Abstract
Venous thromboembolic events (VTEs), especially in the form of portal vein thrombosis (PVT), are common complications of cirrhosis and are associated with significant morbidity. These patients can also be easily tipped toward bleeding because of deficiencies in procoagulant factors and pharmacokinetic and pharmacodynamic changes that occur during disease progression. Therefore, the understanding of how to use pharmacotherapy to treat VTE is a key to success in achieving VTE resolution without potentiating adverse bleeding events (AEs). Based on a review of the literature and the authors’ clinical experience, it was determined that unfractionated heparin (UFH), low molecular weight heparin (LMWH), fondaparinux, argatroban, warfarin, and direct oral anticoagulants all have evidence of use in patients with cirrhosis and VTE. However, the available literature is mostly limited to retrospective studies and case reports. There appears to be a paucity of prospective, randomized trials that compare the available pharmacotherapy at typical and adjusted doses. Overall, the decision as to the choice of agent and dose prescribed for anticoagulant therapy should include assessment on clot burden, bleeding risk, drug-drug/disease interactions, and the risk of presence of AEs.
1. Introduction
Cirrhosis represents the final common pathway of chronic liver injury, which is marked by progressive hepatic fibrosis, nodular regeneration, and architectural distortion of liver parenchyma. It arises from various etiologies, including chronic viral hepatitis (particularly hepatitis B and C), alcohol-associated liver disease, metabolic dysfunction-associated steatohepatitis (MASH), autoimmune hepatitis, and inherited disorders such as Wilson disease and hereditary hemochromatosis. Hepatic dysfunction affects multiple physiological systems as the disease advances, notably, hemostasis, drug metabolism, and renal clearance.
Hematologic disturbances are particularly prominent in cirrhosis. The liver’s impaired synthetic function reduces the production of procoagulant and anticoagulant factors, creating a fragile and rebalanced hemostatic state. As a result, patients are susceptible to both bleeding and thrombotic events [1]. Venous thromboembolism (VTE) is a recognized complication, with portal vein thrombosis (PVT) being the most common, occurring in approximately 10% to 15% of patients—especially those with more advanced liver disease [2,3]. Systemic thromboembolic events, including deep vein thrombosis (DVT) and pulmonary embolism (PE), also occur in hospitalized cirrhotic patients, with the reported incidence rates ranging from 0.5% to 6.3% [4].
As cirrhosis progresses, alterations in hepatic and renal drug elimination necessitate adjustments in dosing for numerous medications, including anticoagulants [5]. However, evidence-based guidance for anticoagulant use in this population remains limited. Although the U.S. Food and Drug Administration (FDA) provides some recommendations regarding hepatic impairment, these are often non-specific and may not reflect the evolving body of clinical literature.
According to AASLD guidance on PVT in cirrhosis, anticoagulation is recommended in both compensated and decompensated cirrhosis when PVT is acute, progressive, symptomatic, or associated with planned liver transplantation. Given their reversibility and clinical familiarity, low molecular weight heparin (LMWH) and vitamin K antagonists (VKAs) have traditionally been used as first-line agents. AASLD guidance acknowledges growing interest in direct oral anticoagulants (DOACs) but advises caution due to limited safety data in patients with Child–Pugh Class C cirrhosis. In patients with compensated cirrhosis (Child–Pugh A and select B), DOACs may be used with appropriate patient selection and close monitoring [6].
AASLD further emphasizes that anticoagulation should be individualized, weighing the risks of bleeding against the benefits of thrombus resolution or prevention. The decision to initiate anticoagulation must consider portal hypertension, variceal status, platelet count, and renal function. For decompensated patients, pretreatment screening and management of esophageal varices are recommended to mitigate bleeding risk.
This review aims to synthesize the current literature, including recent AASLD recommendations, to guide clinicians in optimizing the use of anticoagulants in cirrhotic patients with VTE. This article discusses the pharmacokinetic and pharmacodynamic changes associated with hepatic dysfunction and proposes dosing strategies tailored to disease severity and clinical context.
2. Materials and Methods
A comprehensive literature search was conducted to evaluate the use of anticoagulant medications in patients with hepatic cirrhosis. A PubMed search on English medication database was performed using combinations of Medical Subject Headings (MeSHs) using keywords cirrhosis, venous thromboembolism, and anticoagulants was performed. Additional papers were found using references from the first round of papers.
The search was restricted to studies published between January 2017 and January 2025, ensuring the inclusion of recent evidence. Only randomized controlled trials (RCTs) and observational studies were considered for inclusion to provide a robust and high-quality evidence base. In instances where limited data were available for certain anticoagulants, case studies and case reports were also reviewed to supplement the available evidence. Studies that investigated the pharmacokinetics, efficacy, safety, and outcomes of anticoagulant therapy in patients with cirrhosis were prioritized. Studies that met the following inclusion criteria were included: clinical trials (RCTs) or observational studies involving adult patients with hepatic cirrhosis; investigations that evaluated the use of anticoagulant; and studies that reported on outcomes related to anticoagulant therapy, including safety, efficacy, adverse events, or thromboembolic events. Studies were excluded if they focused on populations without hepatic cirrhosis, involved non-human studies, or if there was no report on anticoagulant therapy in the context of cirrhosis.
