
Functional Profiling of Antimicrobial Resistance in Rabbit Gut Microbiome



Abstract
1. Introduction
2. Materials and Methods
2.1. Animal and Sample Collection
2.2. DNA Extraction and 16S rRNA Amplicon Sequencing
2.3. Bioinformatic Analysis
3. Results
3.1. Summary of Sequencing Data and Taxonomic Assignment
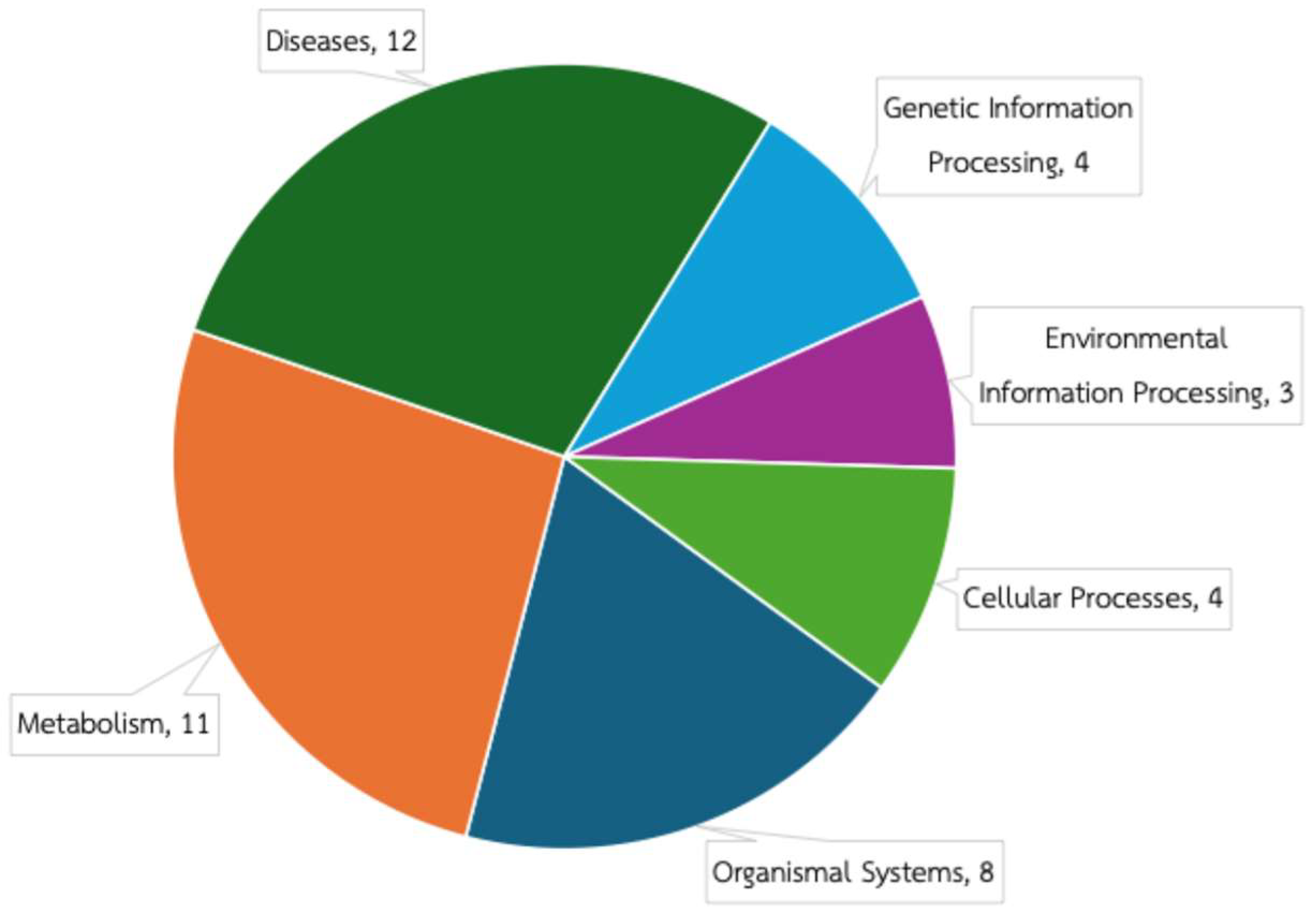
3.2. Predicted Functional Composition of Rabbit Gut Microbiome
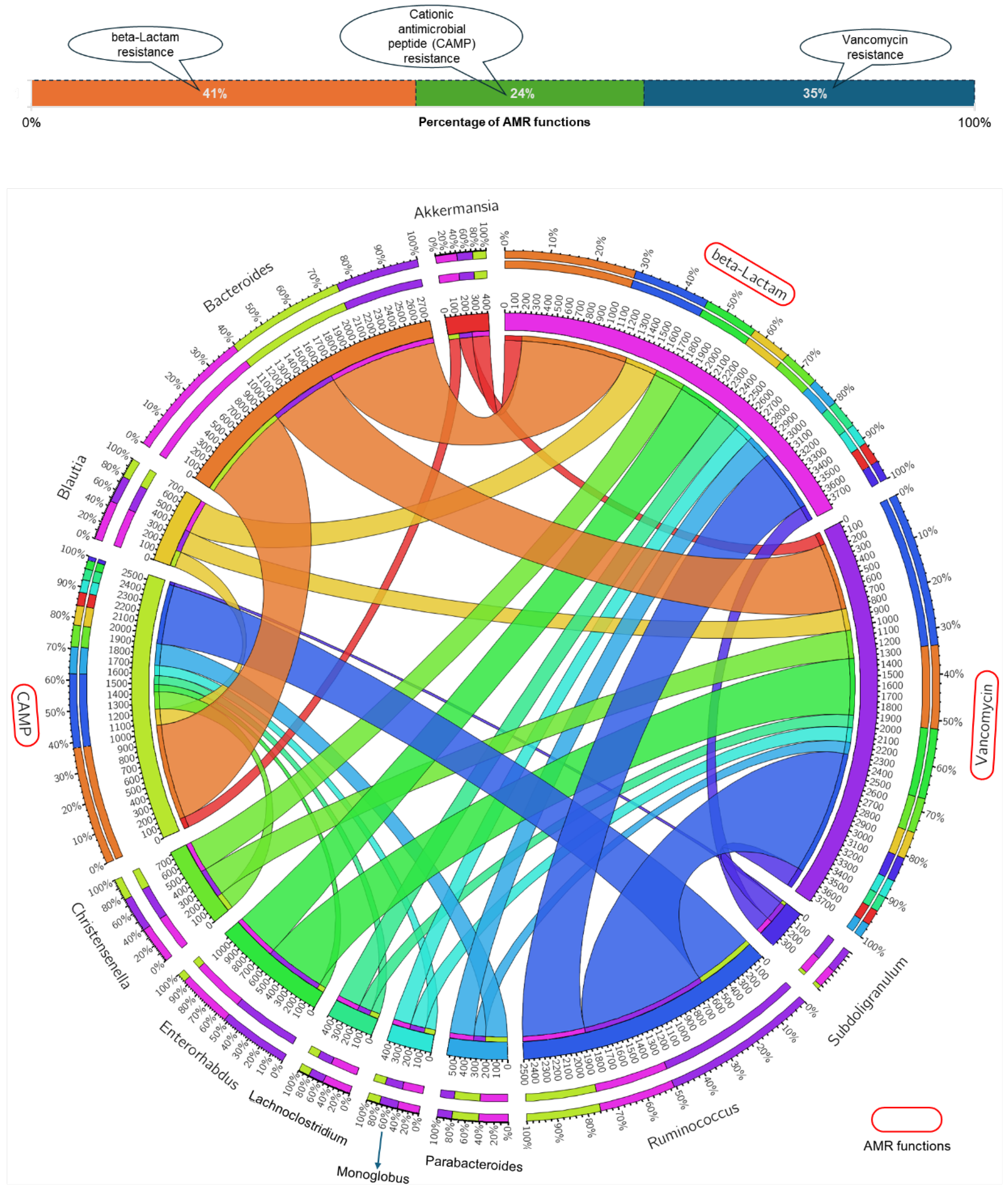
3.3. Gut Microbial Genera and AMR Function Relationship
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AMR | antimicrobial resistance |
| PICRUSt2 | Phylogenetic Investigation of Communities by Reconstruction of Unobserved States 2 |
| NLAC | National Laboratory Animal Center |
| IACUC | Institutional Animal Care and Use Committee |
| NZW | New Zealand White |
| PCR | polymerase chain reaction |
| PE | paired-end |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| ASV | amplicon sequence variant |
| CAMP | cationic antimicrobial peptide |
| ARG | antimicrobial resistance gene |
References
- Murray, C.J.L.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Robles Aguilar, G.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E.; et al. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, J. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations. Wellcome Trust: London, UK, 2016; 84p. [Google Scholar]
- Pandey, S.; Doo, H.; Keum, G.B.; Kim, E.S.; Kwak, J.; Ryu, S.; Choi, Y.; Kang, J.; Kim, S.; Lee, N.R.; et al. Antibiotic resistance in livestock, environment and humans: One Health perspective. J. Anim. Sci. Technol. 2024, 66, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.K.; Hussein, S.; Qurbani, K.; Ibrahim, R.H.; Fareeq, A.; Mahmood, K.A.; Mohamed, M.G. Antimicrobial resistance: Impacts, challenges, and future prospects. J. Med. Surg. Public Health 2024, 2, 100081. [Google Scholar] [CrossRef]
- Carson, C.A.; Weese, J.S. Advances in addressing antimicrobial resistance. Rev. Sci. Tech. 2024, Special Edition, 113–123. [Google Scholar] [CrossRef]
