
Mitochondrial Dysfunction and Glycolytic Shift in the Tumor Microenvironment: Impact on Paclitaxel Efficacy in Cancer Therapy

 , , , , , and
, , , , , and
Abstract
1. Introduction
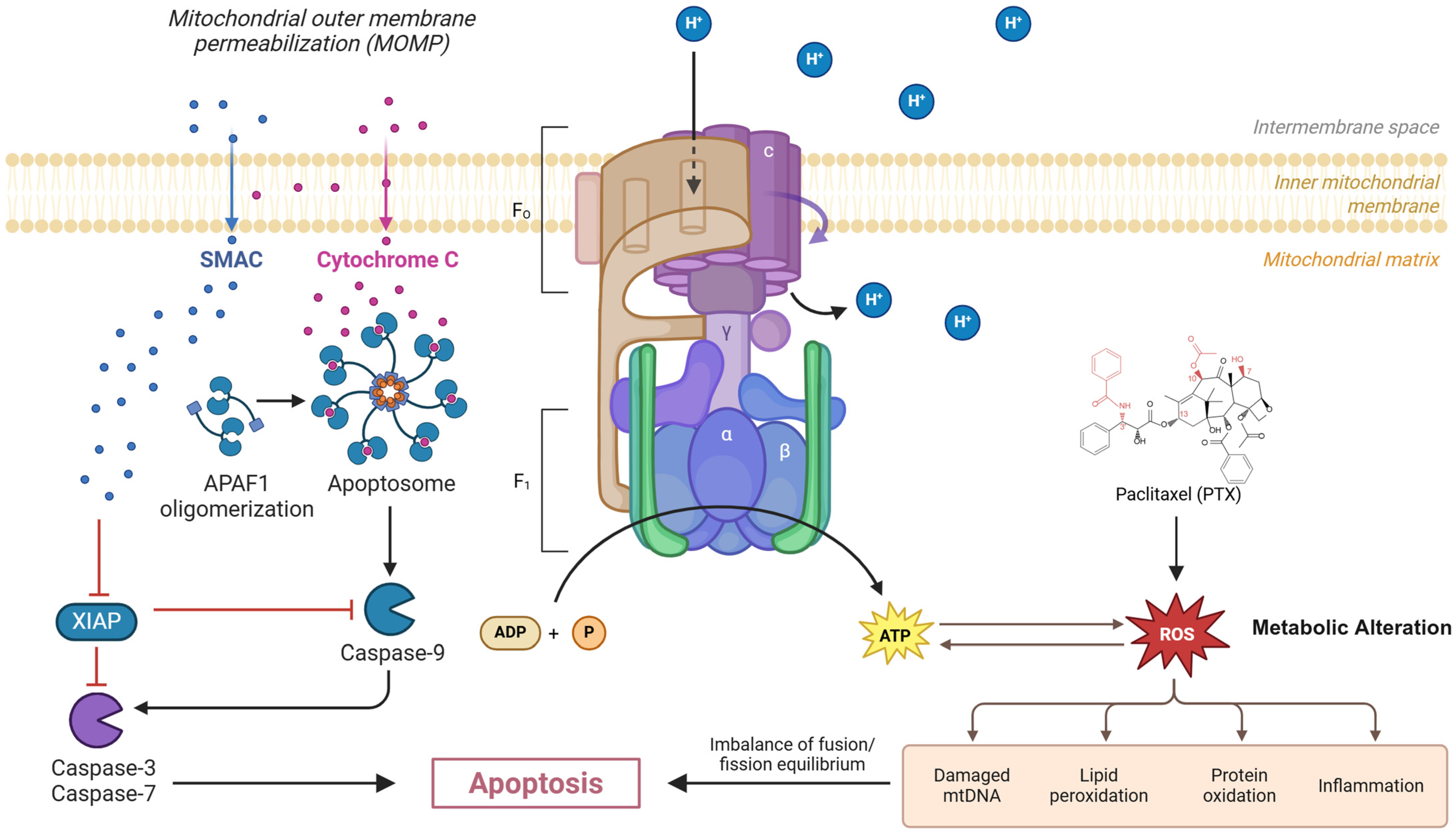
2. Mechanistic Insights
3. Metabolic Drivers of Paclitaxel Resistance in Major Cancers

{kind=link}
{kind=link}
| Metabolic Pathway | Ovarian | Breast (TNBC) | NSCLC | Cross-Cancer Relevance |
|---|---|---|---|---|
| Glycolytic flux | HK2 ↑, PGAM1 ↑ | Baseline glycolysis ↑ | PDK2 ↑ | Universal resistance driver |
| OXPHOS dependence | – | Residual disease ↑ | – | Contextual vulnerability |
| Lipid metabolism | CPT1A ↑, FASN ↑ | – | FASN ↑, CPT1 ↓ | Hypoxia-associated adaptation |
| Hypoxia response | Indirect | Indirect | HIF-1α direct | NSCLC > ovarian/breast |
| Cancer Type | Model System | Metabolic Alteration | Impact on PTX Efficacy | Key Molecular Target/Pathway | Strategy Tested | Outcome | Key Finding | Ref |
|---|---|---|---|---|---|---|---|---|
| Ovarian clear cell carcinoma (OCCC) | OCCC cell lines (resistant vs. parental) [in vitro] | Increased HK2-driven glycolysis (decreased OCR/ATP, increased NADH/NAD+) | PTX resistance (reduced apoptosis) | HK2 | HK2-PROTAC degrader | Reduced glycolysis, partial restoration of PTX sensitivity | HK2 degradation partially restores PTX sensitivity | [13] |
| Triple-negative breast cancer (TNBC) | TNBC cell lines, patient organoids [in vitro/ex vivo] | PTX-induced shift to enhanced glycolysis (decreased OXPHOS) | Enhanced glycolysis correlates with residual disease; chemoresistance | Glycolytic enzymes (general) | Glycolysis inhibitors (e.g., 2-DG) | Markedly improved PTX response in organoids | Glycolysis inhibition improves PTX response | [3] |
| Ovarian carcinoma | SKOV3/SKOV3-TR30 cells [in vitro] | Increased PGAM1 → increased pyruvate/lactate production | SKOV3-TR30 has higher PTX resistance | PGAM1 (glycolysis) | PGAM1 siRNA/inhibitor | Decreased glycolysis, decreased PTX resistance | PGAM1 inhibition decreases PTX resistance | [14] |
| Epithelial ovarian cancer (EOC) | SKOV3-R, A2780-R cells and xenografts [in vitro/vivo] | Increased KHDRBS3 → increased glycolysis (via MIR17HG/CLDN6) | KHDRBS3 upregulated in PTX-R cells; drives resistance | KHDRBS3/CLDN6 | KHDRBS3 siRNA | Decreased glycolysis, restored PTX sensitivity in vitro and in vivo | KHDRBS3 knockdown restores PTX sensitivity | [23] |
| ER-positive breast cancer | MCF7 cells (NgBR high vs. knockdown) [in vitro] | NgBR increased → increased HIF-1α, increased glycolysis | NgBR expression promotes PTX resistance | NgBR/HIF-1α | NgBR knockdown | Decreased glycolysis, increased PTX-induced apoptosis | NgBR knockdown increases PTX sensitivity | [21] |
| Various (osteosarcoma, NSCLC) | Mouse xenografts (MV522, MG63) [in vivo] | Hypoxia → increased glycolysis (Warburg phenotype) | Glycolytic tumors resistant; combination needed | Glycolysis pathway | 2-Deoxy-D-glucose (2-DG) + PTX | Significantly slower tumor growth vs. PTX alone | Glycolysis inhibitor (2-DG) enhances PTX efficacy in vivo | [22] |
