Abstract
The landscape of cancer therapy has gained major impetus through the development of materials capable of selectively targeting cancer cells while sparing normal cells. Synthetic peptides are appealing as scaffolds for the creation of such materials. They are small in size, amenable to chemical synthesis and functionalization, and possess diverse chemical and structural space for modulating targeting properties. Here, we review some fundamental insights into the design, discovery, and evolution of peptide-based targeting agents, with a particular focus on two types of cancer cell targets: unique/overexpressed surface receptors and abnormal physiological properties. We highlight the cutting-edge strategies from the literature of the last two decades that demonstrate innovative approaches to constructing receptor-specific cyclic binders and stimulus-responsive targeting materials. Additionally, we discuss potential future directions for advancing this field, with the aim of pushing the frontiers of targeted cancer therapy forward.
1. Introduction
Cancer remains one of the leading causes of death worldwide, with 20 million new cases expected annually by 2025. This underscores the need for highly effective cancer therapies [1]. Surgery, radiation therapy, chemotherapy, and combinations of these therapies dominate clinical practice. However, they often result in severe side effects and unintended toxicity due to their non-selective action against normal cells and tissues [2,3,4]. Immunotherapy is a transformative approach in this field by deploying our body’s own immune system as a personalized medicine to fight against cancer [5,6]. This can be achieved by targeting inhibitory pathways with immune checkpoint inhibitors, or activating pathways with chimeric antigen receptor immune cells or cell engagers. Compared to chemotherapy or radiation, immunotherapy typically has better action specificity with fewer side effects. In some cases, immunotherapy can induce durable responses, contrasting with conventional therapies where cancer may recur more frequently. Clearly, in almost all cancer therapies, it is critical to ensure therapeutic activity against cancer cells while sparing normal cells from harm. Paul Ehrlich’s “magic bullet” concept in the 1890s laid the foundation for targeted therapies by emphasizing the importance of selective-targeting capabilities [7]. Developing molecules or materials that specifically target cancer cells has thus become a highly rewarding endeavor. Cancer cells have a large number of unique or overexpressed surface receptors compared to normal cells. These proteins serve as prime molecular targets for the design or discovery of receptor-specific binding molecules. These binders can facilitate targeted cargo delivery, enable immune checkpoint inhibition, and recruit immune and effector cells, thereby advancing both cancer therapy and diagnostics. In addition, cancer cells exhibit aberrant physiological properties such as overexpressed enzymes, elevated redox potentials, and acidic pH conditions. Effectively targeting these features, as opposed to the receptor-based approaches, is also critical for distinguishing cancer cells from normal cells in therapeutic interventions.
Monoclonal antibodies (mAbs) are widely used because of their exceptional binding specificity and high affinity for cell-surface receptors, making them valuable for generating cell-targeting binders. However, their biological production is costly and can lead to variability in potency between batches. The large size of mAbs may also pose challenges in vivo, such as inadequate pharmacokinetics and limited tissue penetration [8,9,10,11]. In contrast, nucleic acid aptamers—a short, single strand of DNA or RNA—function like chemical antibodies. Like antibodies, aptamers can specifically recognize molecular targets based on defined nucleotide sequences and conformations. The SELEX random library technique has been used to generate numerous high-affinity aptamers that selectively bind to various cellular targets [12,13,14]. Aptamer-based antagonists and aptamer–drug/toxin conjugates have been developed for cancer treatment, as reviewed in detail elsewhere [15,16,17].
Here, we highlight peptides as promising candidates for cancer cell-targeting agents. Compared to mAbs, peptides offer distinct advantages: they are small in size, amenable to chemical synthesis and functionalization, exhibit minimal batch-to-batch variability, possess low immunogenicity, and have an extended shelf life [18,19,20]. Peptides also offer greater chemical diversity for modulating targeting properties compared to nucleic acid aptamers, thanks to the wide range of natural and unnatural amino acids available. Their diverse secondary and higher order structures further expand the scope for tailoring chemical distribution and folding or assembly behaviors. As shown in recent reviews, peptides have been increasingly explored for cancer treatments and diagnostics [20,21,22,23,24]. Previous reviews have systematically discussed their therapeutic effects in different cell types and ways to improve their in vivo efficacy. However, limited attention has been paid to the approaches for molecular design and the search for cancer cell-targeting peptides. Practical methods to correlate peptide sequence-dependent folding and assembly behaviors with cancer-related physiological properties remain elusive [25,26,27]. This mini-review illustrates selected studies to show cutting-edge strategies in the discovery and design of peptide-based binders and dynamic peptide materials, and their receptors and physiological signals, respectively. By leveraging the rich molecular codes encoded in peptides, this review promises to reliably and predictably design targeting agents, advancing targeted cancer therapy.