- Masebo, N.T.; Giovanna, M.; Flavia, S.D.R.; Laura, A.; Damiano, C.; Marco, D.P.; Andrea, B.; Eliana, S.; Marilena, B.; Joaquin, H.B.; et al. Evaluation of antimicrobial and non-steroidal anti-inflammatory treatments for BRD on health and welfare in fattening bulls: A cross-sectional study. Vet. Q. 2024, 44, 1–11. [Google Scholar] [CrossRef]
- Samreen; Ahmad, I.; Malak, H.A.; Abulreesh, H.H. Environmental antimicrobial resistance and its drivers: A potential threat to public health. J. Glob. Antimicrob. Resist. 2021, 27, 101–111. [Google Scholar] [CrossRef]
- Wilcox, R.S.; Marenda, M.S.; Devlin, J.M.; Wilks, C.R. Antimicrobial use in laboratory rodent facilities in Australia and New Zealand—A cross-sectional survey of veterinarians and facility managers. PLoS ONE 2024, 19, e0292908. [Google Scholar] [CrossRef]
- Narver, H.L. Antimicrobial stewardship in laboratory animal facilities. J. Am. Assoc. Lab. Anim. Sci. 2017, 56, 6–10. [Google Scholar]
- Desselberge, U. The mammalian gut microbiome, immune responses and disease: From observational to causal relationships. J. Cell. Immunol. 2020, 2, 294–300. [Google Scholar]
- Shi, Y.; Peng, G.; Gebremariam, A.A.; Iqbal, M.M.; Daemi, H.B.; Khan, M.A.; Ullah, R.; Wang, D. Analytical insights, modulation and compositional dynamics of the feline gut microbiota: A review. Animal Diseases 2024, 4, 36. [Google Scholar] [CrossRef]
- Blackmer-Raynolds, L.; Sampson, T.R. Overview of the Gut Microbiome. Semin. Neurol. 2023, 43, 518–529. [Google Scholar] [CrossRef] [PubMed]
- Pierre, J.F.; Leone, V.A. Chapter 37—The microbiome and health. In Present Knowledge in Nutrition, 11th ed.; Marriott, B.P., Birt, D.F., Stallings, V.A., Yates, A.A., Eds.; Academic Press: Cambridge, MA, USA, 2020; pp. 605–624. [Google Scholar]
- Mapara, M.; Thomas, B.S.; Bhat, K.M. Rabbit as an animal model for experimental research. Dent. Res. J. (Isfahan) 2012, 9, 111–118. [Google Scholar] [CrossRef]
- El-Sabrout, K.; Sherasiya, A.; Ahmad, S.; Aggag, S.; Nannoni, E.; Cavallini, D.; Buonaiuto, G. Environmental Enrichment in Rabbit Husbandry: Comparative Impacts on Performance and Welfare. Animals 2024, 14, 2367. [Google Scholar] [CrossRef] [PubMed]