3. Results
3.1. Pharmacokinetic Changes Arising from Cirrhosis
Pharmacokinetic alterations in cirrhosis encompass changes in drug absorption, distribution, metabolism, and excretion (ADME). Typically, these deviations from normal pharmacokinetic processes become more pronounced as the severity of cirrhosis increases [7,8].
3.1.1. Absorption
Several alterations in upper gastrointestinal (GI) physiology have been observed in patients with cirrhosis, which may influence drug absorption. Notable changes include the presence of esophogeal varices and ulcers, increased intestinal permeability, delayed gastric emptying, and reduced intestinal motility, all of which may variably impact the pharmacokinetics of orally administered medications [8]. Impaired GI motility is relevant in the context of delayed-release formulations, as prolonged GI transit time can enhance drug exposure. However, specific recommendations for adjusting pharmacotherapy remain unclear.
First-pass metabolism is dependent on hepatic drug extraction, uptake into hepatocytes, the metabolic capacity of hepatic enzymes, and hepatic blood flow. In individuals without cirrhosis, drugs with high hepatic extraction are substantially metabolized during their first pass through the liver, resulting in reduced systemic bioavailability. In contrast, cirrhotic patients exhibit impaired hepatic function due to a reduction in both the number and functionality of hepatocytes and metabolic enzymes, leading to decreased hepatic conversion and elevated systemic drug concentrations. Moreover, the development of portosystemic shunting in cirrhosis further reduces hepatic drug metabolism by diverting blood flow away from functional hepatocytes [9,10].
3.1.2. Distribution
In patients with cirrhosis, fluid retention and the development of ascites contribute to third spacing, which can lead to an increased volume of distribution (Vd) for hydrophilic medications. This expansion of Vd may, in turn, result in reduced drug clearance and a prolonged elimination half-life for certain agents [9]. Additionally, hypoalbuminemia is commonly observed in cirrhosis due to impaired hepatic protein synthesis and malnutrition. Consequently, the free (unbound) fraction of highly protein-bound drugs may be elevated, potentially enhancing pharmacologic effects or toxicity [9]. While these hemodynamic and pharmacokinetic alterations are clinically significant, they have not yet been directly incorporated into standardized drug dosing adjustments.
3.1.3. Metabolism
Hepatic metabolism represents one of the most profoundly affected aspects of drug disposition in patients with cirrhosis. Phase I cytochrome P450 (CYP) enzyme activity generally declines in a manner that correlates with disease severity; however, the extent and rate of decline differ among individual isoenzymes [11,12]. For example, CYP2D6 and, in particular, CYP2E1 tend to retain their metabolic capacity until advanced stages of cirrhosis, whereas CYP2C19 activity is markedly reduced early in the disease course (Figure 1) [11,12].
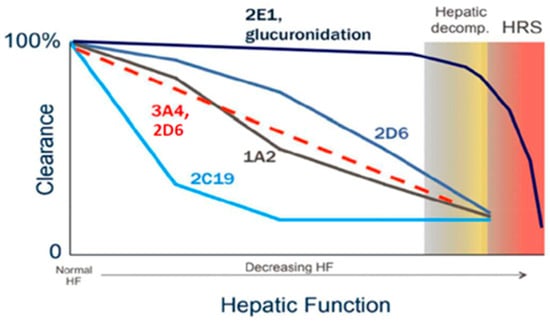
Figure 1.
Relative changes in Phase I and Phase II metabolism as cirrhosis progresses [11,12].
In contrast, Phase II metabolic pathways, such as sulfoxidation and glucuronidation, are relatively preserved until the later stages of cirrhosis. This preservation is attributed to both extra-hepatic glucuronidation—occurring in organs such as the intestine and kidneys—and compensatory upregulation of uridine 5′-diphospho-glucuronosyltransferase (UGT) enzymes in residual functional hepatocytes. As a result, glucuronidation remains largely intact in patients with mild to moderate hepatic impairment [13].
3.1.4. Excretion: Biliary and Renal
As cirrhosis advances, progressive obstruction of bile canaliculi occurs, leading to impaired bile flow and, ultimately, clinically significant cholestasis accompanied by hyperbilirubinemia. These pathophysiologic changes are associated with a reduction in the clearance of drugs eliminated via biliary excretion [14]. However, due to the variable and unpredictable nature of cholestatic impairment, standardized dose adjustments for biliary-excreted medications based solely on the presence of cholestasis are not currently recommended.
In addition to hepatic effects, cirrhosis induces systemic hemodynamic alterations that significantly impact renal function and drug elimination. Elevated portal vein pressure promotes systemic vasodilation, resulting in decreased peripheral vascular resistance and reduced effective arterial blood volume. In response, compensatory activation of the renin–angiotensin–aldosterone system (RAAS) occurs, leading to sodium and water retention, efferent arteriolar vasoconstriction mediated by angiotensin II, and a subsequent decline in glomerular filtration rate (GFR) [7,15].
Despite a progressive reduction in GFR with advancing cirrhosis, serum creatinine concentrations often fail to accurately reflect renal function in this population. This discrepancy is attributable to diminished hepatic synthesis of creatine—the precursor to creatinine—due to impaired nitrogen metabolism. Moreover, progressive loss of skeletal muscle mass, driven by metabolic, hormonal, and nutritional disturbances, further reduces creatinine generation. Together, these factors contribute to an overestimation of the true GFR when relying on serum creatinine as a surrogate marker [7,15,16]. As a result, reliance on unadjusted serum creatinine may underestimate renal impairment, potentially leading to accumulation and toxicity of renally cleared medications.