2. Receptor-Specific Binders
2.1. Linear Binders
Cancer cells possess oncogenic aberrations that promote abnormal proliferation, migration, and the evasion of immune surveillance [28,29]. In particular, unique/overexpressed membrane receptors have become prime targets for distinguishing cancer cells from their normal counterparts [20,30,31,32]. Early efforts in developing receptor-specific binding molecules centered on structure-guided design by exploring natural proteins that engage with target receptors. For example, integrins play a central role in cell adhesion to the extracellular matrix (ECM), facilitating cellular motility and invasion [33]. The well-known RGD peptide was originally derived from the sequence of fibronectin, an abundant ECM protein [34,35]. RGD has been identified as a key interacting motif with integrin heterodimers, such as α5β1, αVβ3, αVβ6, and αIIβ3. Consequently, a variety of synthetic RGD peptides and their derivatives have been constructed as potent binders for integrin-overexpressed cancer cells in malignancies, such as melanoma, glioblastoma, and breast, prostate, and ovarian malignancies [36,37,38]. This approach is time-consuming and highly dependent on the availability of high-resolution structural information on the protein-receptor complex. In contrast, techniques such as phage display [39], and mRNA display [40] enable the screening of large numbers of random peptides against nearly any given molecular target. For example, the overexpression of human epidermal growth factor receptor 2 (HER2) on cancer cells triggers receptor homodimerization and clustering. This activates downstream MAPK and PI3K pathways to drive cell proliferation, growth, and anti-apoptosis [41]. Quinn and coworkers used a random 6-amino-acid peptide bacteriophage display library to find the HER2-specific peptide (KCCYSL) with a dissociation constant (KD) in the range of hundreds of micromolar [42]. In a separate study, Sioud and colleagues used a phage display biopanning technique to identify a HER2 binder (LTVSPWY) with a remarkable KD value in the nanomolar range [43].
2.2. Cyclic Binders
Cyclic peptides dominate in cell-related molecular binders based on the literature of the last two decades [44,45]. Studies suggest that cyclization of linear RGD peptides can significantly increase their binding affinity to αVβ3 receptors from several micromolar to nanomolar levels [46,47]. The increase is attributed to reduced chain flexibility, which facilitates peptide–target engagements by minimizing entropic loss during binding. In addition, cyclization of many linear peptide-based binders has resulted in improved proteolytic resistance during in vivo cancer cell targeting. This is due to constrained backbone conformations that impede access to protease catalytic sites. Clearly, cyclization has emerged as a facile route to enhancing the targeting performance of peptide-based binders.
Numerous bioactive cyclic peptides found in nature maintain their three-dimensional structures through intramolecular disulfide bonds. Wu and coworkers were inspired to develop a series of cysteine-rich motifs, such as CXC and CPPC, to direct peptide cyclization. These motifs enable the programmable formation of intramolecular disulfide bonds, leading to single-, bi-, and multi-cyclic architectures with predictable topologies (Figure 1A) [48,49]. In addition to the disulfide-based approach, various chemical conjugation and crosslinking strategies have been developed, allowing the creation of cyclic scaffolds with exceptional efficiency [50]. The screening library (such as phage [39] and mRNA display [40]) has been extensively used to search for unnatural cyclic binders that target given molecular entities. For example, Heinis, Winter and coworkers presented a strategy using phage display to screen and isolate cyclic binders for various molecular targets [45,51]. Cysteine-containing peptides presented on phage tips were cyclized using thiol-reactive chemical linkers, resulting in redox-stable conformations as opposed to disulfide-bridged cyclic structures (Figure 1B) [52]. Suga and coworkers established the random nonstandard peptide integrated discovery (RaPID) platform to generate the library of thioether-closed macrocyclic peptides containing non-proteinogenic amino acids (Figure 1C) [53,54]. This platform integrates genetic code reprogramming using a flexible in vitro translation (FIT) system with mRNA display, greatly expanding the repertoire of cyclic binders [55]. An increasing number of cyclic peptides with nanomolar and even picomolar binding affinities have been generated for a wide range of targets, including cancer cell-surface receptors.
