar] [CrossRef]
- Evans, M.J.; Van Winkle, L.S.; Fanucchi, M.V.; Plopper, C.G. Cellular and molecular characteristics of basal cells in airway epithelium. Exp. Lung Res. 2001, 27, 401–415. [Google Scholar] [CrossRef]
- Jeffery, P.K.; Reid, L. New observations of rat airway epithelium: A quantitative and electron microscopic study. J. Anat. 1975, 120, 295–320. [Google Scholar]
- Wirkes, A.; Jung, K.; Ochs, M.; Mühlfeld, C. Allometry of the mammalian intracellular pulmonary surfactant system. J. Appl. Physiol. 2010, 109, 1662–1669. [Google Scholar] [CrossRef]
- Mason, R.J.; Crystal, R.G. Pulmonary Cell Biology. Am. J. Respir. Crit. Care Med. 1998, 157, S72–S81. [Google Scholar] [CrossRef] [PubMed]
- Plasschaert, L.W.; Žilionis, R.; Choo-Wing, R.; Savova, V.; Knehr, J.; Roma, G.; Klein, A.M.; Jaffe, A.B. A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature 2018, 560, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Montoro, D.T.; Haber, A.L.; Biton, M.; Vinarsky, V.; Lin, B.; Birket, S.E.; Yuan, F.; Chen, S.; Leung, H.M.; Villoria, J.; et al. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 2018, 560, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Gruenert, D.C.; Basbaum, C.B.; Welsh, M.J.; Li, M.; Finkbeiner, W.E.; Nadel, J.A. Characterization of human tracheal epithelial cells transformed by an origin-defective simian virus 40. Proc. Natl. Acad. Sci. USA 1988, 85, 5951–5955. [Google Scholar] [CrossRef] [PubMed]
- Gruenert, D.C.; Willems, M.; Cassiman, J.J.; Frizzell, R.A. Established cell lines used in cystic fibrosis research. J. Cyst. Fibros. 2004, 3, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Cozens, A.L.; Yezzi, M.J.; Chin, L.; Simon, E.M.; Finkbeiner, W.E.; Wagner, J.A.; Gruenert, D.C. Characterization of immortal cystic fibrosis tracheobronchial gland epithelial cells. Proc. Natl. Acad. Sci. USA 1992, 89, 5171–5175. [Google Scholar] [CrossRef] [PubMed]
- Reddel, R.R.; Ke, Y.; Gerwin, B.I.; McMenamin, M.G.; Lechner, J.F.; Su, R.T.; Brash, D.E.; Park, J.-B.; Rhim, J.S.; Harris, C.C. Transformation of Human Bronchial Epithelial Cells by Infection with SV40 or Adenovirus-12 SV40 Hybrid Virus, or Transfection via Strontium Phosphate Coprecipitation with a Plasmid Containing SV40 Early Region Genes. Cancer Res. 1988, 48, 1904–1909. [Google Scholar] [PubMed]
- Haghi, M.; Ong, H.X.; Traini, D.; Young, P. Across the pulmonary epithelial barrier: Integration of physicochemical properties and human cell models to study pulmonary drug formulations. Pharmacol. Ther. 2014, 144, 235–252. [Google Scholar] [CrossRef] [PubMed]
- Zabner, J.; Karp, P.; Seiler, M.; Phillips, S.L.; Mitchell, C.J.; Saavedra, M.; Welsh, M.; Klingelhutz, A.J. Development of cystic fibrosis and noncystic fibrosis airway cell lines. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 284, L844–L854. [Google Scholar] [CrossRef]
- Bhowmick, R.; Gappa-Fahlenkamp, H. Cells and Culture Systems Used to Model the Small Airway Epithelium. Lung 2016, 194, 419–428. [Google Scholar] [CrossRef]