To address this limitation, some investigators have proposed adjusting the measured serum creatinine by multiplying it by a factor of 1.5 in patients with decompensated cirrhosis [7]. This correction aligns with findings from studies employing reference measures such as inulin clearance or cystatin C-based estimates, which have demonstrated that conventional methods may overestimate the GFR by approximately 1.5- to 2-fold in cirrhotic populations [7,16].
3.2. Pharmacologic Agents Used for VTE Treatment and Prevention
3.2.1. Unfractionated Heparin
Unfractionated heparin (UFH) has demonstrated a reasonable safety and efficacy profile in patients with cirrhosis and remains a commonly used anticoagulant in this population. UFH exerts its anticoagulant effect primarily by binding to antithrombin (AT), which enhances the inactivation of several activated coagulation factors, most notably thrombin (factor IIa) and factor Xa. The metabolic pathways responsible for UFH clearance are not fully elucidated but appear to involve depolymerization and desulfation by the reticuloendothelial system. In healthy individuals, the plasma half-life of UFH ranges from approximately 30 to 90 min, though this is dose-dependent and increases with escalating doses. In patients with hepatic impairment, the half-life may be altered—typically decreased in general liver dysfunction but paradoxically prolonged in advanced cirrhosis. At therapeutic doses, UFH is rapidly cleared via non-renal mechanisms, primarily through enzymatic degradation by heparinases and desulfatases. However, at higher doses, renal excretion may also contribute to elimination [17].
Therapeutic monitoring of UFH commonly involves laboratory assays such as activated partial thromboplastin time (aPTT), antifactor Xa (aXa), and antifactor IIa (aIIa) levels. However, in patients with decompensated cirrhosis, the utility of these assays is significantly limited due to altered coagulation physiology. Strong evidence indicates that aPTT values may be falsely elevated, while aXa levels may be artifactually low in this population, complicating the interpretation and titration of UFH therapy [18,19,20]. Although less extensively studied, preliminary data suggest that aIIa levels may also be falsely low in cirrhosis [21].
In light of these limitations, some authors have proposed modifications to standard UFH infusion protocols for patients with cirrhosis. While the development of a dedicated UFH nomogram for cirrhosis may not be strictly necessary, it has been suggested that targeting the lower end of the therapeutic aXa range may help mitigate bleeding risk in this vulnerable population [18]. Additionally, reliance on aPTT for UFH monitoring is discouraged, given its tendency to be disproportionately elevated in cirrhotic patients, potentially leading to inappropriate dose adjustments. Although both aXa/aIIa and aPTT assays are influenced by cirrhosis-associated coagulopathy, the magnitude of distortion appears to be significantly less for the anti-Xa and anti-IIa assays [18].
3.2.2. Low Molecular Weight Heparin
Similar to unfractionated heparin (UFH), low molecular weight heparins (LMWHs) have demonstrated both safety and efficacy in patients with cirrhosis. Although routine monitoring of LMWH is generally not required in the cirrhotic population, antifactor Xa (aXa) level monitoring remains an available option and is typically recommended in specific clinical scenarios, such as obesity, renal insufficiency, pregnancy, and prolonged treatment durations.
LMWHs exert their anticoagulant effect through binding to antithrombin (AT), with a primary mechanism of inhibiting factor Xa. Enoxaparin, the most commonly utilized LMWH, has a volume of distribution of approximately 4.2 L and undergoes hepatic metabolism through desulfation and depolymerization. In contrast to UFH, enoxaparin has a significantly longer elimination half-life—ranging from 4.5 to 7 h—that is not dose-dependent. LMWHs are primarily renally excreted, and enoxaparin clearance is reduced by at least 30% in individuals with a creatinine clearance below 30 mL/min [22].
In patients with cirrhosis, the reliability of aXa monitoring is limited by disease-related alterations in the coagulation cascade, particularly the reduced circulating levels of antithrombin. This may lead to underestimation of anticoagulant activity, thereby complicating dose titration decisions [17]. Consequently, routine use of aXa levels for dose adjustment in decompensated cirrhosis is not recommended, as it may result in inappropriate dose escalation and increased bleeding risk. Nonetheless, aXa measurements may have utility in select cases, such as in the diagnostic evaluation of unexplained bleeding during LMWH therapy [20].
3.2.3. Fondaparinux
Fondaparinux (FPX) exerts its anticoagulant effects by selectively binding to antithrombin, which is similar to heparinoids. However, due to its lack of antigenicity, FPX represents a suitable alternative for patients who have developed heparin-induced thrombocytopenia (HIT).
Pharmacokinetically, fondaparinux exhibits a volume of distribution ranging from 7 to 11 L and demonstrates an elimination half-life of approximately 17 to 21 h in healthy individuals. Renal excretion accounts for up to 77% of the drug in its unchanged form. In patients with impaired renal function, elimination is significantly prolonged, necessitating appropriate dose adjustments [23].