Peptide-based binders facilitate cancer therapy by guiding the delivery of chemotherapeutics, radioisotopes, and other cytotoxic agents. They also serve to inhibit and antagonize cell-surface receptors as well as intracellular proteins [56,57,58]. Among them, many binder-based drugs and peptide–drug/toxin conjugates have been approved or are under clinical evaluation (Table 1). For instance, romidepsin [59], a bicyclic peptide isolated from natural fermentation products, has received FDA approval for the treatment of cutaneous T-cell lymphoma. This prodrug features an intrapeptide disulfide bond that is reduced within the cell. The active peptide form is then released to specifically target and inhibit histone deacetylase enzymes. Another notable advancement is 177Lu DOA-TATE [60], the first FDA-approved radiopharmaceutical for the treatment of gastroenteropancreatic neuroendocrine tumors. The molecule conjugates a radionuclide to octreotide, and a cyclic peptide and somatostatin analogue. The octreotide selectively delivers ionizing radiation to cancer cells bearing overexpressed somatostatin receptors. Similarly, 177Lu-AB-3PRGD is in a Phase I trial to determine its effectiveness in various advanced solid tumors (NCT06375564) [61]. This conjugate employs a dimeric RGD peptide with 3 PEG4 linkers for high-avidity multivalent targeting of αVβ3 on cancer cells. In addition to these targeting peptides derived from natural protein sources, screened synthetic binders are also being explored in preclinical or clinical trials. A notable example is the bicycle toxin conjugate (BTC) BT8009, which combines a nectin-4 targeting bicyclic peptide with the cytotoxin monomethyl auristatin E [62]. This conjugate has shown promising anticancer activity in patients with advanced or metastatic malignancies, including urothelial cancer (NCT04561362, NCT06225596). Comprehensive lists of binder-based therapeutics in clinical trials and approved for marketing have been presented elsewhere [63,64]. Concurrent with the rapid development of cancer immunotherapy, peptide-based binders have also been harnessed as cell engagers to orchestrate cancer elimination using immune effector cells. For example, Wang and coauthors designed a bispecific triblock peptide AKMGEGGWGANDY-GNNQQNY-RGD to facilitate interactions between T cells and cancer cells [65]. The first and third blocks of the peptide selectively targeted CD3 on T cells and integrins on MCF-7 cells, respectively. Upon RGD-integrin interaction, the fibril-forming sequence (the second block) drove the clustering of the cell-surface peptides, activating T cells and culminating in the cytolysis of cancer cells.
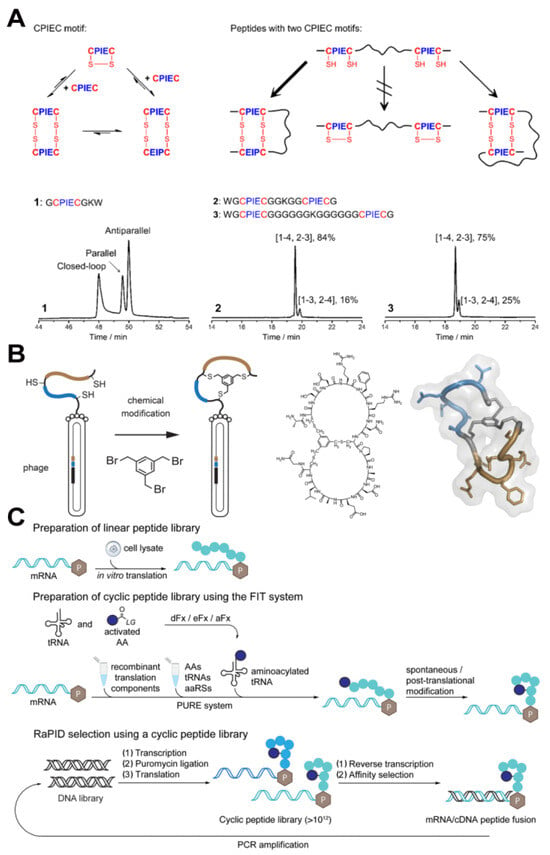
Figure 1.
(A) Schematics of disulfide pairing of CPIEC motifs and the oxidation of peptides containing two CPIEC motifs [49]. Reprinted (adapted) with permission from 49. Copyright 2023 American Chemical Society. (B) Phage selection of bicyclic peptides using a thiol-reactive chemical linker [52]. Reprinted (adapted) with permission from 52. Copyright 2017 American Chemical Society. (C) mRNA selection of cyclic peptides on the RaPID platform [54]. Reprinted (adapted) with permission from 54. Copyright 2019 American Chemical Society.

Table 1.
Representative cell-targeting peptides that have been approved or are in clinical trials.
3. Physiological Stimulus–Responsive Peptide Assemblies
In addition to targeting cell-surface receptors, the aberrant physiological characteristics inherent to cancer cells can be an alternative type of cancer cell target [69,70]. Tumor microenvironments, both extracellular and intracellular, exhibit overexpressed enzymes, elevated redox potentials, and acidic pH conditions [71,72]. Aiming at these physiological attributes, targeting cancer cells require approaches distinct from the binder-receptor interaction mode. A straightforward strategy to create the targeting materials is to use these physiological traits to alter the physicochemical properties of self-assembling peptides (i.e., hydrophobicity, hydrophilicity, size, and charge state). As demonstrated by the following examples, this provides a facile route to correlate the changes in folding and or assembly behaviors of the peptides with cancer cell-specific cues (Table 2).
