The Impact of Chronic Mild Stress and Agomelatine Treatment on the Expression Level and Methylation Status of Genes Involved in Tryptophan Catabolic Pathway in PBMCs and Brain Structures

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
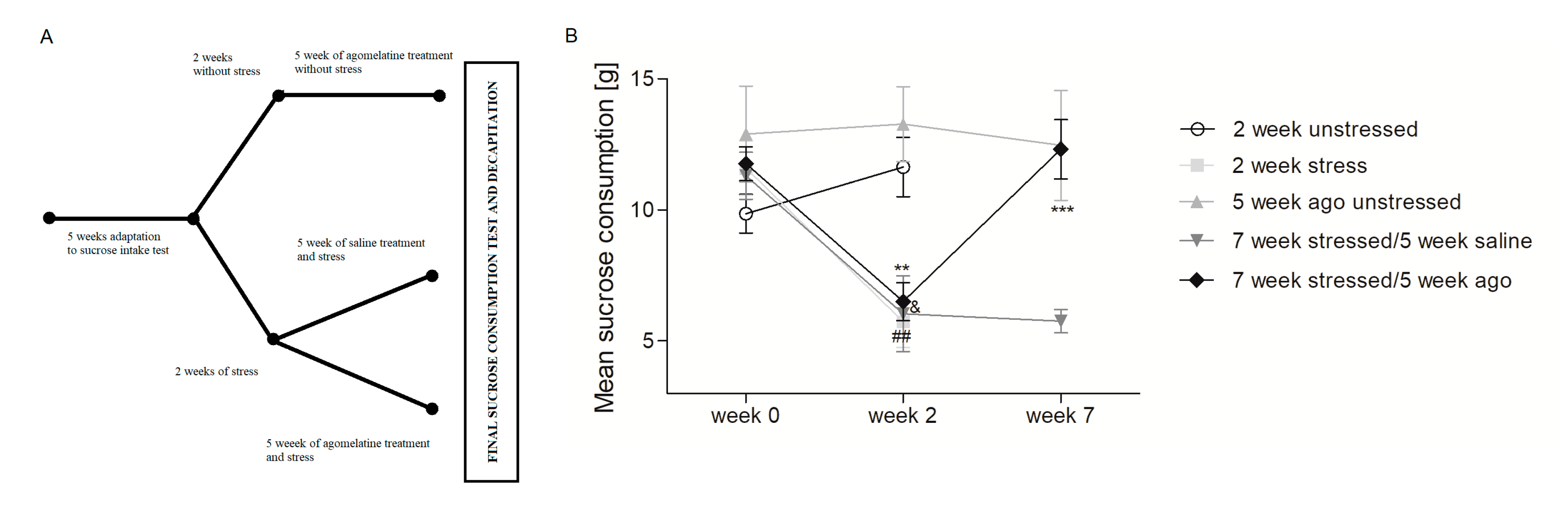
2.2. Chronic Mild Stress (CMS) Procedure