Although data on the use of fondaparinux in patients with hepatic impairment, particularly cirrhosis, are limited, current U.S. Food and Drug Administration (FDA) guidance indicates that no dosage modification is required in individuals with Child–Pugh Class A or B cirrhosis. Pharmacokinetic analyses in Child–Pugh Class B patients reveal a 22% reduction in maximum concentration (Cmax) and a 39% decrease in area under the concentration–time curve (AUC). However, no dosing recommendations are available for patients with Child–Pugh Class C cirrhosis [22].
A retrospective study evaluating the treatment of portal vein thrombosis (PVT) in cirrhotic patients across Child–Pugh Classes A through C compared fondaparinux to low molecular weight heparin (LMWH). Patients were treated with LMWH or FPX at therapeutic dosage and reduction was considered in selected cases: platelets <50 × 109/L, GFR <30 mL/min for LMWH and <50 mL/min for FPX, or body weight <50 kg for FPX. Additionally, those considered to be at high risk of bleeding received reduced doses, though the definition of high bleed risk and how doses were selected were not well defined. The findings suggested a higher rate of complete recanalization with fondaparinux compared to LMWH, although bleeding rates were higher in the FPX group. However, no statistical significance was found between the groups for either outcome, indicating that fondaparinux may be a safe and effective anticoagulant option in this patient population [24].
Fondaparinux thus presents a potentially valuable therapeutic alternative in patients with cirrhosis and a history of HIT. Nevertheless, its heavy reliance on renal clearance and prolonged half-life in individuals with reduced glomerular filtration rates (GFRs) raise concerns regarding unrecognized renal dysfunction. Consequently, assessment of renal function using cystatin C may be prudent prior to initiating fondaparinux in patients with decompensated cirrhosis [7].
3.2.4. Argatroban
Argatroban, a direct thrombin inhibitor, is commonly utilized in the management of HIT. It undergoes hepatic metabolism primarily through hydroxylation and aromatization, with minor contributions from cytochrome P450 enzymes, particularly CYP3A4/5. In individuals with moderate hepatic impairment, clearance is reduced approximately fourfold, while the elimination half-life increases by approximately threefold [25].
In light of these pharmacokinetic alterations, the FDA recommends a reduced initial dose of 0.5 mcg/kg/min in patients with Child–Pugh Class B or C cirrhosis compared to the standard starting dose of 1 mcg/kg/min [25]. Therapeutic monitoring typically involves measurement of aPTT or anti-IIa activity. However, as previously noted, these monitoring parameters can be complex and less reliable in the setting of hepatic dysfunction.
A case report involving a patient with Child–Pugh Class C cirrhosis demonstrated that, even with an initial dose reduction to 0.2 mcg/kg/min due to concurrent comorbidities, the patient’s aPTT remained supratherapeutic. After further dose reductions, a maintenance dose of 0.05 mcg/kg/min was ultimately required to achieve the desired aPTT range [26]. Such cases underscore the need for further investigation into optimal dosing strategies for patients with advanced hepatic impairment. In one study, a starting dose of 0.25 mcg/kg/min in Child–Pugh Class C patients was associated with a shorter median time to target aPTT compared to the FDA-recommended initial dose, suggesting that current guidance may overestimate dosing requirements in this population [26].
A single center observational study aimed to evaluate the effectiveness and safety of empiric versus nomogram-based dosing strategies for argatroban and bivalirudin in patients with suspected heparin-induced thrombocytopenia (HIT). In their study, 25% of patients had hepatic impairment (Total bilirubin >2 mg/dL or Child–Pugh Class B or C). Additionally, 41% of patients had SCr values ≥1.5 mg/dL. In patients who received the new protocol, those with moderate hepatic dysfunction received an initial dose of 0.5 mcg/kg/min, and those with severe hepatic dysfunction received an initial dose of 0.25 mcg/kg/min as compared to the typical initial dose of 1.5 mcg/kg/min. The protocol-derived initial dosing recommendations obtained a goal aPTT value in the majority of patients, with only 13% of patients having a subtherapeutic initial aPTT measurement. There were no significant differences in thromboembolism, bleeding, or mortality between the protocol and historical control groups; however, the study was not designed or powered for these outcomes [27].
These findings indicate that existing FDA recommendations are largely derived from data on patients with moderate hepatic dysfunction and may not be fully applicable to those with decompensated cirrhosis. A lower initial dosing approach may be more appropriate in this subgroup.
An additional consideration in the management of argatroban therapy in cirrhotic patients is the timing of follow-up laboratory assessments after dose adjustments. Given the prolonged half-life of the drug in this population, a standard four-hour interval between dose changes and subsequent measurement of anti-IIa activity may be insufficient. A more conservative approach may involve extending this interval to six hours in patients with Child–Pugh Class A or B cirrhosis and to eight hours or more in those with Class C. Anecdotal clinical experience suggests that some patients with advanced cirrhosis may require even longer durations to achieve steady-state anti-IIa levels following a dose modification.
3.2.5. Warfarin
Warfarin undergoes cytochrome P450 (CYP)-dependent hepatic metabolism, with the S-enantiomer primarily metabolized by CYP2C9 and the R-enantiomer by CYP1A2, CYP3A4, and CYP2C19. Additionally, warfarin is highly protein-bound, with approximately 99% of the drug bound to plasma proteins. Given these pharmacokinetic properties, it is unsurprising that hepatic dysfunction, such as that seen in cirrhosis, significantly alters warfarin metabolism and response.