| NSCLC (A549-R) | A549-PTX cells [in vitro] | Increased PDK2 → enhanced glycolysis, inhibited PDH | PDK2 overexpression in PTX-R cells | PDK2 | PDK2 siRNA; DCA (PDK inhibitor) | Decreased glycolysis, increased apoptosis; DCA + PTX synergy | PDK2 inhibition/DCA restores PTX sensitivity | [15] |
3.1. Ovarian Cancer: Glycolytic and Lipid Metabolism Dominance
3.2. Breast Cancer: Metabolic Heterogeneity Across Subtypes
3.3. NSCLC: Hypoxia Induced Metabolic Rewiring
3.4. TME Metabolic Crosstalk
| Cancer Type | Model System | Metabolic Alteration | Impact on PTX Efficacy | Key Molecular Target/Pathway | Strategy Tested | Outcome | Key Finding | Ref |
|---|---|---|---|---|---|---|---|---|
| NSCLC (A549) | A549 vs. A549-PTX cells [in vitro] | Increased mitochondrial fusion (Mfn ↑, Fis1 ↓), Decreased membrane potential, impaired OXPHOS | Correlated with PTX resistance | Mito. fusion/fission (Mfn, Fis1) | Observational | Correlated with reduced PTX sensitivity | Mitochondrial dysfunction correlates with PTX resistance | [4] |
| TNBC PDX models | Patient-derived xenografts (BL1 subtype) [in vivo] | High OXPHOS gene signature in resistant tumors | High OXPHOS signature → worse PTX outcome | Mito. ETC complexes | OXPHOS inhibitor (IACS-10759) ± combos (e.g., CDK4i) | Tumor growth stabilized; combos enhance response | OXPHOS inhibition stabilizes tumors, combos enhance PTX efficacy | [25] |
| General cancer (incl. TNBC) | Human cancer cell lines [in vitro] | Paclitaxel → mitochondrial dysfunction in OXPHOS cells (increased ROS, cytochrome c release) | OXPHOS cells sensitive; glycolytic cells less affected | Mitochondrial complex I/III | Observational | Oxidative cells sensitive to Taxol; glycolytic cells less affected | OXPHOS dependency correlates with PTX sensitivity | [9] |
| Ovarian cancer | Br22i cell clones [in vitro] | Platinum-induced mtDNA mutations → decreased OXPHOS, disrupted tubulin | mtDNA mutations confer PTX resistance | mtDNA-encoded ETC proteins | Observational | mtDNA mutations confer PTX resistance | Acquired mtDNA defects cause PTX resistance | [18] |
| TNBC persistent cells | MDA-MB-231 persistent post-chemo [in vitro] | Increased reliance on pyruvate-driven OXPHOS (TCA upregulated) | Chemo-persistent cells less PTX-sensitive | Mito. pyruvate carrier (MPC) | MPC inhibitor (UK-5099) | Decreased OXPHOS, re-sensitized cells to chemotherapy | Inhibiting pyruvate import re-sensitizes persistent cells | [39] |
| NSCLC (hypoxia-resistant) | A549 hypoxia-selected cells [in vitro] | Hypoxia: Increased FASN/ADRP, decreased CPT1 (increased lipid uptake, decreased oxidation) | Hypoxia-induced cells resist PTX (G2/M arrest hampered) | HIF-1α/FASN/CPT1 (FA metabolism) | FV-429 (wogonin analog) | Reprogrammed FA metabolism, restored PTX sensitivity | Targeting hypoxia-induced lipid metabolism restores PTX sensitivity | [28] |