- Abd El-Aziz, A.; Elfadadny, A.; Abo Ghanima, M.; Cavallini, D.; Fusaro, I.; Giammarco, M.; Buonaiuto, G.; El-Sabrout, K. Nutritional Value of Oregano-Based Products and Its Effect on Rabbit Performance and Health. Animals 2024, 14, 3021. [Google Scholar] [CrossRef] [PubMed]
- Chamtim, P. Fecal Microbiome Analysis of New Zealand White Rabbits. In Proceedings of the the 2nd Thailand Bioinformatics Research Network Conference (TBRN 2025), Centara Riverside Hotel, Chiang Mai, Thailand, 22–24 January 2025; pp. 38–47. [Google Scholar]
- Ramirez, J.; Guarner, F.; Fernandez, L.B.; Maruy, A.; Sdepanian, V.L.; Cohen, H. Antibiotics as Major Disruptors of Gut Microbiota. Front. Cell. Infect. Microbiol. 2020, 10, 572912. [Google Scholar] [CrossRef]
- Chowdhury, G.; Ramamurthy, T. Influence of Antimicrobials on the Gut Microbiota. In Antimicrobial Resistance: Global Challenges and Future Interventions; Thomas, S., Ed.; Springer Singapore: Singapore, 2020; pp. 53–79. [Google Scholar]
- Paul, D.; Das, B. Chapter One—Gut microbiome in the emergence of antibiotic-resistant bacterial pathogens. In Progress in Molecular Biology and Translational Science; Das, B., Singh, V., Eds.; Academic Press: Cambridge, MA, USA, 2022; Volume 192, pp. 1–31. [Google Scholar]
- McInnes, R.S.; McCallum, G.E.; Lamberte, L.E.; van Schaik, W. Horizontal transfer of antibiotic resistance genes in the human gut microbiome. Curr. Opin. Microbiol. 2020, 53, 35–43. [Google Scholar] [CrossRef]
- Hu, X.; Wang, F.; Yang, S.; Yuan, X.; Yang, T.; Zhou, Y.; Li, Y. Rabbit microbiota across the whole body revealed by 16S rRNA gene amplicon sequencing. BMC Microbiol 2021, 21, 312. [Google Scholar] [CrossRef]
- Do, T.T.; Tamames, J.; Stedtfeld, R.D.; Guo, X.; Murphy, S.; Tiedje, J.M.; Walsh, F. Antibiotic Resistance Gene Detection in the Microbiome Context. Microb. Drug Resist. 2017, 24, 542–546. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Malinowska, A.M.; Kok, D.E.; Steegenga, W.T.; Hooiveld, G.J.E.J.; Chmurzynska, A. Human gut microbiota composition and its predicted functional properties in people with western and healthy dietary patterns. Eur. J. Nutr. 2022, 61, 3887–3903. [Google Scholar] [CrossRef]