Although the international normalized ratio (INR) is routinely used to monitor warfarin therapy, its reliability in cirrhotic patients is compromised due to the frequent presence of an elevated baseline INR associated with hepatic insufficiency [20,28]. The existing literature suggests that warfarin may be initiated in patients with a baseline INR of less than 1.5; however, there is a lack of robust evidence to guide optimal therapeutic INR targets specifically for patients with cirrhosis. Consequently, clinicians often default to conventional INR goals used in the general population [28,29].
A key consideration in this population is the reduced dose requirement for warfarin. This is likely due to decreased synthesis of vitamin-K-dependent clotting factors, resulting in fewer available targets for warfarin to antagonize. Moreover, once an INR becomes supratherapeutic, the time to normalization tends to be prolonged in cirrhotic individuals compared to the general population. In these cases, the utility of vitamin K for reversal may be limited, as impaired hepatic synthetic function leads to inadequate production of clotting factor precursors, reducing the effectiveness of vitamin-K-mediated correction [27].
In situations involving hemorrhagic complications, both fresh frozen plasma (FFP) and four-factor prothrombin complex concentrate (4F-PCC) have demonstrated efficacy and safety as rapid reversal agents for warfarin-induced anticoagulation in patients with liver disease [30,31].
3.2.6. Direct Oral Anticoagulants
Direct oral anticoagulants (DOACs) are increasingly being considered as alternatives to enoxaparin and warfarin for anticoagulation in various clinical settings. Rivaroxaban, apixaban, and edoxaban exert their anticoagulant effects via direct inhibition of factor Xa, while dabigatran acts by binding directly to the active site of thrombin.
Among the DOACs, apixaban and rivaroxaban have the most robust clinical and pharmacokinetic data in patients with hepatic impairment, followed by dabigatran. Apixaban is approximately 75% hepatically metabolized, primarily via the cytochrome P450 enzyme CYP3A4/5. According to FDA labeling, no dose adjustment is required for patients with Child–Pugh Class A or B cirrhosis, although use is not recommended in those with Class C cirrhosis [32,33]. Pharmacokinetic studies have shown that the area under the concentration–time curve (AUC) for apixaban in subjects with mild and moderate hepatic impairment is comparable to healthy individuals, with AUC ratios of 1.03 (0.80–1.32) and 1.09 (0.85–1.41), respectively [34].
Rivaroxaban is approximately 65% hepatically metabolized via CYP3A4/5 and CYP2J2. FDA guidance advises against the use of rivaroxaban in patients with Child–Pugh Class B or C cirrhosis [35]. Pharmacokinetic evaluations have demonstrated a 1.15-fold increase in AUC in patients with mild hepatic impairment (CPS A) and a 2.27-fold increase in those with moderate impairment (CPS B) relative to healthy controls [36].
Dabigatran is eliminated approximately 20% via hepatic pathways and undergoes complete hydrolysis by plasma and hepatic esterases, as well as hepatic glucuronidation to active isomers [36]. Currently, pharmacokinetic data specific to cirrhotic populations are limited. A small cohort trial comparing healthy individuals to individuals with Child–Pugh B cirrhosis found no meaningful difference in drug exposure; however, the results of this study are limited due to its small sample size [37].
A retrospective study comparing the safety and efficacy of apixaban, rivaroxaban, and dabigatran in patients with splanchnic vein thrombosis found that patients with cirrhosis were more frequently prescribed reduced DOAC doses compared to non-cirrhotic controls. Average daily doses in cirrhotic vs. non-cirrhotic cohorts were 15 mg vs. 20 mg for rivaroxaban, 5 mg vs. 7.5 mg for apixaban, and 165 mg vs. 220 mg for dabigatran, respectively. Despite these differences, no significant disparities were observed in safety or efficacy outcomes between the two groups [38].
Additional studies have explored both standard and reduced dosing regimens in cirrhotic patients. One comparative study reported that 10 mg of rivaroxaban taken twice daily was more effective and associated with fewer major bleeding events than warfarin titrated to an INR goal of 2–2.5, though the time in range was not reported [39]. In a case series involving 35 patients with Child–Pugh Class B cirrhosis, participants received either 2.5 mg of apixaban twice daily or 110 mg of dabigatran twice daily following initial treatment with low molecular weight heparin for 2 to 6 months. Over a median follow-up period of 50 months, the study documented three thrombotic events (2.2% per year), two major bleeding episodes (1.5% per year), and three clinically relevant non-major bleeding events (2.2% per year), which may indicate that reduced dose apixaban and dabigatran could be effective and safe in VTE prophlaxis in patients with Child–Pugh Class B cirrhosis. However, the generalizability of these results is limited, as there was a lack of a control group, and the causes of cirrhosis differed significantly between the two groups [39].