| Breast cancer (4T1 in mice) | 4T1 tumor with PTX-albumin [in vivo] | Targeting TAM mitochondrial metabolism in lung metastasis microenvironment | Chemo-resistant lung metastases microenvironment | Mitochondrial complex I (via TAM) | TPP-TAM (mito-targeted AMPK activator) + PTX@Alb | Enhanced PTX uptake, T cell infiltration, and tumor killing | Targeting TAM mitochondrial metabolism enhances PTX efficacy in metastasis | [40] |
| Ovarian cancer (A2780) | A2780-PTX resistant line [in vitro] | Increased CPT1A, increased FASN, increased SCD (enhanced lipid synthesis/β-oxidation) | Lipid-rich metabolism drives PTX resistance | CPT1A, FASN, SCD (Lipid metabolism) | Inhibitors of CPT1A, SCD, FASN | Decreased viability, increased apoptosis; sensitized to PTX | Inhibiting lipid metabolism sensitizes to PTX | [41] |
4. Integrated Resistance Mechanisms in the TME
5. Metabolic Targeting Strategies
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Smith, E.R.; Huang, M.; Schlumbrecht, M.P.; George, S.H.L.; Xu, X.X. Rationale for combination of paclitaxel and CDK4/6 inhibitor in ovarian cancer therapy—Non-mitotic mechanisms of paclitaxel. Front. Oncol. 2022, 12, 907520. [Google Scholar] [CrossRef] [PubMed]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029. [Google Scholar] [CrossRef] [PubMed]
- Derouane, F.; Desgres, M.; Moroni, C.; Ambroise, J.; Berlière, M.; Van Bockstal, M.R.; Galant, C.; van Marcke, C.; Vara-Messler, M.; Hutten, S.J.; et al. Metabolic adaptation towards glycolysis supports resistance to neoadjuvant chemotherapy in early triple negative breast cancers. Breast Cancer Res. 2024, 26, 29. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Li, R.; Chen, R.; Liu, J. Altered mitochondrial dynamics, biogenesis, and functions in the paclitaxel-resistant lung adenocarcinoma cell line A549/Taxol. Med. Sci. Monit. 2020, 26, e918216-1. [Google Scholar] [CrossRef]
- Rong, G.; Kang, H.; Wang, Y.; Hai, T.; Sun, H. Candidate Markers That Associate with Chemotherapy Resistance in Breast Cancer through the Study on Taxotere-Induced Damage to Tumor Microenvironment and Gene Expression Profiling of Carcinoma-Associated Fibroblasts (CAFs). PLoS ONE 2013, 8, e70960. [Google Scholar] [CrossRef]
- Feng, B.; Wu, J.; Shen, B.; Jiang, F.; Feng, J. Cancer-associated fibroblasts and resistance to anticancer therapies: Status, mechanisms, and countermeasures. Cancer Cell Int. 2022, 22, 166. [Google Scholar] [CrossRef]
- Peng, X.; Gong, F.; Chen, Y.; Jiang, Y.; Liu, J.; Yu, M.; Zhang, S.; Wang, M.; Xiao, G.; Liao, H. Autophagy promotes paclitaxel resistance of cervical cancer cells: Involvement of Warburg effect activated hypoxia-induced factor 1-α-mediated signaling. Cell Death Dis. 2014, 5, e1367. [Google Scholar] [CrossRef]