- Falzon, M.; McMahon, J.B.; Schuller, H.M.; Boyd, M.R. Metabolic Activation and Cytotoxicity of 4-Ipomeanol in Human Non-Small Cell Lung Cancer Lines. Cancer Res. 1986, 46, 3484–3489. [Google Scholar] [PubMed]
- Cozens, A.L.; Yezzi, M.J.; Kunzelmann, K.; Ohrui, T.; Chin, L.; Eng, K.; Finkbeiner, W.E.; Widdicombe, J.H.; Gruenert, D.C. CFTR expression and chloride secretion in polarized immortal human bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 1994, 10, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Forbes, B.; Shah, A.; Martin, G.P.; Lansley, A.B. The human bronchial epithelial cell line 16HBE14o- as a model system of the airways for studying drug transport. Int. J. Pharm. 2003, 257, 161–167. [Google Scholar] [CrossRef]
- Bur, M.; Lehr, C.-M. Pulmonary cell culture models to study the safety and efficacy of innovative aerosol medicines. Expert Opin. Drug Deliv. 2008, 5, 641–652. [Google Scholar] [CrossRef]
- Goncz, K.K.; Feeney, L.; Gruenert, D.C. Differential sensitivity of normal and cystic fibrosis airway epithelial cells to epinephrine. Br. J. Pharmacol. 1999, 128, 227–233. [Google Scholar] [CrossRef]
- Ehrhardt, C.; Collnot, E.-M.; Baldes, C.; Becker, U.; Laue, M.; Kim, K.-J.; Lehr, C.-M. Towards an in vitro model of cystic fibrosis small airway epithelium: Characterisation of the human bronchial epithelial cell line CFBE41o. Cell Tissue Res. 2006, 323, 405–415. [Google Scholar] [CrossRef]
- Lieber, M.; Todaro, G.; Smith, B.; Szakal, A.; Nelson-Rees, W. A continuous tumor-cell line from a human lung carcinoma with properties of type II alveolar epithelial cells. Int. J. Cancer 1976, 17, 62–70. [Google Scholar] [CrossRef]
- Rothen-Rutishauser, B.M.; Kiama, S.G.; Gehr, P. A Three-Dimensional Cellular Model of the Human Respiratory Tract to Study the Interaction with Particles. Am. J. Respir. Cell Mol. Biol. 2005, 32, 281–289. [Google Scholar] [CrossRef]
- de Souza Carvalho, C.; Daum, N.; Lehr, C.-M. Carrier interactions with the biological barriers of the lung: Advanced in vitro models and challenges for pulmonary drug delivery. Adv. Drug Deliv. Rev. 2014, 75, 129–140. [Google Scholar] [CrossRef]
- Kreft, M.E.; Jerman, U.D.; Lasič, E.; Hevir-Kene, N.; Rižner, T.L.; Peternel, L.; Kristan, K. The characterization of the human cell line Calu-3 under different culture conditions and its use as an optimized in vitro model to investigate bronchial epithelial function. Eur. J. Pharm. Sci. 2015, 69, 1–9. [Google Scholar] [CrossRef]
- Grainger, C.I.; Greenwell, L.L.; Lockley, D.J.; Martin, G.P.; Forbes, B. Culture of Calu-3 Cells at the Air Interface Provides a Representative Model of the Airway Epithelial Barrier. Pharm. Res. 2006, 23, 1482–1490. [Google Scholar] [CrossRef] [PubMed]
- Wan, H.; Winton, H.L.; Soeller, C.; Stewart, G.A.; Thompson, P.J.; Gruenert, D.C.; Cannell, M.B.; Garrod, D.R.; Robinson, C. Tight junction properties of the immortalized human bronchial epithelial cell lines Calu-3 and 16HBE14o-. Eur. Respir. J. 2000, 15, 1058–1068. [Google Scholar] [CrossRef] [PubMed]