3.1. Enzyme-Responsive Materials
Elevated levels of enzymes in both the extra- and intracellular environments of cancer have been documented, including alkaline phosphatases, matrix metalloproteinases (MMPs), cathepsin B, carbonic anhydrases, and many others [73,74,75]. The catalytic abilities of these enzymes to cleave chemical bonds have been leveraged to link peptide assembly behavior to the presence of enriched enzymes from cancer cells [73,76,77]. Xu and colleagues have pioneered the construction of enzyme-instructed peptide self-assembly systems [78,79]. They first showed the use of alkaline phosphatase (ALP) to induce molecular self-assembly [80]. By dephosphorylating an FMOC-tyrosine phosphate, the balance of molecular hydrophobicity and hydrophilicity was altered, resulting in the transformation of the precursor into a hydrogenator. This innovative strategy was subsequently used to direct peptide fibrillization in the pericellular space of cancer cells [81]. Surface and secretory ALPs from HeLa and MES-SA cancer cells catalytically dephosphorylated a naphthalene-capped peptide, DFDFpDY. With enhanced hydrophobicity and decreased charge repulsion, the resultant tripeptide DFDFDY self-assembled into hydrogel-like structures selectively surrounding cancer cells. This action effectively inhibited cancer cell growth and metastasis by impeding cellular mass exchange and inducing apoptosis (Figure 2A).
Recently, Yang, Gao, and their colleagues extended the concept of ALP-responsive peptide assembly structures to develop a bis-specific cell engager (Supra-BiCE) for cancer immunotherapy [82]. The Supra-BiCE platform comprised two self-assembly peptides: Ada-GDFDFpDYG conjugated to a DPPA-1 peptide DNDYDSDKDPDTDDDRDQDYDHDF to target PD-L1 on cancer cells and a DTBP-3 peptide, GGDYDTDFDHDWDHDRDLDNDP. The two peptide segments selectively targeted the Ig and ITIM domains on T and NK cells, respectively. Upon ALP dephosphorylation, the nanoribbons formed by the co-assembly of these two peptides underwent a morphology transformation into long nanofibers. This structural alternation enhanced the binding affinity of the assemblies to both immune and cancer cells, consequently activating T and NK cells via checkpoint blockade. In vivo studies demonstrated that this in situ peptide assembly effectively suppressed colon carcinoma models in mice by facilitating the targeting, enrichment, and retention of T and NK cells at cancer cell sites.
Enzymatic cleavage peptide sequences by enzymes represents another prevalent approach in the design of enzyme-responsive peptide assemblies. For example, Lou and coworkers designed a peptide-conjugated probe (DMFA), capable of undergoing matrix metalloproteinase-2 (MMP-2)-induced morphological changes for cancer therapy [83]. As shown in Figure 2B, DMFA comprised three consecutive segments: a charged, amphiphilic α-helical peptide with the amino-acid sequence of GRFKRFRKKFKKLFKKLSPVIPLLHL, an MMP-2 cleavable peptide PLGLAG, and an amyloid-forming sequence KLVFF. DMFA formed nanoparticles with the α-helical peptide exposed to the aqueous phase. Following cleavage by overexpressed MMP-2 on cancer cells, the resultant α-helical portion disrupted the phospholipid bilayers of the cells, while the KLVFF fragment self-assembled into extracellular nanofibers. This morphological transformation of peptide assemblies has demonstrated efficacy in suppressing tumor growth and metastasis by facilitating Ca2+ influx and disrupting Na+/K+-ATPase, consequently inhibiting the PI3K-Akt signaling pathway.
In addition to extracellular enzymes within the pericellular space, intracellular enzymes also represent promising targets for peptide assembly-based therapies. For example, Liang and coworkers reported an apoptosis-amplified assembly of a peptide analogue for cancer photodynamic therapy (PDT) (Figure 2C) [84]. They synthesized a peptide-porphyrin conjugate, Ac-DEVDD-TPP, by incorporating a caspase-3 cleavable sequence DEVD to a TPP-based PDT photosensitizer. Upon cleavage by endogenous caspase-3, the conjugate yielded D-TPP, which self-assembled into nanofibrils around the mitochondrion. This located assembly enabled the in situ generation of singlet oxygen 1O2 upon laser irradiation, inducing mitochondrion damage and triggering cell apoptosis. Concurrently, more generated caspase-3 continued to convert the conjugate into nanofibrils, thereby amplifying cancer cell apoptosis. Similarly, Wang and colleagues demonstrated the conversion of drug-appended peptide nanoparticles into nanofibers via overexpressed lysosomal protease, cathepsin B, in cancer cells [85]. As depicted in Figure 2D, the peptide-based prodrug was synthesized by conjugating an integrin-targeting peptide RGD to the C-terminus of a chemodrug camptothecin (CPT)-appended β-sheet forming peptide LVFF, linked by an enzyme-cleavable sequence GFLG and a hydrophilic PEG chain. The resultant amphiphilic peptide conjugates self-assembled into nanoparticles. The small size of the particles (~30 nm) enabled the internalization of the assemblies into HeLa cell lysosomes via integrin-assisted cellular endocytosis. Subsequent shedding of the hydrophilic PEG-RGD segment by cathepsin B increased the hydrophobicity of the molecules, leading to the formation of CPT-LVFF nanofibers in cancer cells. These preformed fibrous drugs then acted as seeds to promote drug accumulation in cancer cells, effectively inhibiting cancer progression.