2.3. Specimen Collection
2.4. Real-Time PCR
2.5. Methylation-Sensitive High-Tesolution Melting (MS-HRM) PCR
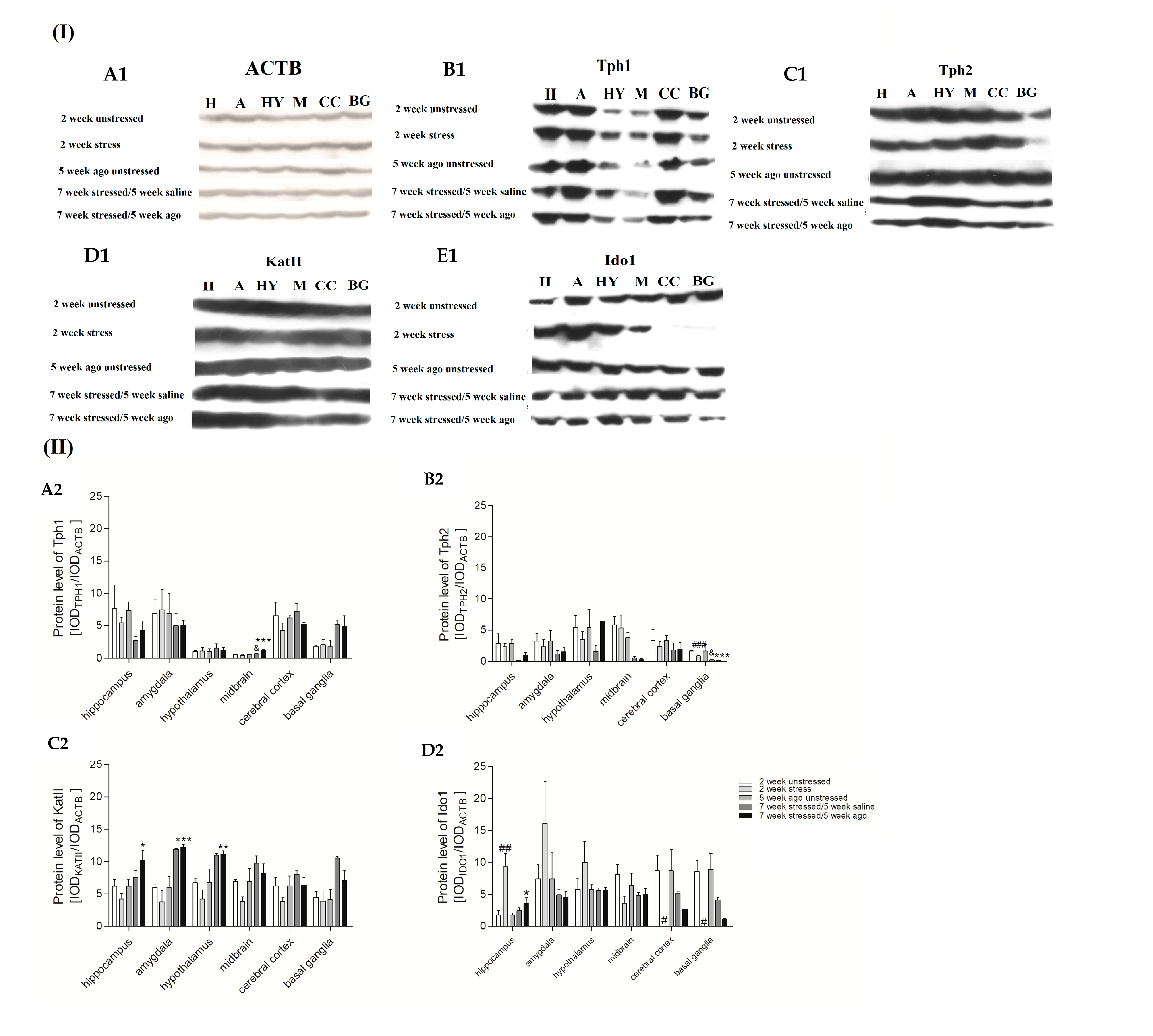
2.6. Western Blot
2.7. Statistical Analysis
3. Results
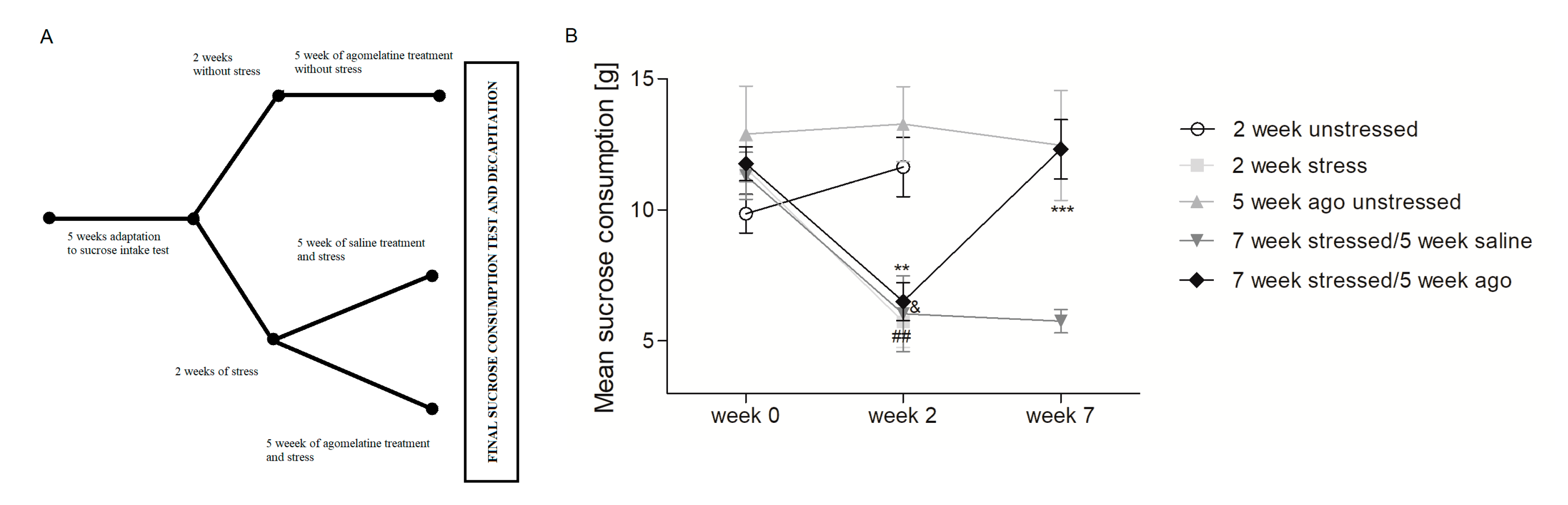
3.1. Agomelatine Impact on the Sucrose Intake of Rats Exposed to Chronioc Mild Stress