- Zhou, X.; An, B.; Lin, Y.; Ni, Y.; Zhao, X.; Liang, X. Molecular mechanisms of ROS-modulated cancer chemoresistance and therapeutic strategies. Biomed. Pharmacother. 2023, 165, 115036. [Google Scholar] [CrossRef]
- Penjweini, R.; Link, K.A.; Qazi, S.; Mattu, N.; Zuchowski, A.; Vasta, A.; Sackett, D.L.; Knutson, J.R. Low dose Taxol causes mitochondrial dysfunction in actively respiring cancer cells. J. Biol. Chem. 2025, 301, 108450. [Google Scholar] [CrossRef]
- Avolio, R.; Matassa, D.S.; Criscuolo, D.; Landriscina, M.; Esposito, F. Modulation of Mitochondrial Metabolic Reprogramming and Oxidative Stress to Overcome Chemoresistance in Cancer. Biomolecules 2020, 10, 135. [Google Scholar] [CrossRef]
- Lv, L.; Yang, S.; Zhu, Y.; Zhai, X.; Li, S.; Tao, X.; Dong, D. Relationship between metabolic reprogramming and drug resistance in breast cancer. Front. Oncol. 2022, 12, 942064. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Cui, Y.; Xu, S.; Wu, X.; Huang, Y.; Zhou, W.; Wang, S.; Fu, Z.; Xie, H. Altered glycolysis results in drug-resistant in clinical tumor therapy. Oncol. Lett. 2021, 21, 369. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.Y.; Gu, S.Y.; Lin, Y.H.; Shih, J.H.; Lin, J.H.; Chou, T.Y.; Lee, Y.C.; Chang, S.F.; Lang, Y.D. Paclitaxel-resistance facilitates glycolytic metabolism via Hexokinase-2-regulated ABC and SLC transporter genes in ovarian clear cell carcinoma. Biomed. Pharmacother. 2024, 180, 117452. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Zhang, X.; Zhang, S.; Xu, S.; Chen, X.; Zhou, C.; Xi, Y.; Xie, X.; Lu, W. PGAM1 Promotes Glycolytic Metabolism and Paclitaxel Resistance via Pyruvic Acid Production in Ovarian Cancer Cells. Front. Biosci.—Landmark 2022, 27, 262. [Google Scholar] [CrossRef]
- Sun, H.; Zhu, A.; Zhou, X.; Wang, F. Suppression of pyruvate dehydrogenase kinase-2 re-sensitizes paclitaxel-resistant human lung cancer cells to paclitaxel. Oncotarget 2017, 8, 52642–52650. [Google Scholar] [CrossRef]
- Zong, Y.; Li, H.; Liao, P.; Chen, L.; Pan, Y.; Zheng, Y.; Zhang, C.; Liu, D.; Zheng, M.; Gao, J. Mitochondrial dysfunction: Mechanisms and advances in therapy. Signal Transduct. Target. Ther. 2024, 9, 124. [Google Scholar] [CrossRef]
- van Gisbergen, M.W.; Voets, A.M.; Starmans, M.H.W.; de Coo, I.F.M.; Yadak, R.; Hoffmann, R.F.; Boutros, P.C.; Smeets, H.J.M.; Dubois, L.; Lambin, P. How do changes in the mtDNA and mitochondrial dysfunction influence cancer and cancer therapy? Challenges, opportunities and models. Mutat. Res. Rev. Mutat. Res. 2015, 764, 16–30. [Google Scholar] [CrossRef]
- Girolimetti, G.; Guerra, F.; Iommarini, L.; Kurelac, I.; Vergara, D.; Maffia, M.; Vidone, M.; Amato, L.B.; Leone, G.; Dusi, S.; et al. Platinum-induced mitochondrial DNA mutations confer lower sensitivity to paclitaxel by impairing tubulin cytoskeletal organization. Hum. Mol. Genet. 2017, 26, 2961–2974. [Google Scholar] [CrossRef]