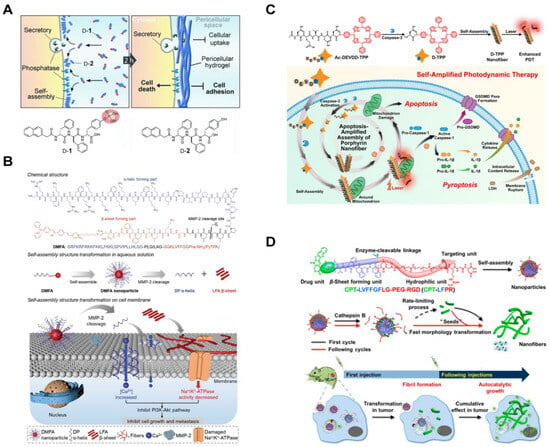
Figure 2.
(A) Enzyme-catalyzed formation of pericellular hydrogels/nanoparticles to induce cell death [81]. Reproduced with permission from 81, © 2014 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. (B) Scheme of the peptide-coupled probe DMFA with division-induced changes on cell membrane morphology [83]. Reproduced with permission from 83, © 2023 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. (C) Self-amplification of Ac-DEVDD-TPP by caspase-3 activation enhances PDT mechanism and induction of apoptosis to enhance pyroptosis generated by porphyrin nanofibers via apoptosis-amplified assembly [84]. Reprinted (adapted) with permission from 84. Copyright 2023 American Chemical Society. (D) Schematic representation of enzyme-triggered morphological transformation and autocatalytic growth of nanofibers [85]. Reprinted (adapted) with permission from 85. Copyright 2019 American Chemical Society.
3.2. Redox-Responsive Materials
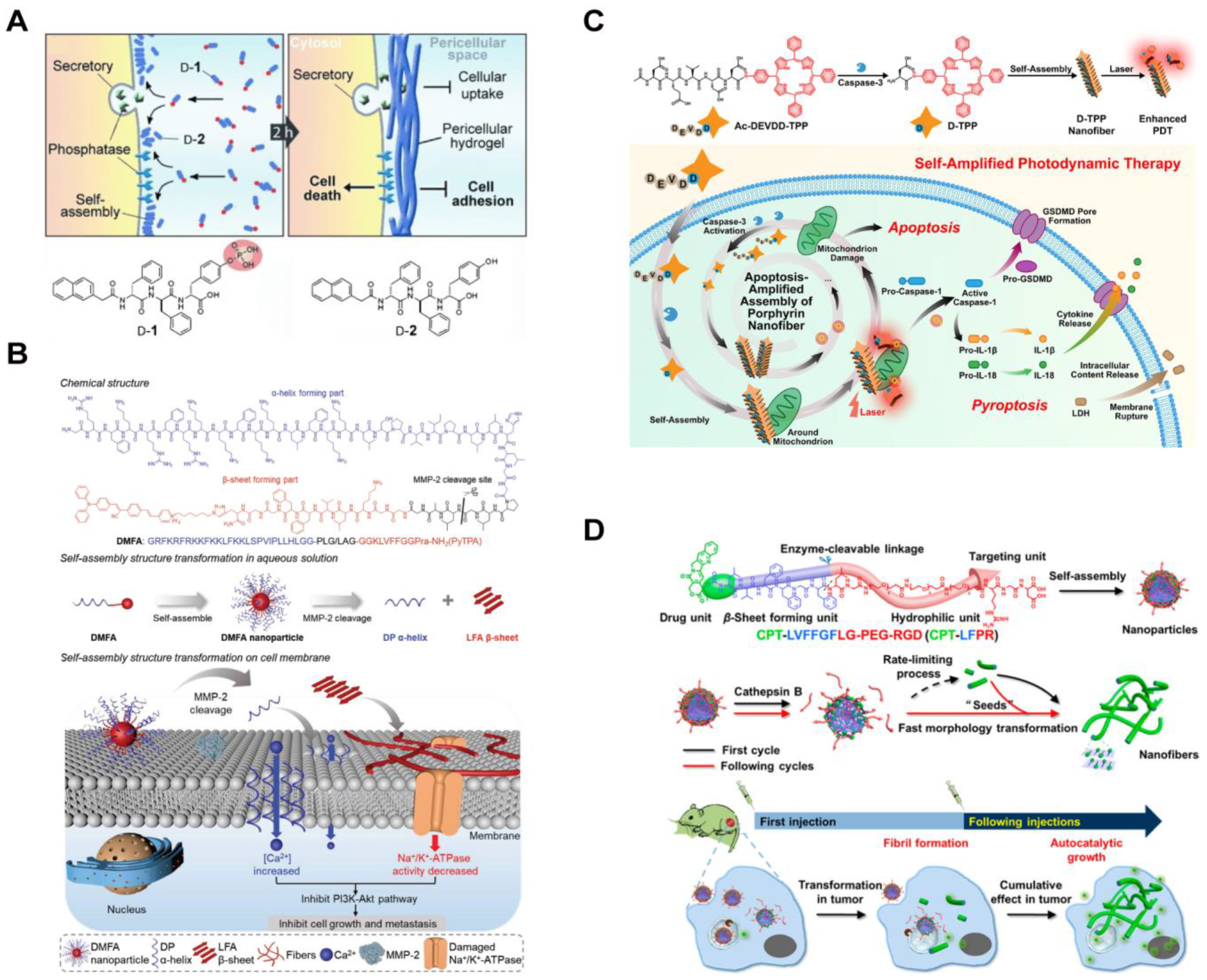
The aberrant redox environment is another characteristic feature of cancer cells. These cells produce higher basal levels of reactive oxygen species (ROS) compared to normal cells, through the mitochondrial respiratory chain and nicotinamide adenine dinucleotide phosphate oxidase [86,87,88,89]. Wang, Qiao, and coworkers constructed assemblies of a polymer–peptide conjugate (PPC) to target the excessive ROS generated in cancer cell mitochondria [90]. Poly (vinyl alcohol) (PVA) was coupled with β-sheet forming peptide KLVFF, tethered with PEG via ROS-cleavable thioketal bond, and the mitochondria-targeting peptide (KLAKLAK)2, respectively (Figure 3A). The resultant amphiphilic PPCs self-assembled into nanoparticles capable of cellular entry through endocytosis. Upon ROS-induced thioketal cleavage, which increased the molecular hydrophobicity, the conjugates transformed into long fibers inside the cells. This increased exposure of the KLAK peptide along the fibers facilitated enhanced multivalent interactions between the assemblies and mitochondria, inducing organelle dysfunction-based cytotoxicity against cancer cells. The ROS-responsive materials exhibited tumor suppression efficacy in mice.