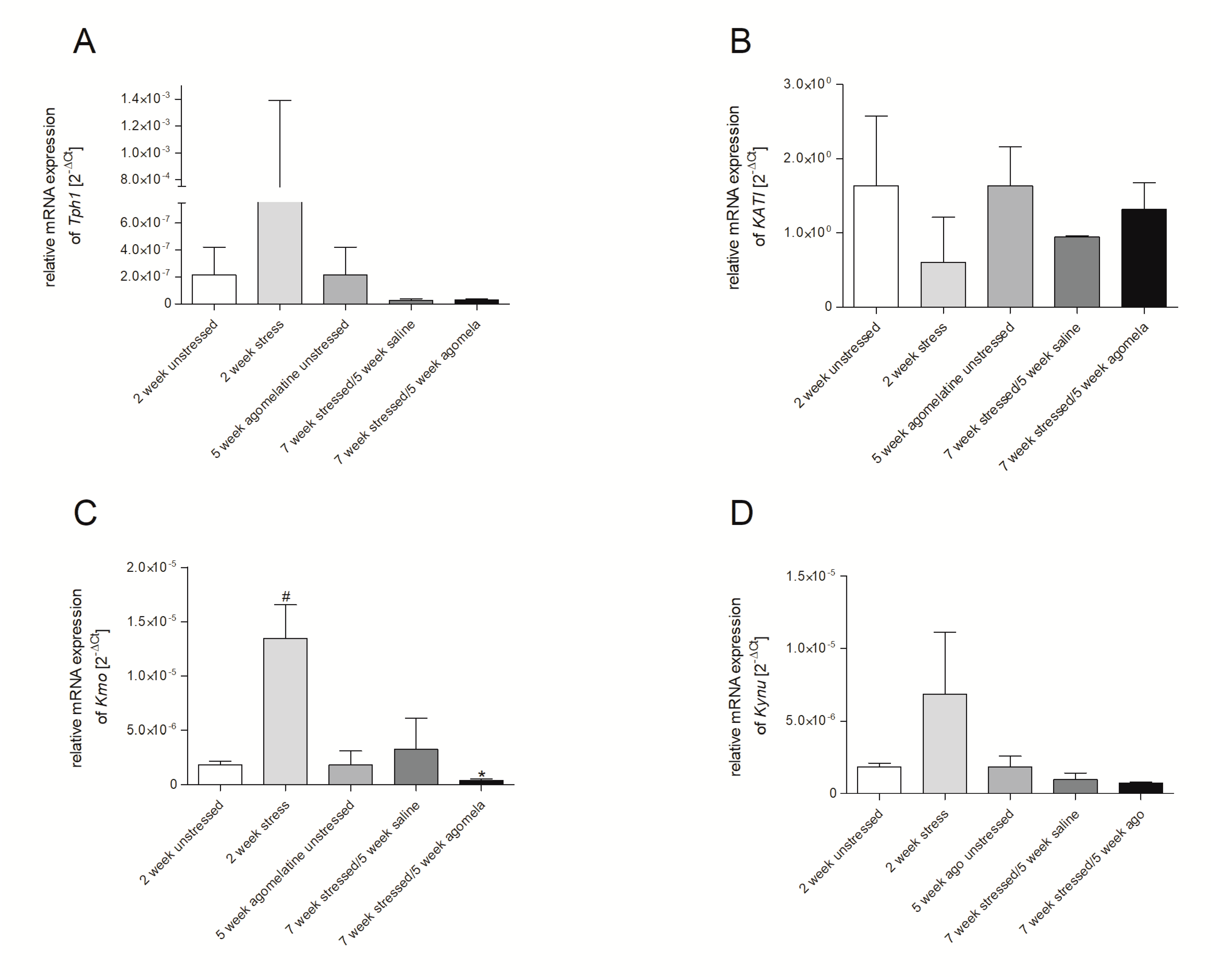
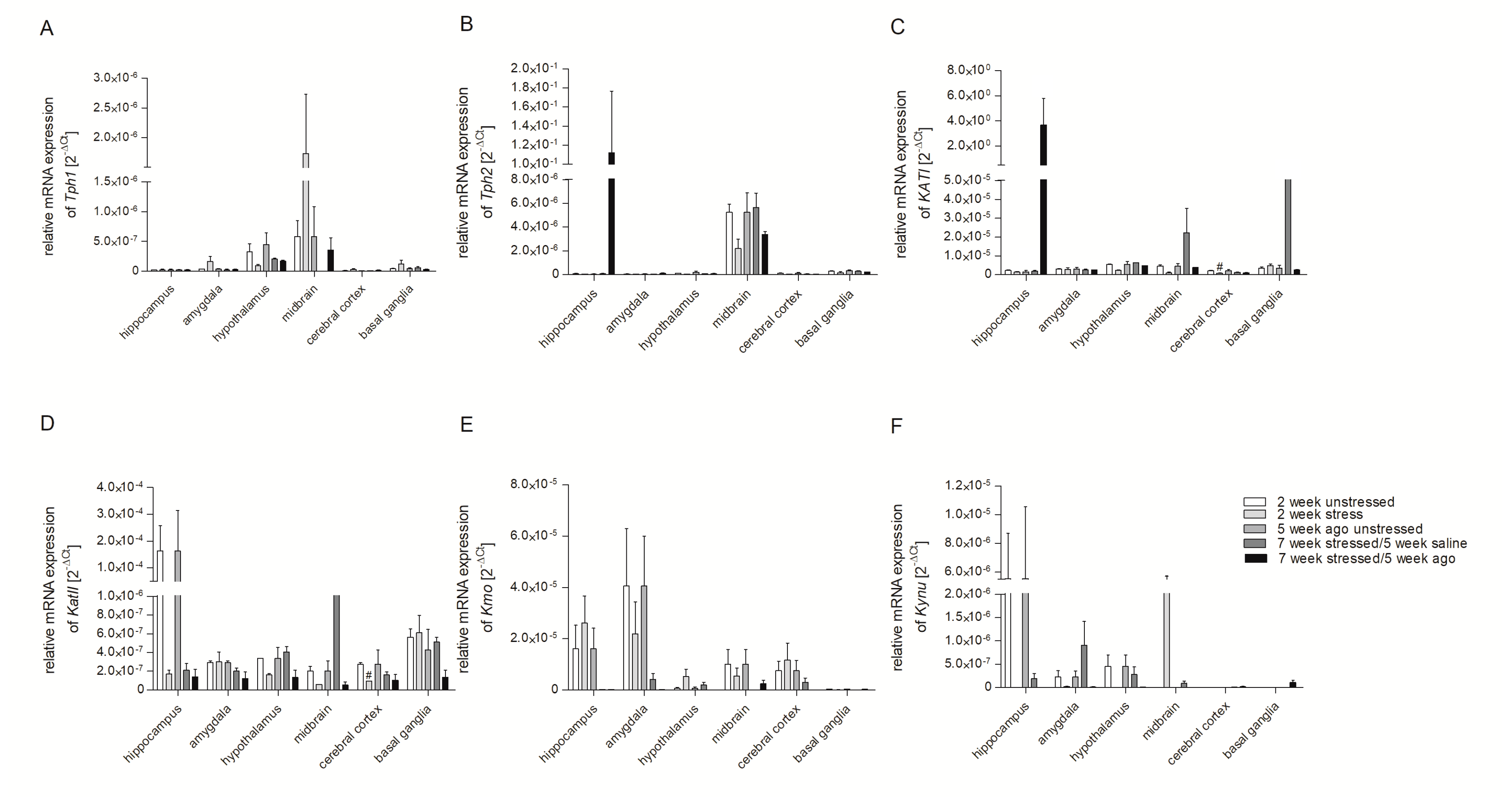
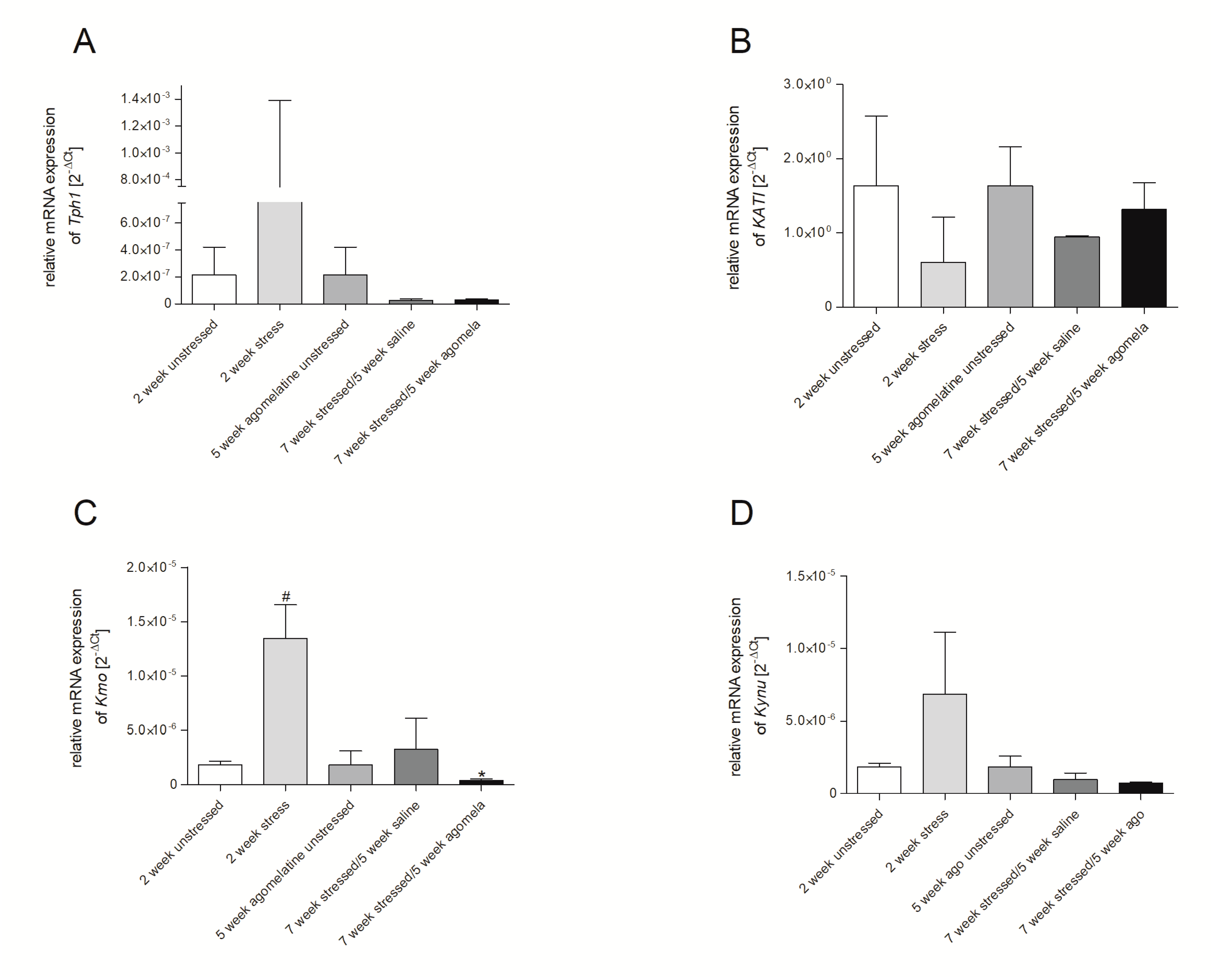
3.2. mRNA Expression of Genes Involved in Tryptophan Catabolic Pathway in PBMCs and Brain Structures
3.3. Aagomelatine Impact on the mRNA Expression Level in PBMCs and Brain Structures