Taken together, the available evidence suggests that DOACs may be used safely and effectively in patients with Child–Pugh Class A and B cirrhosis, provided that dosing is adjusted to account for pharmacokinetic alterations. Given the long half-life of dabigatran and its reliance on renal excretion, its use beyond Child–Pugh Class A is not recommended, especially in the setting of possible subclinical renal impairment. For patients with Class A cirrhosis, the standard dosing of apixaban and rivaroxaban is generally appropriate. In Class B cirrhosis, apixaban may still be used at usual doses, whereas a dose reduction of rivaroxaban (e.g., from 20 mg to 15 mg or from 15 mg to 10 mg) may be warranted based on the observed doubling of AUC in this population. Furthermore, in either case, it may be prudent to omit or shorten the initial loading phases (typically 7 or 21 days) to mitigate bleeding risk, pending further clinical data.
4. Discussion
The complex pathophysiological changes associated with cirrhosis should be considered when evaluating the pharmacokinetics and pharmacodynamics of anticoagulant therapy. Cirrhosis, particularly in its decompensated stages, leads to significant pharmacokinetic alterations, which may influence both the efficacy and safety profile of anticoagulants. These changes are multifactorial and include impaired hepatic enzyme activity, reduced hepatic blood flow, altered protein binding due to hypoalbuminemia, increased volume of distribution from ascites and fluid retention, and reduced renal function masked by deceptively normal serum creatinine levels.
Many anticoagulants undergo extensive hepatic metabolism, particularly via Phase I cytochrome P450 (CYP) enzymes, which are variably affected in cirrhosis (Figure 2). Drugs with high first-pass metabolism may have increased systemic bioavailability due to reduced hepatic extraction and portosystemic shunting. Additionally, impaired renal clearance can lead to drug accumulation. These factors contribute to increased drug exposure, prolonged half-life, and a heightened risk of bleeding complications in this patient population.
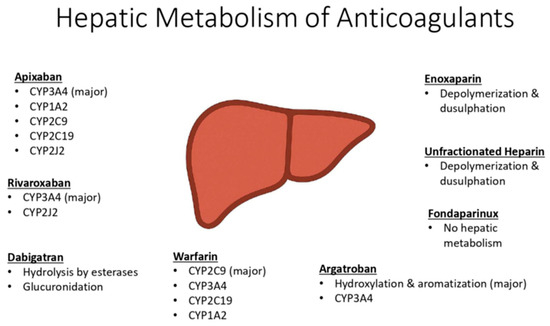
Figure 2.
Hepatic metabolism of anticoagulants.
While the FDA often includes generalized cautions regarding use in hepatic impairment, specific dose adjustment recommendations are frequently absent, leaving clinicians with limited regulatory direction. This gap underscores the need for expert interpretation of available pharmacokinetic data and clinical studies, particularly in the context of cirrhosis-related physiology.
The present review aims to synthesize the available evidence on anticoagulant use in cirrhosis and provide practical, clinically oriented guidance. This includes recommendations for individualized dosing, therapeutic drug monitoring, and the selection of anticoagulants with more favorable metabolic and excretory profiles in patients with varying degrees of hepatic impairment (Table 1). Additional research is needed to more comprehensively evaluate the safety and efficacy of anticoagulant therapy in patients with hepatic cirrhosis, particularly in those with decompensated disease.

Table 1.
Summary of recommendations for dosing of anticoagulation in liver cirrhosis.
Based on current available data, the authors’ conclusion is that for patients with mild to moderate cirrhosis, UFH, LMWH, apixaban, and rivaroxaban are preferred for pharmacotherapy in the majority barring patient specific factors that would preclude them from these options. Preference is given to apixaban over rivaroxaban due to more available evidence supporting its use. Regardless of choice, evaluating clot burden and bleed risk should be utilized to determine if loading doses should be reduced in Child–Pugh B cirrhosis. DOACs have little evidence supporting their use in those with severe cirrhosis. In those with severe cirrhosis, UFH and LMWH likely remain the safest and most effective options. Suggestions to approach to antithrombotic therapy in the most safe and effective way is to promote interdisciplinary interaction between hepatologists and pharmacists and consider the creation of risk stratification tools to aide in the decision-making process.
Author Contributions
Both authors contributed substantially to the research, writing, and editing of this paper. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Kujovich, J.L. Coagulopathy in liver disease: A balancing act. Hematol. Am. Soc. Hematol. Educ. Program. 2015, 2015, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, A.; Puri, K.; Liangpunsakul, S. Deep vein thrombosis and pulmonary embolism in cirrhotic patients: Systematic review. World J. Gastroenterol. 2014, 20, 5737–5745. [Google Scholar] [CrossRef]
- Xian, J.; Tang, Y.; Shao, H.; Wang, X.; Zhang, M.; Xing, T. Effect of portal vein thrombosis on the prognosis of patients with cirrhosis without a liver transplant: A systematic review and meta-analysis. Medicine 2021, 100, e25439. [Google Scholar] [CrossRef] [PubMed]
- Buresi, M.; Hull, R.; Coffin, C.S. Venous thromboembolism in cirrhosis: A review of the literature. Can. J. Gastroenterol. 2012, 26, 905–908. [Google Scholar] [CrossRef]
- Mukhtar, N.A.; Khalili, M. Liver Disease. In Pathophysiology of Disease: An Introduction to Clinical Medicine, 8th ed.; Hammer, G.D., McPhee, S.J., Eds.; McGraw-Hill Education: New York, NY, USA, 2019; Available online: http://accessmedicine.mhmedical.com/content.aspx?aid=1156658705 (accessed on 10 January 2023).