- Shi, Y.; Gahagan, A.C.; Morrison, M.J.; Gregorich, E.; Lapen, D.R.; Chen, W. Stratified Effects of Tillage and Crop Rotations on Soil Microbes in Carbon and Nitrogen Cycles at Different Soil Depths in Long-Term Corn, Soybean, and Wheat Cultivation. Microorganisms 2024, 12, 1635. [Google Scholar] [CrossRef] [PubMed]
- Sahin Dogan, S.; Kocabaş, A. Profiling the genes associated with osmoadaptation and their variation by seasonally in Tuz Lake. Commun. Fac. Sci. Univ. Ank. Ser. C Biol. 2023, 32, 174–191. [Google Scholar] [CrossRef]
- Wang, S.; Wang, Y.; Liu, H.; Li, X.; Zhao, J.; Dong, Z.; Li, J.; Kaka, N.A.; Nazar, M.; Shao, T. Using PICRUSt2 to explore the functional potential of bacterial community in alfalfa silage harvested at different growth stages. Chem. Biol. Technol. Agric. 2022, 9, 98. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 3. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Holmes, S.P. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 2017, 11, 2639–2643. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]
- Abachin, E.; Poyart, C.; Pellegrini, E.; Milohanic, E.; Fiedler, F.; Berche, P.; Trieu-Cuot, P. Formation of D-alanyl-lipoteichoic acid is required for adhesion and virulence of Listeria monocytogenes. Mol. Microbiol. 2002, 43, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Nishi, H.; Komatsuzawa, H.; Fujiwara, T.; McCallum, N.; Sugai, M. Reduced content of lysyl-phosphatidylglycerol in the cytoplasmic membrane affects susceptibility to moenomycin, as well as vancomycin, gentamicin, and antimicrobial peptides, in Staphylococcus aureus. Antimicrob. Agents Chemother. 2004, 48, 4800–4807. [Google Scholar] [CrossRef]
- Cullen, T.W.; Schofield, W.B.; Barry, N.A.; Putnam, E.E.; Rundell, E.A.; Trent, M.S.; Degnan, P.H.; Booth, C.J.; Yu, H.; Goodman, A.L. Gut microbiota. Antimicrobial peptide resistance mediates resilience of prominent gut commensals during inflammation. Science 2015, 347, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.J.; Kim, S.A.; Yang, S.H.; Kim, D.H.; Cheng, Y.Y.; Kang, J.I.; Lee, S.Y.; Han, N.S. Integrated genome-based assessment of safety and probiotic characteristics of Lactiplantibacillus plantarum PMO 08 isolated from kimchi. PLoS ONE 2022, 17, e0273986. [Google Scholar] [CrossRef] [PubMed]