- Basheeruddin, M.; Qausain, S. Hypoxia-Inducible Factor 1-Alpha (HIF-1α): An Essential Regulator in Cellular Metabolic Control. Cureus 2024, 16, e63852. [Google Scholar] [CrossRef]
- Škubník, J.; Pavlíčková, V.S.; Ruml, T.; Rimpelová, S. Autophagy in cancer resistance to paclitaxel: Development of combination strategies. Biomed. Pharmacother. 2023, 161, 114458. [Google Scholar] [CrossRef]
- Liu, C.; Li, S.; Zhang, X.; Jin, C.; Zhao, B.; Li, L.; Miao, Q.R.; Jin, Y.; Fan, Z. Nogo-B receptor increases glycolysis and the paclitaxel resistance of estrogen receptor-positive breast cancer via the HIF-1α-dependent pathway. Cancer Gene Ther. 2023, 30, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Maschek, G.; Savaraj, N.; Priebe, W.; Braunschweiger, P.; Hamilton, K.; Tidmarsh, G.F.; De Young, L.R.; Lampidis, T.J. 2-Deoxy-D-glucose Increases the Efficacy of Adriamycin and Paclitaxel in Human Osteosarcoma and Non-Small Cell Lung Cancers in Vivo. Cancer Res. 2004, 64, 31–34. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Qiu, L.; Feng, H.; Zhang, H.; Yu, H.; Du, Y.; Wu, H.; Zhu, S.; Ruan, Y.; Jiang, H. KHDRBS3 promotes paclitaxel resistance and induces glycolysis through modulated MIR17HG/CLDN6 signaling in epithelial ovarian cancer. Life Sci. 2022, 293, 120328. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Liu, Z.; Wang, T.; Zhao, P.; Liu, M.; Wang, Y.; Zhao, W.; Yuan, Y.; Li, S. Resensitizing Paclitaxel-Resistant Ovarian Cancer via Targeting Lipid Metabolism Key Enzymes CPT1A, SCD and FASN. Int. J. Mol. Sci. 2023, 24, 16503. [Google Scholar] [CrossRef]
- Evans, K.W.; Yuca, E.; Scott, S.S.; Zhao, M.; Arango, N.P.; Pico, C.X.C.; Saridogan, T.; Shariati, M.; Class, C.A.; Bristow, C.A.; et al. Oxidative phosphorylation is a metabolic vulnerability in chemotherapy-resistant triple-negative breast cancer. Cancer Res. 2021, 81, 5572–5581. [Google Scholar] [CrossRef]
- Bakleh, M.Z.; Zen, A.H. The Distinct Role of HIF-1α and HIF-2α in Hypoxia and Angiogenesis. Cells 2025, 14, 673. [Google Scholar] [CrossRef]
- Valli, A.; Rodriguez, M.; Moutsianas, L.; Fischer, R.; Fedele, V.; Huang, H.L.; Van Stiphout, R.; Jones, D.; Mccarthy, M.; Vinaxia, M.; et al. Hypoxia induces a lipogenic cancer cell phenotype via HIF1α-dependent and -independent pathways. Oncotarget 2014, 6, 1920. [Google Scholar] [CrossRef]
- Guo, Y.; Yang, L.; Guo, W.; Wei, L.; Zhou, Y. FV-429 enhances the efficacy of paclitaxel in NSCLC by reprogramming HIF-1α-modulated FattyAcid metabolism. Chem. Biol. Interact. 2021, 350, 109702. [Google Scholar] [CrossRef]
- Yu, T.; Yang, G.; Hou, Y.; Tang, X.; Wu, C.; Wu, X.A.; Guo, L.; Zhu, Q.; Luo, H.; Du, Y.E.; et al. Cytoplasmic GPER translocation in cancer-associated fibroblasts mediates cAMP/PKA/CREB/glycolytic axis to confer tumor cells with multidrug resistance. Oncogene 2017, 36, 2131–2145. [Google Scholar] [CrossRef]