- Stentebjerg-Andersen, A.; Vedsted Notlevsen, I.; Birger, B.; Uhd Nielsen, C. Calu-3 cells grown under AIC and LCC conditions: Implications for dipeptide uptake and transepithelial transport of substances. Eur. J. Pharm. Biopharm. 2011, 78, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Ong, H.X.; Traini, D.; Young, P.M. Pharmaceutical applications of the Calu-3 lung epithelia cell line. Expert Opin. Drug Deliv. 2013, 10, 1287–1302. [Google Scholar] [CrossRef] [PubMed]
- Florea, B.I.; Meaney, C.; Junginger, H.E.; Borchard, G. Transfection efficiency and toxicity of polyethylenimine in differentiated Calu-3 and nondifferentiated COS-1 cell cultures. AAPS PharmSci 2002, 4, 1–11. [Google Scholar] [CrossRef]
- Hein, S.; Bur, M.; Schaefer, U.F.; Lehr, C.-M. A new Pharmaceutical Aerosol Deposition Device on Cell Cultures (PADDOCC) to evaluate pulmonary drug absorption for metered dose dry powder formulations. Eur. J. Pharm. Biopharm. 2011, 77, 132–138. [Google Scholar] [CrossRef]
- Jeong, M.H.; Kim, H.R.; Bang, I.J.; Yoo, S.H.; Lee, S.J.; Lee, K.H.; Chung, K.H. In vitro model for predicting acute inhalation toxicity by using a Calu-3 epithelium cytotoxicity assay. J. Pharmacol. Toxicol. Methods 2019, 98, 106576. [Google Scholar] [CrossRef]
- Lam, E.; Ramke, M.; Groos, S.; Warnecke, G.; Heim, A. A differentiated porcine bronchial epithelial cell culture model for studying human adenovirus tropism and virulence. J. Virol. Methods 2011, 178, 117–123. [Google Scholar] [CrossRef]
- O’Boyle, N.; Sutherland, E.; Berry, C.C.; Davies, R.L. Temporal dynamics of ovine airway epithelial cell differentiation at an air-liquid interface. PLoS ONE 2017, 12, e0181583. [Google Scholar] [CrossRef]
- Lam, H.C.; Choi, A.M.K.; Ryter, S.W. Isolation of Mouse Respiratory Epithelial Cells and Exposure to Experimental Cigarette Smoke at Air Liquid Interface. J. Vis. Exp. 2011. [Google Scholar] [CrossRef]
- Cozens, D.; Sutherland, E.; Marchesi, F.; Taylor, G.; Berry, C.C.; Davies, R.L. Temporal differentiation of bovine airway epithelial cells grown at an air-liquid interface. Sci. Rep. 2018, 8, 14893. [Google Scholar] [CrossRef]
- Lin, H.; Li, H.; Cho, H.-J.; Bian, S.; Roh, H.-J.; Lee, M.-K.; Kim, J.S.; Chung, S.-J.; Shim, C.-K.; Kim, D.-D. Air-Liquid Interface (ALI) Culture of Human Bronchial Epithelial Cell Monolayers as an in vitro Model for Airway Drug Transport Studies. J. Pharm. Sci. 2007, 96, 341–350. [Google Scholar] [CrossRef]
- Hall-Stoodley, L.; Watts, G.; Crowther, J.E.; Balagopal, A.; Torrelles, J.B.; Robison-Cox, J.; Bargatze, R.F.; Harmsen, A.G.; Crouch, E.C.; Schlesinger, L.S. Mycobacterium tuberculosis Binding to Human Surfactant Proteins A and D, Fibronectin, and Small Airway Epithelial Cells under Shear Conditions. Infect. Immun. 2006, 74, 3587–3596. [Google Scholar] [CrossRef]
- Kode, A.; Yang, S.-R.; Rahman, I. Differential effects of cigarette smoke on oxidative stress and proinflammatory cytokine release in primary human airway epithelial cells and in a variety of transformed alveolar epithelial cells. Respir. Res. 2006, 7, 132. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Kalydeco (ivacaftor): EMA/473279/2012. Available online: http://www.accessdata.fda.gov/drugsatfda_docs/label/2012/203188s001lbl.pdf (accessed on 22 June 2020).