- Northup, P.G.; Garcia-Pagan, J.C.; Garcia-Tsao, G.; Intagliata, N.M.; Superina, R.A.; Roberts, L.N.; Lisman, T.; Valla, D.C. Vascular Liver Disorders, Portal Vein Thrombosis, and Procedural Bleeding in Patients With Liver Disease: 2020 Practice Guidance by the American Association for the Study of Liver Diseases. Hepatology 2021, 73, 366–413. [Google Scholar] [CrossRef] [PubMed]
- Scappaticci, G.B.; Regal, R.E. Cockcroft-Gault Revisited: New De-Liver-Ance on Recommendations for Use in Cirrhosis. World J. Hepatol. 2017, 9, 131–138. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5295146/ (accessed on 28 June 2022). [CrossRef]
- Krähenbühl, S.; Reichen, J. Pharmacokinetics and Pharmacodynamics in Cirrhosis. Medicine 2002, 30, 24–27. [Google Scholar] [CrossRef]
- Zoratti, C.; Moretti, R.; Rebuzzi, L.; Albergati, I.V.; Di Somma, A.; Decorti, G.; Di Bella, S.; Crocè, L.S.; Giuffrè, M. Antibiotics and Liver Cirrhosis: What the Physicians Need to Know. Antibiotics 2021, 11, 31. [Google Scholar] [CrossRef]
- Bollinger, J.E. The Impact of Liver Disease on Drug Metabolism. In Surgical Procedures on the Cirrhotic Patient; Eghtesad, B., Fung, J., Eds.; Springer: Cham, Swizerland, 2017. [Google Scholar] [CrossRef]
- Frye, R.; Zgheib, N.; Matzke, G.; Chavesgnecco, D.; Rabinovitz, M.; Shaikh, O.; Branch, R. Liver disease selectively modulates cytochrome P450--mediated metabolism. Clin. Pharmacol. Ther. 2006, 80, 235–245. [Google Scholar] [CrossRef]
- Duthaler, U.; Bachmann, F.; Suenderhauf, C.; Grandinetti, T.; Pfefferkorn, F.; Haschke, M.; Hruz, P.; Bouitbir, J.; Krähenbühl, S. Liver Cirrhosis Affects the Pharmacokinetics of the Six Substrates of the Basel Phenotyping Cocktail Differently. Clin. Pharmacokinet. 2022, 61, 1039–1055. [Google Scholar] [CrossRef]
- Elbekai, R.; Korashy, H.; El-Kadi, A. The Effect of Liver Cirrhosis on the Regulation and Expression of Drug Metabolizing Enzymes. Curr. Drug Metab. 2005, 5, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Verbeeck, R.K. Pharmacokinetics and dosage adjustment in patients with hepatic dysfunction. Eur. J. Clin. Pharmacol. 2008, 64, 1147–1161. [Google Scholar] [CrossRef]
- Teneva, B.H. Pathogenesis and assessment of renal function in patients with liver cirrhosis. Folia. Med. 2012, 54, 5–13. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Carrier, P.; Debette-Gratien, M.; Loustaud-Ratti, V. Serum creatinine in cirrhotic patients: A cornerstone. AME Med. J. 2018, 3, 109. [Google Scholar] [CrossRef]
- Heparin Sodium and Dextrose; Package Insert; Hospira Inc.: Lake Forest, IL, USA, 2022. Available online: https://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=eede8a0c-5ae6-4166-84b3-12081405f08e (accessed on 10 January 2023).
- Franz, N.D.; Brancaccio, A.; Robinson, A.C.; Regal, R.E. Cirrhosis, Thrombosis, Finding FaXts about Doses: Dosing of Unfractionated Heparin for Venous Thromboembolism in Cirrhosis. Ann. Pharmacother. 2020, 54, 450–456. [Google Scholar] [CrossRef]
- Potze, W.; Arshad, F.; Adelmeijer, J.; Blokzijl, H.; Berg, A.P.v.D.; Porte, R.J.; Lisman, T. Routine coagulation assays underestimate levels of antithrombin-dependent drugs but not of direct anticoagulant drugs in plasma from patients with cirrhosis. Br. J. Haematol. 2013, 163, 666–673. [Google Scholar] [CrossRef]
- Ha, N.B.; Regal, R.E. Anticoagulation in patients with cirrhosis: Caught between a rock liver and a hard place. Ann. Pharmacother. 2016, 50, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Uemoto, R.; Sekine, A.; Mitsui, Y.; Masuda, S.; Yamagami, H.; Kurahashi, K.; Yoshida, S.; Otoda, T.; Yuasa, T.; et al. Plasma Heparin Cofactor II Activity Is Inversely Associated with Hepatic Fibrosis of Non-Alcoholic Fatty Liver Disease in Patients with Type 2 Diabetes Mellitus. J. Atheroscler. Thromb. 2022; ahead of print. [Google Scholar] [CrossRef]
- Enoxaparin Sodium; Package Insert; Amphastar Pharmaceuticals, Inc.: Rancho Cucamonga, CA, USA, 2022. Available online: https://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=ab8118dc-aca8-478b-8290-a468cbe36ae1 (accessed on 10 January 2023).