- Tavares-Valente, D.; Baltazar, F.; Moreira, R.; Queirós, O. Cancer cell bioenergetics and pH regulation influence breast cancer cell resistance to paclitaxel and doxorubicin. J. Bioenerg. Biomembr. 2013, 45, 467–475. [Google Scholar] [CrossRef]
- Qiao, Y.; Liu, Y.; Ran, R.; Zhou, Y.; Gong, J.; Liu, L.; Zhang, Y.; Wang, H.; Fan, Y.; Fan, Y.; et al. Lactate metabolism and lactylation in breast cancer: Mechanisms and implications. Cancer Metastasis Rev. 2025, 44, 48. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, F.; Cui, J.Y.; Chen, L.; Chen, Y.T.; Liu, B.W. CAFs enhance paclitaxel resistance by inducing EMT through the IL-6/JAK2/STAT3 pathway. Oncol. Rep. 2018, 39, 2081–2090. [Google Scholar] [CrossRef] [PubMed]
- Qin, Q.; Yu, R.; Eriksson, J.E.; Tsai, H.I.; Zhu, H. Cancer-associated fibroblasts in pancreatic ductal adenocarcinoma therapy: Challenges and opportunities. Cancer Lett. 2024, 591, 216859. [Google Scholar] [CrossRef] [PubMed]
- Gunaydin, G. CAFs Interacting with TAMs in Tumor Microenvironment to Enhance Tumorigenesis and Immune Evasion. Front. Oncol. 2021, 11, 668349. [Google Scholar] [CrossRef]
- Wang, W.; Cheng, B.; Yu, Q. Cancer-associated fibroblasts as accomplices to confer therapeutic resistance in cancer. Cancer Drug Resist. 2022, 5, 889. [Google Scholar] [CrossRef]
- Xiao, L.; Wang, Q.; Peng, H. Tumor-associated macrophages: New insights on their metabolic regulation and their influence in cancer immunotherapy. Front. Immunol. 2023, 14, 1157291. [Google Scholar] [CrossRef]
- He, Z.; Chen, D.; Wu, J.; Sui, C.; Deng, X.; Zhang, P.; Chen, Z.; Liu, D.; Yu, J.; Shi, J.; et al. Yes associated protein 1 promotes resistance to 5-fluorouracil in gastric cancer by regulating GLUT3-dependent glycometabolism reprogramming of tumor-associated macrophages. Arch. Biochem. Biophys. 2021, 702, 108838. [Google Scholar] [CrossRef]
- Wang, S.; Wang, J.; Chen, Z.; Luo, J.; Guo, W.; Sun, L.; Lin, L. Targeting M2-like tumor-associated macrophages is a potential therapeutic approach to overcome antitumor drug resistance. npj Precis. Oncol. 2024, 8, 31. [Google Scholar] [CrossRef]
- Winter, M.; Eldjoudi, A.N.; Guette, C.; Hondermarck, H.; Bourette, R.P.; Fovez, Q.; Laine, W.; Ghesquiere, B.; Adriaenssens, E.; Kluza, J.; et al. Mitochondrial adaptation decreases drug sensitivity of persistent triple negative breast cancer cells surviving combinatory and sequential chemotherapy. Neoplasia 2023, 46, 100949. [Google Scholar] [CrossRef]
- Zhou, Z.; Luo, W.; Zheng, C.; Wang, H.; Hu, R.; Deng, H.; Shen, J. Mitochondrial metabolism blockade nanoadjuvant reversed immune-resistance microenvironment to sensitize albumin-bound paclitaxel-based chemo-immunotherapy. Acta Pharm. Sin. B 2024, 14, 4087–4101. [Google Scholar] [CrossRef]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy-Kanniappan, S.; Geschwind, J.F.H. Tumor glycolysis as a target for cancer therapy: Progress and prospects. Mol. Cancer 2013, 12, 152. [Google Scholar] [CrossRef] [PubMed]