- dos Santos, C.I.; Campos, C.D.L.; Nunes-Neto, W.R.; do Carmo, M.S.; Nogueira, F.A.B.; Ferreira, R.M.; Costa, E.P.S.; Gonzaga, L.F.; Araújo, J.M.M.; Monteiro, J.M.; et al. Genomic Analysis of Limosilactobacillus fermentum ATCC 23271, a Potential Probiotic Strain with Anti-Candida Activity. J. Fungi 2021, 7, 794. [Google Scholar] [CrossRef]
- Ahmed, M.O.; Baptiste, K.E. Vancomycin-Resistant Enterococci: A Review of Antimicrobial Resistance Mechanisms and Perspectives of Human and Animal Health. Microb. Drug Resist. 2017, 24, 590–606. [Google Scholar] [CrossRef]
- Boolchandani, M.; D’Souza, A.W.; Dantas, G. Sequencing-based methods and resources to study antimicrobial resistance. Nat. Rev. Genet. 2019, 20, 356–370. [Google Scholar] [CrossRef]
- Lamanna, M.; Muca, E.; Buonaiuto, G.; Formigoni, A.; Cavallini, D. From posts to practice: Instagram’s role in veterinary dairy cow nutrition education—How does the audience interact and apply knowledge? A survey study. J. Dairy Sci. 2025, 108, 1659–1671. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Sequencing Feature | Average | Total |
|---|---|---|
| Raw sequencing data (PE reads) | 80,135 ± 72 | 240,405 |
| Clean sequencing data (PE reads) | 73,103 ± 295 | 219,310 |
| Denoised sequencing data (PE reads) | 72,648 ± 316 | 217,945 |
| Non-chimeric sequencing data (PE reads) | 43,473 ± 3069 | 130,419 |
| Number of ASVs | 905 ± 110 | 2427 |
| Phylum | Family | Average Relative Abundance |
|---|---|---|
| Acidobacteriota | 0.001 ± 0.001% | |
| unclassified_Subgroup_17 | 0.001 ± 0.001% | |
| Actinobacteriota | 5.485 ± 3.969% | |
| Atopobiaceae | 4.418 ± 3.395% | |
| Bifidobacteriaceae | 0.001 ± 0.002% | |
| Corynebacteriaceae | 0.004 ± 0.004% | |
| Eggerthellaceae | 0.963 ± 0.532% | |
| Frankiaceae | 0.001 ± 0.001% | |
| Streptomycetaceae | 0.003 ± 0.002% | |
| unclassified_Actinobacteria | 0.002 ± 0.003% | |
| unclassified_Actinobacteriota | 0.044 ± 0.057% | |
| unclassified_Coriobacteriales | 0.05 ± 0.024% | |
| Bacteroidota | 17.114 ± 7.175% | |