Cancer cells possess antioxidants such as glutathione (GSH) to regulate ROS levels. As a result, a highly reductive environment can be exploited to design GSH-responsive therapeutic assemblies [91,92,93,94]. For example, Yu and colleagues demonstrated a GSH-triggered self-sorting assembly of two peptides in cancer cells [95]. As shown in Figure 3B, a fiber-forming peptide amphiphile E3C16 was linked to a hydrophilic peptide sequence EEEEEE via a disulfide bond. The resultant E3C16E6 formed irregular nanostructures. Additionally, seleno-methionine residues were incorporated into a fiber-forming bola-amphiphile EVM to prevent 1D self-assembly of EVMSeO by oxidizing the selenide to selenoxide groups. Upon cellular internalization of two peptides, intracellular GSH cleaved the disulfide bond of E3C16E6 and reduced the selenoxide group of EVMSeO. As a result, E3C16 and EVMSe self-assembled into self-sorted nanofibers. The twisted E3C16 fibers located around the Golgi apparatus due to the reduced thiol from the peptides, targeting the cysteine-rich proteins on the organelle. Meanwhile, the EVMSe fibers grew near the endoplasmic reticulum (ER) due to a p-toluene sulfonamide (Ts) moiety that conferred ER-targeting capability. These mechanisms culminated in combinatorial organelle dysfunction and subsequent cancer cell death.
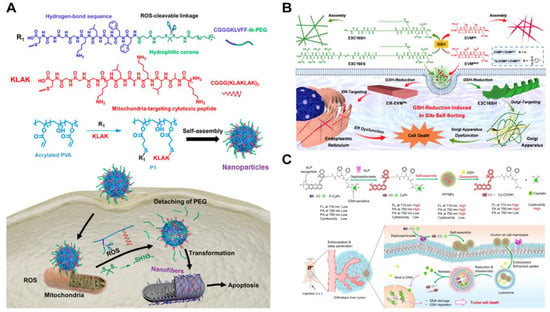
Figure 3.
(A) Synthesis pathway of ROS-sensitive PPCs and morphological transformation of mitochondrial positions [90]. Reprinted (adapted) with permission from Ref. [90]. Copyright 2019 American Chemical Society. (B) Schematic representation of in situ self-sorting peptide assembly within living cells [95]. Reprinted (adapted) with permission from Ref. [95]. Copyright 2022 American Chemical Society. (C) Overall design and proposed mechanism of P-CyPt in cancer therapy [96]. Reproduced with permission from Ref. [96], Copyright © 2023.