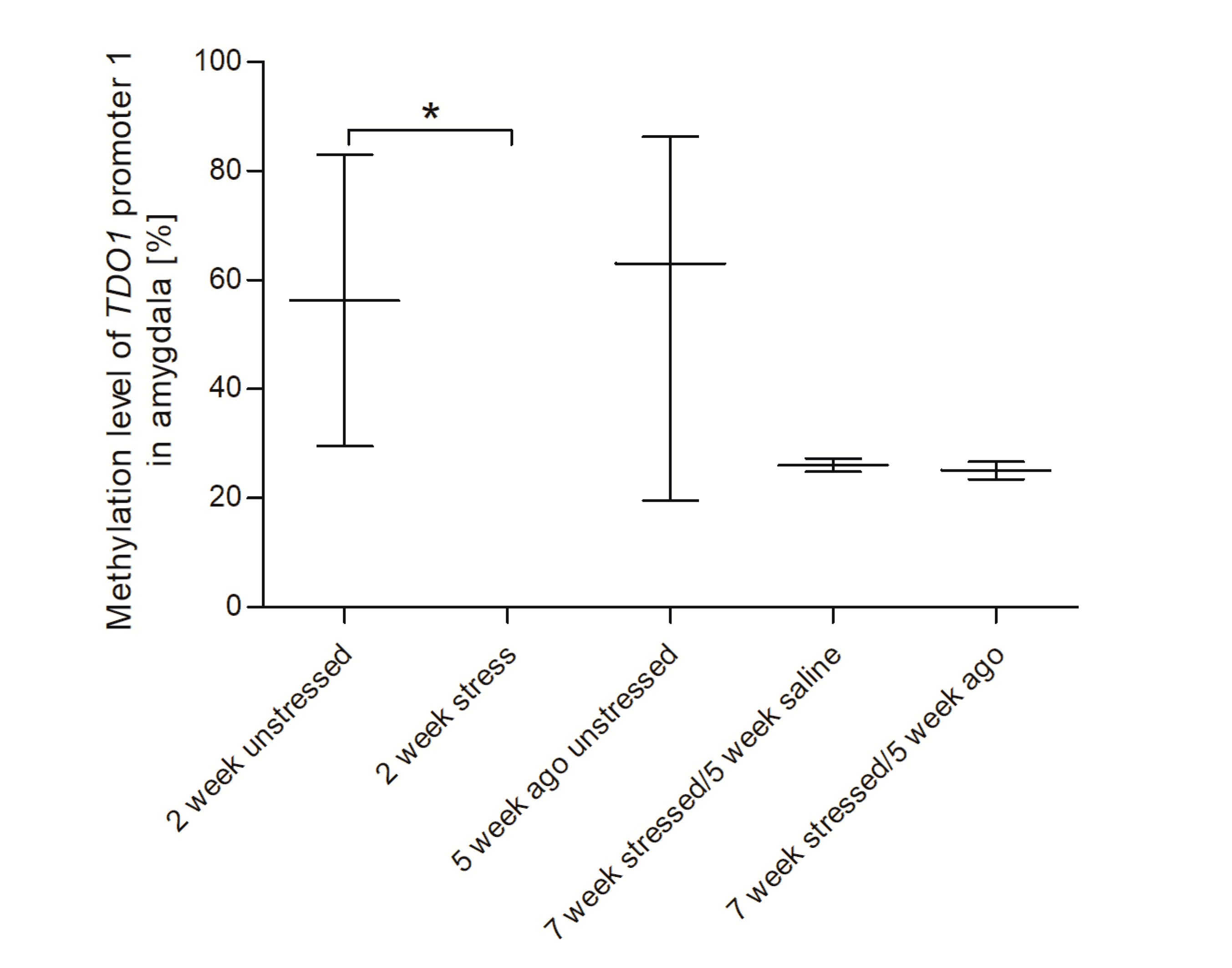
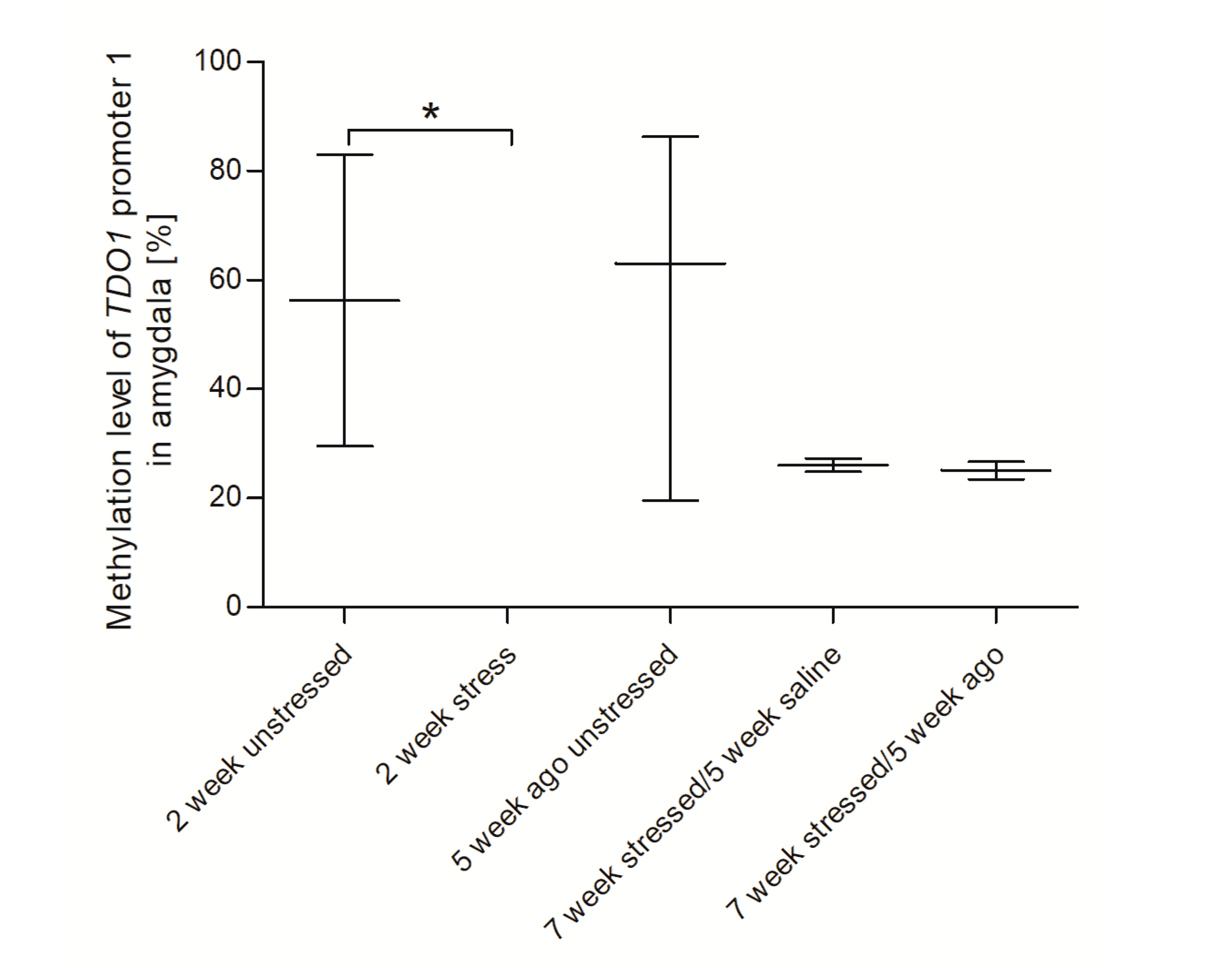
3.4. Methylation Status of Promoter Region of Genes Encoding Tryptophan Catabolic Pathway Enzymes in PBMCs and Brain Structures
3.5. Agomelatine Impact on the Methylation Status of Gene Promoters in PBMCs and Brain Structures