- Tatode, A.; Agrawal, P.R.; Taksande, J.; Qutub, M.; Premchandani, T.; Umekar, M.; Danao, K. Role of folate receptor and CD44 in targeting of docetaxel and paclitaxel fabricated conjugates for efficient cancer therapy. J. Med. Surg. Public Health 2025, 5, 100163. [Google Scholar] [CrossRef]
- Zheng, C.; Luo, W.; Liu, Y.; Chen, J.; Deng, H.; Zhou, Z.; Shen, J. Killing three birds with one stone: Multi-stage metabolic regulation mediated by clinically usable berberine liposome to overcome photodynamic immunotherapy resistance. Chem. Eng. J. 2023, 454, 140164. [Google Scholar] [CrossRef]
- Zhou, Z.; Liu, Y.; Jiang, X.; Zheng, C.; Luo, W.; Xiang, X.; Qi, X.; Shen, J. Metformin modified chitosan as a multi-functional adjuvant to enhance cisplatin-based tumor chemotherapy efficacy. Int. J. Biol. Macromol. 2023, 224, 797–809. [Google Scholar] [CrossRef]
- Akter, R.; Awais, M.; Boopathi, V.; Ahn, J.C.; Yang, D.C.; Kang, S.C.; Yang, D.U.; Jung, S.K. Inversion of the Warburg Effect: Unraveling the Metabolic Nexus between Obesity and Cancer. ACS Pharmacol. Transl. Sci. 2024, 7, 560. [Google Scholar] [CrossRef]
- Mishra, R.; Haldar, S.; Placencio, V.; Madhav, A.; Rohena-Rivera, K.; Agarwal, P.; Duong, F.; Angara, B.; Tripathi, M.; Liu, Z.; et al. Stromal epigenetic alterations drive metabolic and neuroendocrine prostate cancer reprogramming. J. Clin. Investig. 2018, 128, 4472–4484. [Google Scholar] [CrossRef]
- Zhang, D.; Wang, Y.; Shi, Z.; Liu, J.; Sun, P.; Hou, X.; Zhang, J.; Zhao, S.; Zhou, B.P.; Mi, J. Metabolic Reprogramming of Cancer-Associated Fibroblasts by IDH3α Downregulation. Cell Rep. 2015, 10, 1335–1348. [Google Scholar] [CrossRef]
- LaGory, E.L.; Giaccia, A.J. The Ever Expanding Role of HIF in Tumour and Stromal Biology. Nat. Cell Biol. 2016, 18, 356. [Google Scholar] [CrossRef]
- Kim, I.; Choi, S.; Yoo, S.; Lee, M.; Kim, I.S. Cancer-Associated Fibroblasts in the Hypoxic Tumor Microenvironment. Cancers 2022, 14, 3321. [Google Scholar] [CrossRef]
- Karimova, A.F.; Khalitova, A.R.; Suezov, R.; Markov, N.; Mukhamedshina, Y.; Rizvanov, A.A.; Huber, M.; Simon, H.U.; Brichkina, A. Immunometabolism of tumor-associated macrophages: A therapeutic perspective. Eur. J. Cancer 2025, 220, 115332. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Zhang, N.; Yan, T.; Wei, J.; Hao, L.; Sun, C.; Zhao, H.; Jiang, S. Lactate-mediated metabolic reprogramming of tumor-associated macrophages: Implications for tumor progression and therapeutic potential. Front. Immunol. 2025, 16, 1573039. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Hong, J.; Sun, L.; Wei, H.; Gong, W.; Wang, S.; Zhu, J. Glycolysis regulation in tumor-associated macrophages: Its role in tumor development and cancer treatment. Int. J. Cancer 2024, 154, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Cunha, A.; Rocha, A.C.; Barbosa, F.; Baião, A.; Silva, P.; Sarmento, B.; Queirós, O. Glycolytic Inhibitors Potentiated the Activity of Paclitaxel and Their Nanoencapsulation Increased Their Delivery in a Lung Cancer Model. Pharmaceutics 2022, 14, 2021. [Google Scholar] [CrossRef]