| Bacteroidaceae | 13.386 ± 3.744% | |
| Marinifilaceae | 0.199 ± 0.167% | |
| Muribaculaceae | 2.435 ± 3.59% | |
| Prevotellaceae | 0.003 ± 0.003% | |
| Rikenellaceae | 0.527 ± 0.12% | |
| Tannerellaceae | 0.556 ± 0.411% | |
| unclassified_Bacteroidales | 0.004 ± 0.007% | |
| unclassified_Bacteroidia | 0.003 ± 0.002% | |
| Chloroflexi | 0.001 ± 0.001% | |
| uncultured_Chloroflexi_bacterium | 0.001 ± 0.001% | |
| Cyanobacteria | 0.02 ± 0.017% | |
| unclassified_Gastranaerophilales | 0.011 ± 0.019% | |
| uncultured_rumen_bacterium | 0.009 ± 0.016% | |
| Desulfobacterota | 0.007 ± 0.009% | |
| Desulfovibrionaceae | 0.007 ± 0.009% | |
| Firmicutes | 75.446 ± 2.369% | |
| [Clostridium]_methylpentosum_group | 0.09 ± 0.06% | |
| [Eubacterium]_coprostanoligenes_group | 0.719 ± 0.226% | |
| Anaerofustaceae | 0.016 ± 0.014% | |
| Anaerovoracaceae | 0.179 ± 0.177% | |
| Butyricicoccaceae | 0.025 ± 0.023% | |
| Christensenellaceae | 1.15 ± 0.22% | |
| Defluviitaleaceae | 0.018 ± 0.027% | |
| Desulfurisporaceae | 0.001 ± 0.001% | |
| Erysipelatoclostridiaceae | 0.005 ± 0.007% | |
| Erysipelotrichaceae | 1.295 ± 0.859% | |
| Eubacteriaceae | 24.806 ± 13.583% | |
| Exiguobacteraceae | 0.002 ± 0.001% | |
| Hungateiclostridiaceae | 0.054 ± 0.068% | |
| Lachnospiraceae | 16.639 ± 4.871% | |
| Lactobacillaceae | 0.001 ± 0.001% | |
| Limnochordaceae | 0.001 ± 0.001% | |
| Monoglobaceae | 1.846 ± 0.879% | |
| Oscillospiraceae | 6.212 ± 2.285% | |
| Peptococcaceae | 0.2 ± 0.084% | |
| Ruminococcaceae | 17.244 ± 5.909% | |
| Syntrophomonadaceae | 0.001 ± 0.002% | |
| UCG_010 | 0.927 ± 0.981% | |
| UCG_011 | 0.005 ± 0.005% | |
| unclassified_Bacilli | 0.001 ± 0.002% | |
| unclassified_Clostridia | 1.21 ± 0.358% | |
| unclassified_Clostridia_UCG_014 | 1.989 ± 1.446% | |
| unclassified_Clostridia_vadinBB60_group | 0.208 ± 0.165% | |
| unclassified_Erysipelotrichales | 0.003 ± 0.005% | |
| unclassified_Firmicutes | 0.08 ± 0.058% | |
| unclassified_Lachnospirales | 0.003 ± 0.003% | |
| unclassified_Oscillospirales | 0.073 ± 0.041% | |
| unclassified_RF39 | 0.126 ± 0.044% | |
| uncultured_Clostridiales_bacterium | 0.005 ± 0.002% | |