- Ishikawa, S.; Matsumura, K.; Kitamura, N.; Ishimori, K.; Takanami, Y.; Ito, S. Application of a direct aerosol exposure system for the assessment of biological effects of cigarette smoke and novel tobacco product vapor on human bronchial epithelial cultures. Regul. Toxicol. Pharmacol. 2018, 96, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Firth, A.L.; Menon, T.; Parker, G.S.; Qualls, S.J.; Lewis, B.M.; Ke, E.; Dargitz, C.T.; Wright, R.; Khanna, A.; Gage, F.H.; et al. Functional Gene Correction for Cystic Fibrosis in Lung Epithelial Cells Generated from Patient iPSCs. Cell Rep. 2015, 12, 1385–1390. [Google Scholar] [CrossRef] [PubMed]
- Vaidyanathan, S.; Salahudeen, A.A.; Sellers, Z.M.; Bravo, D.T.; Choi, S.S.; Batish, A.; Le, W.; Baik, R.; de la O, S.; Kaushik, M.P.; et al. High-Efficiency, Selection-free Gene Repair in Airway Stem Cells from Cystic Fibrosis Patients Rescues CFTR Function in Differentiated Epithelia. Cell Stem Cell 2020, 26, 161–171.e4. [Google Scholar] [CrossRef] [PubMed]
- Pezzulo, A.A.; Starner, T.D.; Scheetz, T.E.; Traver, G.L.; Tilley, A.E.; Harvey, B.-G.; Crystal, R.G.; McCray, P.B.; Zabner, J. The air-liquid interface and use of primary cell cultures are important to recapitulate the transcriptional profile of in vivo airway epithelia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 300, L25–L31. [Google Scholar] [CrossRef] [PubMed]
- Foster, K.A.; Avery, M.L.; Yazdanian, M.; Audus, K.L. Characterization of the Calu-3 cell line as a tool to screen pulmonary drug delivery. Int. J. Pharm. 2000, 208, 1–11. [Google Scholar] [CrossRef]
- Mathias, N.R.; Timoszyk, J.; Stetsko, P.I.; Megill, J.R.; Smith, R.L.; Wall, D.A. Permeability Characteristics of Calu-3 Human Bronchial Epithelial Cells: In Vitro—In Vivo Correlation to Predict Lung Absorption in Rats. J. Drug Target. 2002, 10, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Schaefer, N.; Chu, H.W. Air-Liquid Interface Culture of Human and Mouse Airway Epithelial Cells. In Lung Innate Immunity and Inflammation; Methods in Molecular, Biology; Alper, S., Janssen, W., Eds.; Humana Press: New York, NY, USA, 2018; Volume 1809, pp. 91–109. ISBN 978-1-4939-8570-8. [Google Scholar]
- Robinson, D.; Humbert, M.; Buhl, R.; Cruz, A.A.; Inoue, H.; Korom, S.; Hanania, N.A.; Nair, P. Revisiting Type 2-high and Type 2-low airway inflammation in asthma: Current knowledge and therapeutic implications. Clin. Exp. Allergy 2017, 47, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Jiang, Y.; Sheikh, A.S.; Shen, S.; Liu, J.; Jiang, D. Interleukin-13 stimulates MUC5AC expression via a STAT6-TMEM16A-ERK1/2 pathway in human airway epithelial cells. Int. Immunopharmacol. 2016, 40, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Wu, M.M.J. Effects of retinoids on human bronchial epithelial cells: Differential regulation of hyaluronate synthesis and keratin protein synthesis. J. Cell. Physiol. 1986, 127, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Zhao, Y.H.; Chang, M.M. Growth and differentiation of conducting airway epithelial cells in culture. Eur. Respir. J. 1997, 10, 2398–2403. [Google Scholar] [CrossRef]
- McDowell, E.M.; Keenan, K.P.; Huang, M. Effects of vitamin A-deprivation on hamster tracheal epithelium. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1984, 45, 197–219. [Google Scholar] [CrossRef]