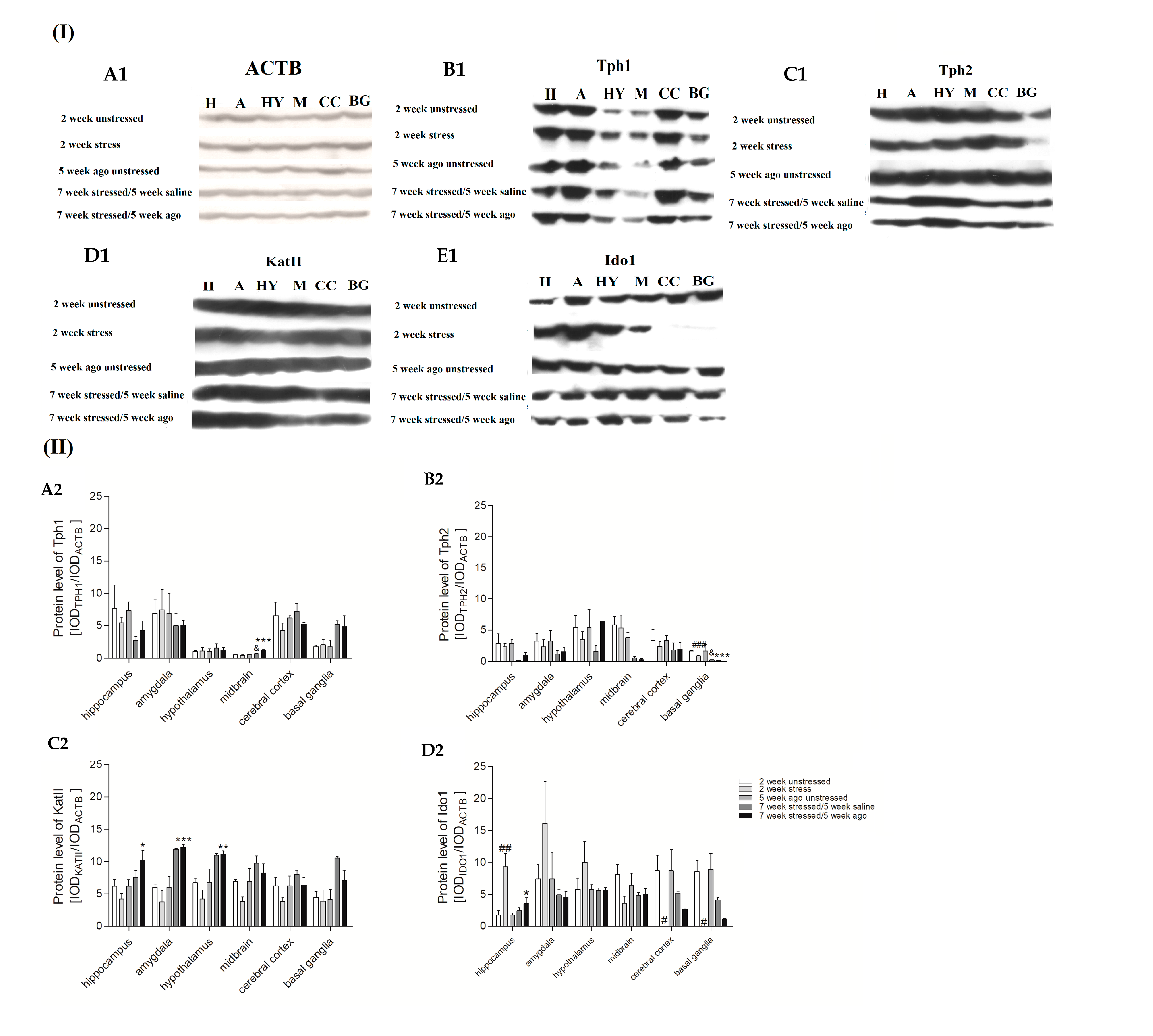
3.6. Protein Expression Level in Brain Structures
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organization—Depression. Available online: https://www.who.int/news-room/fact-sheets/detail/depression (accessed on 16 March 2020).