In addition to morphological changes, intracellular GSH is a key player in inducing the disassembly of therapeutic peptide nanostructures. Ye, Guo, Liu, and coworkers recently demonstrated the controlled self-assembly and disassembly of a peptide-based cisplatin prodrug for cancer therapy [96]. Prodrug P-CyPt consisted of a hydrophobic DFDF dipeptide, a near-infrared (NIR) merocyanine fluorophore capped with an ALP-sensitive phosphate group, and a GSH-reducible cisplatin prodrug (Pt (IV)) (Figure 3C). Pericellular ALP enzymes in the cancer cells catalyzed the conversion of P-CyPt to CyPt, turning on the NIR fluorescence signal and facilitating the assembly of Pt (IV) nanoparticles due to the increased molecular hydrophobicity. Upon internalization of locally formed PtIVNPs, abundant endogenous GSH reduced PtIV into PtII. This promoted disassembly of the resultant Cy-COOH nanoparticles and release of the cytotoxic cisplatin. The ROS-responsive disassembly process showed promising anti-cancer efficacy by increasing the intracellular cisplatin concentration while decreasing the GSH level.
3.3. pH-Responsive Materials
Cancer cells typically exhibit a slightly acidic extracellular environment due to the deregulated metabolism and the accumulation of lactic acid [72,97,98]. Additionally, following endocytosis, the pH value within the endosomes and lysosomes falls within the range of 4–6. These factors have led researchers to extensively explore pH as a physiological stimulus of cancer for the development of responsive therapeutic assemblies [99,100,101]. For example, Weil, Ng, and their coworkers demonstrated a depsipeptide undergoing multistage transformations upon exposure to cancer cells [102]. As shown in Figure 4A, the peptide comprised a self-assembling motif ISA and a cell-penetrating TAT peptide RRRQRRRKKRGY, featuring a pH-dependent boronic acid-salicylhydroxamate crosslinker. Upon endocytosis facilitated by TAT, the acidic environment cleaved the linker, releasing the pro-assembling peptide. Subsequently, elevated or endogenous H2O2 within the cancer cells removed the boronic acid cage, inducing an O→N acyl shift. This shift further generated the ISA motif, prompting self-assembly into intracellular fibrillar structures that triggered cell apoptosis.
In addition to incorporating acid–labile bonds, the protonation of residues in self-assembling peptides is another powerful strategy [100,101]. For example, Wang and coworkers designed a pH-responsive laminin mimetic peptide (LMMP) that specifically formed an occlusion in tumor blood vessels [103]. LMMP was composed of a bispyrene-capped fibril-forming peptide sequence KLVFF, a thrombus-targeting sequence (PEG)8-CREKA, and an His6 sequence designed to respond to the acidic environment of cancer cells (Figure 4B). After intravenous administration, LMMP nanoparticles adhered to the microthrombi in tumor blood vessels and underwent transformation into nanofibers through His protonation, altering the molecule’s hydrophilic–hydrophobic balance. These laminin-like fibers effectively captured red blood cells, leading to rapid occlusion within the tumor blood vessels. The action provided a pH-responsive, tumor blood vessel-specific therapeutic approach for cancer treatment. Similarly, Wu and colleagues exploited residue protonation to make acid-specific targeting materials [104]. They prepared nanospheres by mixing a cytolytic melittin peptide GIGAVLKVLTTGLPALISWIKRKRQQ, an NIR-absorbing photothermal molecule cypate, and a tumor-targeting hyaluronic acid (HA) (Figure 4C). Subsequent exposure to the acidic environment caused the complexes to transform into nanofibers. The pH-induced morphology change facilitated the prolonged retention of the hemolytic melittin peptide in tumor tissues and inhibited the mobility and metastasis of the cancer cells. This transformation was proposed to be associated with the protonation of the peptide amine groups and carboxyl groups in the cypate, which increased peptide hydrophilicity and cypate hydrophobicity. Subsequent laser irradiation further resulted in the formation of smaller nanoparticles, presumably due to the disturbed internal structures of the fibers after the photodegradation of cypate.
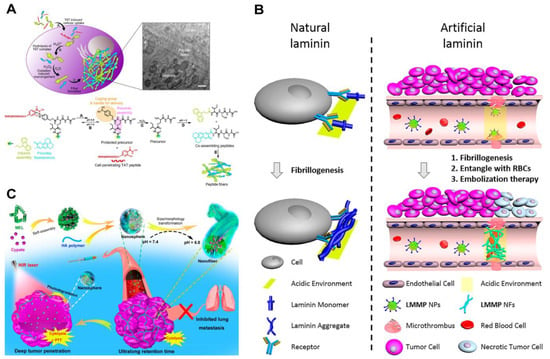
Figure 4.