- Koo, J.S.; Yoon, J.-H.; Gray, T.; Norford, D.; Jetten, A.M.; Nettesheim, P. Restoration of the Mucous Phenotype by Retinoic Acid in Retinoid-Deficient Human Bronchial Cell Cultures: Changes in Mucin Gene Expression. Am. J. Respir. Cell Mol. Biol. 1999, 20, 43–52. [Google Scholar] [CrossRef][Green Version]
- Bouwman-Boer, Y.; Fenton-May, V.; Brun, P.L. Practical Pharmaceutics: An International Guideline for the Preparation, Care and Use of Medicinal Products; Springer: Cham, Switzerland, 2015; ISBN 978-3-319-15814-3. [Google Scholar]
- Sinn, P.L.; Shah, A.J.; Donovan, M.D.; McCray, P.B. Viscoelastic gel formulations enhance airway epithelial gene transfer with viral vectors. Am. J. Respir. Cell Mol. Biol. 2005, 32, 404–410. [Google Scholar] [CrossRef]
- Ravindran, S.; Song, Y.; George, A. Development of Three-Dimensional Biomimetic Scaffold to Study Epithelial-Mesenchymal Interactions. Tissue Eng. Part A 2010, 16, 327–342. [Google Scholar] [CrossRef]
- DeVolder, R.; Kong, H.-J. Hydrogels for in vivo-like three-dimensional cellular studies. Wiley Interdiscip. Rev. Syst. Biol. Med. 2012, 4, 351–365. [Google Scholar] [CrossRef]
- Harrington, H.; Cato, P.; Salazar, F.; Wilkinson, M.; Knox, A.; Haycock, J.W.; Rose, F.; Aylott, J.W.; Ghaemmaghami, A.M. Immunocompetent 3D Model of Human Upper Airway for Disease Modeling and In Vitro Drug Evaluation. Mol. Pharm. 2014, 11, 2082–2091. [Google Scholar] [CrossRef]
- Meenach, S.A.; Tsoras, A.N.; McGarry, R.C.; Mansour, H.M.; Hilt, J.Z.; Anderson, K.W. Development of three-dimensional lung multicellular spheroids in air- and liquid-interface culture for the evaluation of anticancer therapeutics. Int. J. Oncol. 2016, 48, 1701–1709. [Google Scholar] [CrossRef] [PubMed]
- Huh, D.; Hamilton, G.A.; Ingber, D.E. From Three-Dimensional Cell Culture to Organs-on-Chips. Trends Cell Biol. 2011, 21, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Rimington, T.L.; Hodge, E.; Billington, C.K.; Bhaker, S.; Kc, B.; Kilty, I.; Jelinsky, S.; Hall, I.P.; Sayers, I. Defining the inflammatory signature of human lung explant tissue in the presence and absence of glucocorticoid. F1000Research 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Glorieux, S.; Van den Broeck, W.; van der Meulen, K.M.; Van Reeth, K.; Favoreel, H.W.; Nauwynck, H.J. In vitro culture of porcine respiratory nasal mucosa explants for studying the interaction of porcine viruses with the respiratory tract. J. Virol. Methods 2007, 142, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Seeger, W.; Walmrath, D.; Grimminger, F.; Rosseau, S.; Schütte, H.; Krämer, H.-J.; Ermert, L.; Kiss, L. Adult respiratory distress syndrome: Model systems using isolated perfused rabbit lungs. In Methods in Enzymology; Oxygen Radicals in Biological Systems Part C; Academic Press: Cambridge, MA, USA, 1994; Volume 233, pp. 549–584. [Google Scholar]
- Beck-Broichsitter, M.; Gauss, J.; Packhaeuser, C.B.; Lahnstein, K.; Schmehl, T.; Seeger, W.; Kissel, T.; Gessler, T. Pulmonary drug delivery with aerosolizable nanoparticles in an ex vivo lung model. Int. J. Pharm. 2009, 367, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Lau, E.M.T.; Roche, N.A.; Reddel, H.K. Therapeutic approaches to asthma-chronic obstructive pulmonary disease overlap. Expert Rev. Clin. Immunol. 2017, 13, 449–455. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).