- Kessler, R.C.; Berglund, P.; Demler, O.; Jin, R.; Koretz, D.; Merikangas, K.R.; Rush, A.J.; Walters, E.E.; Wang, P.S. The epidemiology of major depressive disorder: Results from the National Comorbidity Survey Replication (NCS-R). JAMA 2003, 289, 3095–3105. [Google Scholar] [CrossRef] [PubMed]
- Capuron, L.; Ravaud, A.; Neveu, P.J.; Miller, A.H.; Maes, M.; Dantzer, R. Association between decreased serum tryptophan concentrations and depressive symptoms in cancer patients undergoing cytokine therapy. Mol. Psychiatr. 2002, 7, 468–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, S.; Fujii, T.; Koga, N.; Hori, H.; Teraishi, T.; Hattori, K.; Noda, T.; Higuchi, T.; Motohashi, N.; Kunugi, H. Plasma L-tryptophan concentration in major depressive disorder: New data and meta-analysis. J. Clin. Psychiatr. 2014, 75, e906–e915. [Google Scholar] [CrossRef]
- Chen, Y.; Xu, H.; Zhu, M.; Liu, K.; Lin, B.; Luo, R.; Chen, C.; Li, M. Stress inhibits tryptophan hydroxylase expression in a rat model of depression. Oncotarget 2017, 8, 63247–63257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wigner, P.; Czarny, P.; Synowiec, E.; Bijak, M.; Białek, K.; Talarowska, M.; Galecki, P.; Szemraj, J.; Sliwinski, T. Association between single nucleotide polymorphisms of TPH1 and TPH2 genes, and depressive disorders. J. Cell. Mol. Med. 2018, 22, 1778–1791. [Google Scholar] [CrossRef] [Green Version]
- Wigner, P.; Czarny, P.; Synowiec, E.; Bijak, M.; Talarowska, M.; Galecki, P.; Szemraj, J.; Sliwinski, T. Variation of genes encoding KAT1, AADAT and IDO1 as a potential risk of depression development. Eur. Psychiatr. 2018, 52, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Maes, M.; Leonard, B.E.; Myint, A.M.; Kubera, M.; Verkerk, R. The new ‘5-HT’ hypothesis of depression: Cell-mediated immune activation induces indoleamine-2,3-dioxygenase, which leads to lower plasma tryptophan and an increased synthesis of detrimental tryptophan catabolites (TRYCATs), both of which contribute to the onset of depression. Prog. Neuro-Psychopharmacol. Biol. Psychiatr. 2011, 35, 702–721. [Google Scholar]
- Maes, M.; Galecki, P.; Verkerk, R.; Rief, W. Somatization, but not depression, is characterized by disorders in the tryptophan catabolite (TRYCAT) pathway, indicating increased indoleamine 2,3-dioxygenase and lowered kynurenine aminotransferase activity. Neuroendocr. Lett. 2011, 32, 264–273. [Google Scholar]
- Lezheiko, T.V.; Golimbet, V.E.; Andryushchenko, A.V.; Melik-Pashayan, A.E.; Mironova, E.V. A study of the association between the kynurenine-3-monooxygenase gene and depression. Zhurnal Nevrol. Psikhiatrii Im. SS Korsakova 2016, 116, 92–95. [Google Scholar] [CrossRef]
- Kwidzinski, E.; Bechmann, I. IDO expression in the brain: A double-edged sword. J. Mol. Med. 2007, 85, 1351–1359. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.F. Quest for biomarkers of treatment-resistant depression: Shifting the paradigm toward risk. Front. Psychiatr. 2013, 18, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Gainetdinov, R.R.; Beaulieu, J.M.; Sotnikova, T.D.; Burch, L.H.; Williams, R.B.; Schwartz, D.A.; Krishnan, K.R.; Caron, M.G. Loss-of-function mutation in tryptophan hydroxylase-2 identified in unipolar major depression. Neuron 2005, 45, 11–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bestor, T.; Laudano, A.; Mattaliano, R.; Ingram, V. Cloning and sequencing of a cDNA encoding DNA methyltransferase of mouse cells. The carboxyl-terminal domain of the mammalian enzymes is related to bacterial restriction methyltransferases. J. Mol. Biol. 1988, 203, 971–983. [Google Scholar] [CrossRef]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Guidotti, A.; Auta, J.; Chen, Y.; Davis, J.M.; Dong, E.; Gavin, D.P.; Grayson, D.R.; Matrisciano, F.; Pinna, G.; Satta, R.; et al. Epigenetic GABAergic targets in schizophrenia and bipolar disorder. Neuropharmacology 2011, 60, 1007–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poulter, M.O.; Du, L.; Weaver, I.C.G.; Palkovits, M.; Faludi, G.; Merali, Z.; Szyf, M.; Anisman, H. GABAA receptor promoter hypermethylation in suicide brain: Implications for the involvement of epigenetic processes. Biol. Psychiatr. 2008, 64, 645–652. [Google Scholar] [CrossRef]
- Melas, P.A.; Rogdaki, M.; Lennartsson, A.; Bjork, K.; Qi, H.; Witasp, A.; Werme, M.; Wegener, G.; Mathe, A.A.; Svenningsson, P.; et al. Antidepressant treatment is associated with epigenetic alterations in the promoter of P11 in a genetic model of depression. Int. J. Neuropsychopharmacol. 2011, 15, 669–679. [Google Scholar] [CrossRef] [Green Version]
- Kessler, R.C.; Demler, O.; Frank, R.G.; Olfson, M.; Pincus, H.A.; Walters, E.E.; Wang, P.; Wells, K.B.; Zaslavsky, A.M. Prevalence and treatment of mental disorders, 1990 to 2003. N. Engl. J. Med. 2005, 352, 2515–2523. [Google Scholar] [CrossRef] [Green Version]
- Parikh, S.V.; Lam, R.W.; CANMAT Depression Work Group. Clinical guidelines for the treatment of depressive disorders, I. Definitions, prevalence, and health burden. Can. J. Psychiatr. 2001, 46, 13S–20S. [Google Scholar]