- Sang, R.; Fan, R.; Deng, A.; Gou, J.; Lin, R.; Zhao, T.; Hai, Y.; Song, J.; Liu, Y.; Qi, B.; et al. Degradation of Hexokinase 2 Blocks Glycolysis and Induces GSDME-Dependent Pyroptosis to Amplify Immunogenic Cell Death for Breast Cancer Therapy. J. Med. Chem. 2023, 66, 8464–8483. [Google Scholar] [CrossRef]
- He, K.; Tao, F.; Lu, Y.; Fang, M.; Huang, H.; Zhou, Y. The Role of HK2 in Tumorigenesis and Development: Potential for Targeted Therapy with Natural Products. Int. J. Med. Sci. 2025, 22, 790. [Google Scholar] [CrossRef]
- Aa, X.; Li, T.; Chen, N.; Wang, H.; Wang, X.; Ma, Y. PGAM1 regulates the glycolytic metabolism of SCs in tibetan sheep and its influence on the development of SCs. Gene 2021, 804, 145897. [Google Scholar] [CrossRef]
- Yap, T.A.; Ahnert, J.R.; Piha-Paul, S.A.; Fu, S.; Janku, F.; Karp, D.D.; Naing, A.; Dumbrava, E.E.I.; Pant, S.; Subbiah, V.; et al. Phase I trial of IACS-010759 (IACS), a potent, selective inhibitor of complex I of the mitochondrial electron transport chain, in patients (pts) with advanced solid tumors. J. Clin. Oncol. 2019, 37, 3014. [Google Scholar] [CrossRef]
- Jiang, W.; Hu, J.W.; He, X.R.; Jin, W.L.; He, X.Y. Statins: A repurposed drug to fight cancer. J. Exp. Clin. Cancer Res. 2021, 40, 241. [Google Scholar] [CrossRef]
- Lemberg, K.M.; Gori, S.S.; Tsukamoto, T.; Rais, R.; Slusher, B.S. Clinical development of metabolic inhibitors for oncology. J. Clin. Investig. 2022, 132, e148550. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Premchandani, T.; Taksande, J.; Tatode, A.; Sheikh, S.; Qutub, M.; Hussain, U.M.; Khan, R.; Umekar, M. Mitochondrial Dysfunction and Glycolytic Shift in the Tumor Microenvironment: Impact on Paclitaxel Efficacy in Cancer Therapy. Clin. Bioenerg. 2025, 1, 5. https://doi.org/10.3390/clinbioenerg1010005
Premchandani T, Taksande J, Tatode A, Sheikh S, Qutub M, Hussain UM, Khan R, Umekar M. Mitochondrial Dysfunction and Glycolytic Shift in the Tumor Microenvironment: Impact on Paclitaxel Efficacy in Cancer Therapy. Clinical Bioenergetics. 2025; 1(1):5. https://doi.org/10.3390/clinbioenerg1010005
Chicago/Turabian StylePremchandani, Tanvi, Jayshree Taksande, Amol Tatode, Sameer Sheikh, Mohammad Qutub, Ujban Md Hussain, Rahmuddin Khan, and Milind Umekar. 2025. "Mitochondrial Dysfunction and Glycolytic Shift in the Tumor Microenvironment: Impact on Paclitaxel Efficacy in Cancer Therapy" Clinical Bioenergetics 1, no. 1: 5. https://doi.org/10.3390/clinbioenerg1010005
APA StylePremchandani, T., Taksande, J., Tatode, A., Sheikh, S., Qutub, M., Hussain, U. M., Khan, R., & Umekar, M. (2025). Mitochondrial Dysfunction and Glycolytic Shift in the Tumor Microenvironment: Impact on Paclitaxel Efficacy in Cancer Therapy. Clinical Bioenergetics, 1(1), 5. https://doi.org/10.3390/clinbioenerg1010005