- Fondaparinux Sodium; Package Insert; Dr. Reddy’s Laboratories Inc.: Princeton, NJ, USA, 2022. Available online: https://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=8a27f341-f612-de72-8c1f-3fd977905de0 (accessed on 10 January 2023).
- Senzolo, M.; Piano, S.; Shalaby, S.; Tonon, M.; Tonello, S.; Zanetto, A.; Sacerdoti, D.; Simioni, P.; Bombonato, G.; Burra, P.; et al. Comparison of Fondaparinux and Low-Molecular-Weight Heparin in the Treatment of Portal Vein Thrombosis in Cirrhosis. Am. J. Med. 2021, 134, 1278–1285.e2. [Google Scholar] [CrossRef]
- Argatroban; Package Insert; Accord Healthcare Inc.: Raleigh, NC, USA, 2022. Available online: https://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=46cdf9e6-839c-49c8-9ee1-c30cfdd9368d (accessed on 10 January 2023).
- Yarbrough, P.M.; Varedi, A.; Walker, A.; Rondina, M.T. Argatroban dose reductions for suspected heparin-induced thrombocytopenia complicated by child-pugh class C liver disease. Ann. Pharmacother. 2012, 46, e30. [Google Scholar] [CrossRef]
- Kiser, T.H.; Mann, A.M.; Trujillo, T.C.; Hassell, K.L. Evaluation of empiric versus nomogram-based direct thrombin inhibitor management in patients with suspected heparin-induced thrombocytopenia. Am. J. Hematol. 2011, 86, 267–272. [Google Scholar] [CrossRef]
- Qamar, A.; Vaduganathan, M.; Greenberger, N.J.; Giugliano, R.P. Oral Anticoagulation in Patients With Liver Disease. J. Am. Coll. Cardiol. 2018, 71, 2162–2175. [Google Scholar] [CrossRef]
- Chung, J.W.; Kim, G.H.; Lee, J.H.; Ok, K.S.; Jang, E.S.; Jeong, S.-H.; Kim, J.-W. Safety, efficacy, and response predictors of anticoagulation for the treatment of nonmalignant portal-vein thrombosis in patients with cirrhosis: A propensity score matching analysis. Clin. Mol. Hepatol. 2014, 20, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Pereira, D.; Liotta, E.; Mahmoud, A.A. The Use of Kcentra® in the Reversal of Coagulopathy of Chronic Liver Disease. J. Pharm. Pract. 2018, 31, 120–125. [Google Scholar] [CrossRef]
- Rassi, A.B.; D’Amico, E.A.; Tripodi, A.; da Rocha, T.R.F.; Migita, B.Y.; Ferreira, C.M.; Carrilho, F.J.; Farias, A.Q. Fresh frozen plasma transfusion in patients with cirrhosis and coagulopathy: Effect on conventional coagulation tests and thrombomodulin-modified thrombin generation. J. Hepatol. 2020, 72, 85–94. [Google Scholar] [CrossRef] [PubMed]
- EliquisEliquis; Package Insert; E.R. Squibb & Sons, LLC.: Princeton, NJ, USA, 2021. Available online: https://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=1523ab32-2c2c-6501-e063-6394a90aa0b3 (accessed on 10 January 2023).
- Frost, C.E.; Ly, V.; Garonzik, S.M. Apixaban Pharmacokinetics and Pharmacodynamics in Subjects with Mild or Moderate Hepatic Impairment. Drugs R. D. 2021, 21, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Xarelto; Package Insert; Janssen Pharmaceuticals, Inc.: Tokyo, Japan, 2022. Available online: https://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=38b11f5d-f0fc-46dc-aef1-ea31872bb621 (accessed on 10 January 2023).
- Kubitza, D.; Roth, A.; Becka, M.; Alatrach, A.; Halabi, A.; Hinrichsen, H.; Mueck, W. Effect of hepatic impairment on the pharmacokinetics and pharmacodynamics of a single dose of rivaroxaban, an oral, direct Factor Xa inhibitor. Br. J. Clin. Pharmacol. 2013, 76, 89–98. [Google Scholar] [CrossRef]
- Dabigatran Etexilate; Package Insert; Ascend Laboratories, LLC.: Princeton, NJ, USA, 2022. Available online: https://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=27e95caf-7938-ef8e-e063-6394a90a6a1a (accessed on 10 January 2023).
- De Gottardi, A.; Trebicka, J.; Klinger, C.; Plessier, A.; Seijo, S.; Terziroli, B.; Magenta, L.; Semela, D.; Buscarini, E.; Langlet, P.; et al. Antithrombotic treatment with direct-acting oral anticoagulants in patients with splanchnic vein thrombosis and cirrhosis. Liver Int. 2017, 37, 694–699. [Google Scholar] [CrossRef]
- Hanafy, A.S.; Abd-Elsalam, S.; Dawoud, M.M. Randomized controlled trial of rivaroxaban versus warfarin in the management of acute non-neoplastic portal vein thrombosis. Vasc. Pharmacol. 2019, 113, 86–91. [Google Scholar] [CrossRef]
- Weronska, A.; Papuga-Szela, E.; Broniatowska, E.; Undas, A. Reduced-dose apixaban and dabigatran in patients with advanced liver cirrhosis and venous thromboembolism: A case series. Pol. Arch. Intern. Med. 2021, 131, 762–765. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).