- Frank, E.; Prien, R.F.; Jarrett, R.B.; Keller, M.B.; Kupfer, D.J.; Lavori, P.W.; Rush, A.J.; Weissman, M.M. Conceptualization and rationale for consensus definitions of terms in major depressive disorder: Remission, recovery, relapse, and recurrence. Arch. Gen. Psychiatr. 1991, 48, 851–856. [Google Scholar] [CrossRef]
- Kennedy, S.H.; Eisfeld, B.S.; Meyer, J.H.; Bagby, R.M. Antidepressants in clinical practice: Limitations of assessment methods and drug response. Human Psychopharmacol. Clin. Exp. 2001, 16, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Vanderkooy, J.D.; Kennedy, S.H.; Bagby, R.M. Antidepressant side effects in depression patients treated in a naturalistic setting: A study of bupropion, moclobemide, paroxetine, sertraline, and venlafaxine. Can. J. Psychiatr. 2002, 47, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Zajecka, J.M. Augmentation strategies to increase antidepressant tolerability. J. Clin. Psychiatr. 2007, 68, 23–27. [Google Scholar]
- Zajecka, J.M. Clinical issues in long-term treatment with antidepressants. J. Clin. Psychiatr. 2000, 61, 20–25. [Google Scholar]
- EMA Valdoxan (Agomelatine). Summary of Product Characteristics; EMA: Amsterdam, The Netherlands, 2008. [Google Scholar]
- Kennedy, S.H.; Eisfeld, B.S. Agomelatine and its therapeutic potential in the depressed patient. Neuropsychiatr. Dis. Treat. 2007, 3, 423–428. [Google Scholar]
- Popoli, M. Agomelatine: Innovative pharmacological approach in depression. CNS Drugs 2009, 23, 27–34. [Google Scholar] [CrossRef]
- De Berardis, D.; Di Iorio, G.; Acciavatti, T.; Conti, C.; Serroni, N.; Olivieri, L.; Cavuto, M.; Martinotti, G.; Janiri, L.; Moschetta, F.S.; et al. The emerging role of melatonin agonists in the treatment of major depression: Focus on agomelatine. CNS Neurol. Disord. Drug Targets 2011, 10, 119–132. [Google Scholar] [CrossRef]
- Martinotti, G.; Sepede, G.; Gambi, F.; Di Iorio, G.; De Berardis, D.; Di Nicola, M.; Onofrj, M.; Janiri, L.; Di Giannantonio, M. Agomelatine versus venlafaxine XR in the treatment of anhedonia in major depressive disorder: A pilot study. J. Clin. Psychopharmacol. 2012, 32, 487–491. [Google Scholar] [CrossRef]
- Guaiana, G.; Gupta, S.; Chiodo, D.; Davies, S.J.; Haederle, K.; Koesters, M. Agomelatine versus other antidepressive agents for major depression. Cochrane Database Syst. Rev. 2013, 12, CD008851. [Google Scholar] [CrossRef] [Green Version]
- Montastruc, F.; Scotto, S.; Vaz, I.R.; Guerra, L.N.; Escudero, A.; Sáinz, M.; Falomir, T.; Bagheri, H.; Herdeiro, M.T.; Venegoni, M.; et al. Hepatotoxicity related to agomelatine and other new antidepressants: A case/noncase approach with information from the Portuguese, French, Spanish, and Italian pharmacovigilance systems. J. Clin. Psychopharmacol. 2014, 34, 327–330. [Google Scholar] [CrossRef]
- Papp, M. Models of affective illness: Chronic mild stress in the rat. Curr. Protoc. Pharmacol. 2012, 57, 5–9. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–8110. [Google Scholar] [CrossRef] [PubMed]
- Wojdacz, T.K.; Dobrovic, A. Methylation-sensitive high resolution melting (MS-HRM): A new approach for sensitive and high-throughput assessment of methylation. Nucleic Acids Res. 2007, 35, e41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojdacz, T.K.; Dobrovic, A.; Hansen, L.L. Methylation-sensitive high-resolution melting. Nat. Protoc. 2008, 3, 1903–1908. [Google Scholar] [CrossRef] [PubMed]
- Wojdacz, T.K.; Borgbo, T.; Hansen, L.L. Primer design versus PCR bias in methylation independent PCR amplifications. Epigenetics 2009, 4, 231–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar]
- Towbin, H.; Staehelin, T.; Gordon, J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: Procedure and some applications. Proc. Natl. Acad. Sci. USA 1979, 76, 4350–4354. [Google Scholar] [CrossRef] [Green Version]
- Gamaro, G.D.; Manoli, L.P.; Torres, I.L.; Silveira, R.; Dalmaz, C. Effects of chronic variate stress on feeding behavior and on monoamine levels in different rat brain structures. Neurochem. Int. 2003, 42, 107–114. [Google Scholar] [CrossRef]
- Bekris, S.; Antoniou, K.; Daskas, S.; Papadopoulou-Daifoti, Z. Behavioural and neurochemical effects induced by chronic mild stress applied to two different rat strains. Behav. Brain Res. 2005, 161, 45–59. [Google Scholar] [CrossRef]
- Réus, G.Z.; Jansen, K.; Titus, S.; Carvalho, A.; Gabbay, V.; Quevedo, J. Kynurenine pathway dysfunction in the pathophysiology and treatment of depression: Evidences from animal and human studies. J. Psychiatr. Res. 2015, 68, 316–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okuda, S.; Nishiyama, N.; Saito, H.; Katsuki, H. 3-Hydroxykynurenine, an endogenous oxidative stress generator, causes neuronal cell death with apoptotic features and region selectivity. J. Neurochem. 1998, 70, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Jayawickrama, G.S.; Nematollahi, A.; Sun, G.; Gorrell, M.D.; Church, W.B. Inhibition of human kynurenine aminotransferase isozymes by estrogen and its derivatives. Sci. Rep. 2017, 7, 17559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baquero, M.; Martin, N. Depressive symptoms in neurodegenerative diseases. World J. Clin. Cases 2015, 3, 682–693. [Google Scholar] [CrossRef] [PubMed]