| uncultured_Erysipelotrichaceae_bacterium | 0.021 ± 0.024% | |
| uncultured_Lachnospiraceae_bacterium | 0.017 ± 0.03% | |
| uncultured_rumen_bacterium | 0.276 ± 0.297% | |
| Gemmatimonadota | 0.002 ± 0.001% | |
| Gemmatimonadaceae | 0.002 ± 0.001% | |
| Patescibacteria | 0.269 ± 0.135% | |
| Saccharimonadaceae | 0.266 ± 0.137% | |
| unclassified_Candidatus_Campbellbacteria | 0.002 ± 0.002% | |
| unclassified_Saccharimonadales | 0.001 ± 0.001% | |
| Planctomycetota | 0.001 ± 0.001% | |
| Pirellulaceae | 0.001 ± 0.001% | |
| Proteobacteria | 0.416 ± 0.211% | |
| Acetobacteraceae | 0.001 ± 0.001% | |
| Alteromonadaceae | 0.001 ± 0.001% | |
| Caulobacteraceae | 0.001 ± 0.001% | |
| Enterobacteriaceae | 0.001 ± 0.002% | |
| Oxalobacteraceae | 0.013 ± 0.008% | |
| Rhizobiaceae | 0.001 ± 0.002% | |
| Solimonadaceae | 0.001 ± 0.001% | |
| Succinivibrionaceae | 0.001 ± 0.001% | |
| TRA3_20 | 0.001 ± 0.001% | |
| unclassified_Burkholderiales | 0.162 ± 0.049% | |
| unclassified_Proteobacteria | 0.002 ± 0.002% | |
| unclassified_Rhodospirillales | 0.231 ± 0.264% | |
| Verrucomicrobiota | 1.241 ± 0.856% | |
| Akkermansiaceae | 1.241 ± 0.856% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Published by MDPI on behalf of the Hellenic Society for Microbiology. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chamtim, P.; Raethong, N.; Thananusak, R. Functional Profiling of Antimicrobial Resistance in Rabbit Gut Microbiome. Acta Microbiol. Hell. 2025, 70, 21. https://doi.org/10.3390/amh70020021
Chamtim P, Raethong N, Thananusak R. Functional Profiling of Antimicrobial Resistance in Rabbit Gut Microbiome. Acta Microbiologica Hellenica. 2025; 70(2):21. https://doi.org/10.3390/amh70020021
Chicago/Turabian StyleChamtim, Pitakthai, Nachon Raethong, and Roypim Thananusak. 2025. "Functional Profiling of Antimicrobial Resistance in Rabbit Gut Microbiome" Acta Microbiologica Hellenica 70, no. 2: 21. https://doi.org/10.3390/amh70020021
APA StyleChamtim, P., Raethong, N., & Thananusak, R. (2025). Functional Profiling of Antimicrobial Resistance in Rabbit Gut Microbiome. Acta Microbiologica Hellenica, 70(2), 21. https://doi.org/10.3390/amh70020021