- Kocki, T.; Wnuk, S.; Kloc, R.; Kocki, J.; Owe-Larsson, B.; Urbanska, E.M. New insight into the antidepressants action: Modulation of kynurenine pathway by increasing the kynurenic acid/3-hydroxykynurenine ratio. J. Neural Transm. (Vienna) 2012, 119, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Laumet, G.; Zhou, W.; Dantzer, R.; Edralin, J.D.; Huo, X.; Budac, D.P.; O’Connor, J.C.; Lee, A.W.; Heijnen, C.J.; Kavelaars, A. Upregulation of neuronal kynurenine 3-monooxygenase mediates depression-like behavior in a mouse model of neuropathic pain. Brain Behav. Immun. 2017, 66, 94–102. [Google Scholar] [CrossRef]
- Wang, B.; Lian, Y.J.; Su, W.J.; Peng, W.; Dong, X.; Liu, L.L.; Gong, H.; Zhang, T.; Jiang, C.L.; Wang, Y.X. HMGB1 mediates depressive behavior induced by chronic stress through activating the kynurenine pathway. Brain Behav. Immun. 2018, 72, 51–60. [Google Scholar] [CrossRef]
- Duda, W.; Curzytek, K.; Kubera, M.; Connor, T.J.; Fagan, E.M.; Basta-Kaim, A.; Trojan, E.; Papp, M.; Gruca, P.; Budziszewska, B.; et al. Interaction of the immune-inflammatory and the kynurenine pathways in rats resistant to antidepressant treatment in model of depression. Int. Immunopharmacol. 2019, 73, 527–538. [Google Scholar] [CrossRef]
- Kocki, T.; Urbańska, E.M.; Kocki, J.; Kloc, R.; Kocka, K.; Olajossy, M.; Owe-Larsson, B. Prolonged therapy with antidepressants increases hippocampal level of kynurenic acid and expression of Kat1 and Kat2 genes. Pharmacol. Rep. 2018, 70, 737–745. [Google Scholar] [CrossRef]
- Bergamini, G.; Mechtersheimer, J.; Azzinnari, D.; Sigrist, H.; Buerge, M.; Dallmann, R.; Freije, R.; Kouraki, A.; Opacka-Juffry, J.; Seifritz, E.; et al. Chronic social stress induces peripheral and central immune activation, blunted mesolimbic dopamine function, and reduced reward-directed behaviour in mice. Neurobiol. Stress 2018, 8, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Jiao, H.; Yan, Z.; Ma, Q.; Li, X.; Jiang, Y.; Liu, Y.; Chen, J. Influence of Xiaoyaosan on depressive-like behaviors in chronic stress-depressed rats through regulating tryptophan metabolism in hippocampus. Neuropsychiatr. Dis. Treat. 2018, 15, 21–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Chen, L.; Lim, G.; Sung, B.; Wang, S.; McCabe, M.F.; Rusanescu, G.; Yang, L.; Tian, Y.; Mao, J. Brain indoleamine 2,3-dioxygenase contributes to the comorbidity of pain and depression. J. Clin. Investig. 2012, 122, 2940–2954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myint, A.M.; Kim, Y.K.; Verkerk, R.; Scharpé, S.; Steinbusch, H.; Leonard, B. Kynurenine pathway in major depression: Evidence of impaired neuroprotection. J. Affect. Disord. 2007, 98, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Wichers, M.C.; Koek, G.H.; Robaeys, G.; Verkerk, R.; Scharpé, S.; Maes, M. IDO and interferon-α-induced depressive symptoms: A shift in hypothesis from tryptophan depletion to neurotoxicity. Mol. Psychiatr. 2005, 10, 538–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, W.; Cai, L.; Yu, Y.; Gao, L.; Xiao, L.; He, Q.; Ren, Z.; Liu, Y. Activation of brain indoleamine 2,3-dioxygenase contributes to epilepsy-associated depressive-like behavior in rats with chronic temporal lobe epilepsy. J. Neuroinflamm. 2014, 11, 41. [Google Scholar] [CrossRef] [Green Version]
- Jacobsen, J.P.; Siesser, W.B.; Sachs, B.D.; Peterson, S.; Cools, M.J.; Setola, V.; Folgering, J.H.; Flik, G.; Caron, M.G. Deficient serotonin neurotransmission and depression-like serotonin biomarker alterations in tryptophan hydroxylase 2 (Tph2) loss-of-function mice. Mol. Psychiatr. 2012, 17, 694–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Li, X.; He, S.; Hu, L.; Guo, J.; Huang, X.; Hu, J.; Qi, Y.; Chen, B.; Shang, D.; et al. Regulation of the kynurenine metabolism pathway by Xiaoyao San and the underlying effect in the hippocampus of the depressed rat. J. Ethnopharmacol. 2018, 214, 13–21. [Google Scholar] [CrossRef]
- Wiborg, O. Chronic mild stress for modeling anhedonia. Cell Tissue Res. 2013, 354, 155–169. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wigner, P.; Synowiec, E.; Jóźwiak, P.; Czarny, P.; Białek, K.; Bijak, M.; Szemraj, J.; Gruca, P.; Papp, M.; Sliwinski, T. The Impact of Chronic Mild Stress and Agomelatine Treatment on the Expression Level and Methylation Status of Genes Involved in Tryptophan Catabolic Pathway in PBMCs and Brain Structures. Genes 2020, 11, 1093. https://doi.org/10.3390/genes11091093
Wigner P, Synowiec E, Jóźwiak P, Czarny P, Białek K, Bijak M, Szemraj J, Gruca P, Papp M, Sliwinski T. The Impact of Chronic Mild Stress and Agomelatine Treatment on the Expression Level and Methylation Status of Genes Involved in Tryptophan Catabolic Pathway in PBMCs and Brain Structures. Genes. 2020; 11(9):1093. https://doi.org/10.3390/genes11091093
Chicago/Turabian StyleWigner, Paulina, Ewelina Synowiec, Paweł Jóźwiak, Piotr Czarny, Katarzyna Białek, Michal Bijak, Janusz Szemraj, Piotr Gruca, Mariusz Papp, and Tomasz Sliwinski. 2020. "The Impact of Chronic Mild Stress and Agomelatine Treatment on the Expression Level and Methylation Status of Genes Involved in Tryptophan Catabolic Pathway in PBMCs and Brain Structures" Genes 11, no. 9: 1093. https://doi.org/10.3390/genes11091093