(A) Functionalized ISA peptide assembly process in cells [102]. Reprinted (adapted) with permission from Ref. [102]. Copyright 2020 American Chemical Society. (B) Schematic diagram of natural laminin and artificial laminin fiber formation [103]. Reprinted (adapted) with permission from Ref. [103]. Copyright 2020 American Chemical Society. (C) Preparation of MEL/Cypate@HA complexes and schematic representation of their successive size/morphology transitions under weakly acidic TME and near-infrared laser irradiation [104]. Reprinted (adapted) with permission from Ref. [104]. Copyright 2019 American Chemical Society.
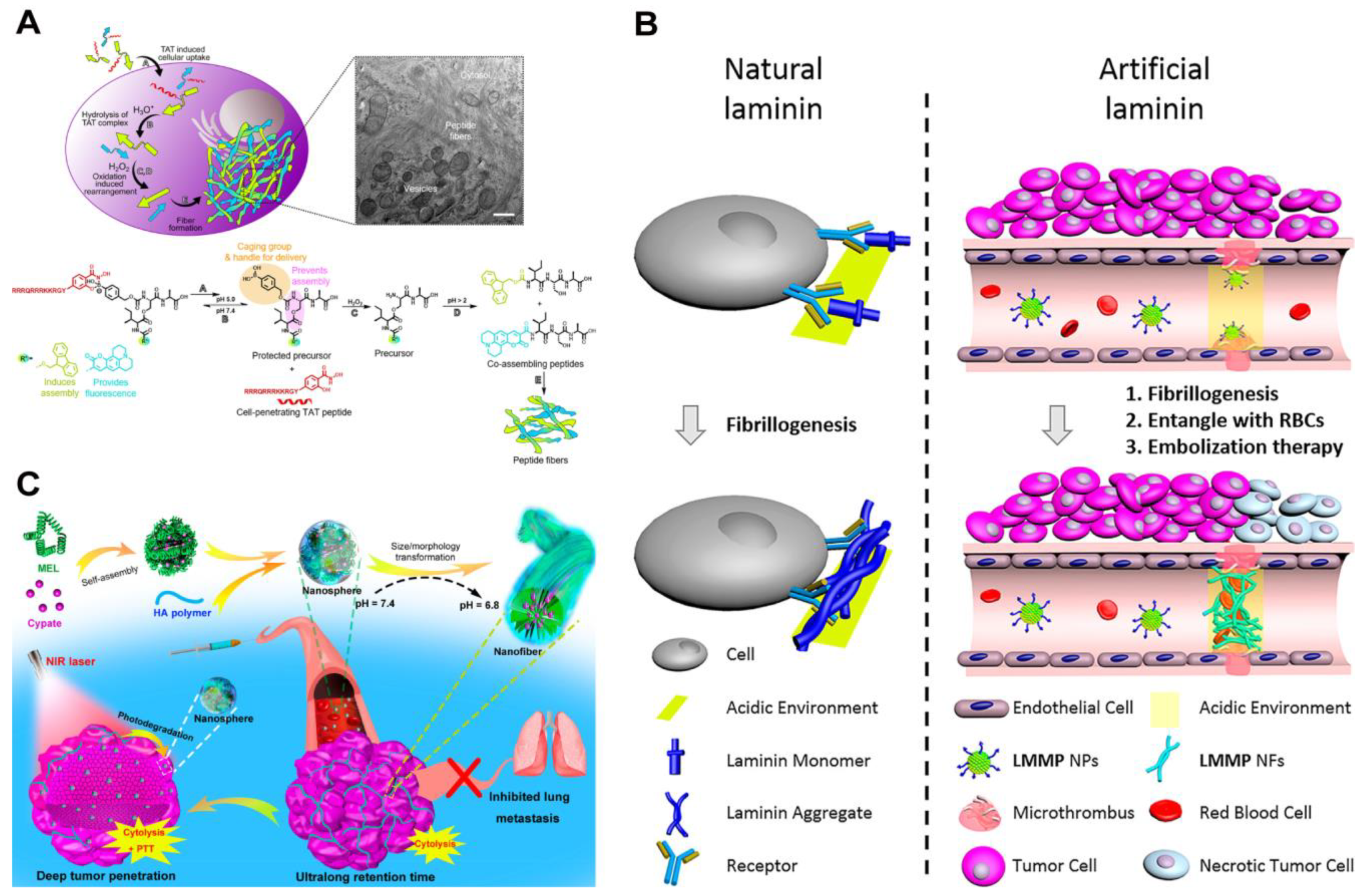
Table 2.
Representative stimulus-responsive targeting materials.
Protonatable residues underlie the mechanism of another widely used family of cell-targeting peptides, the pH low-insertion peptide (pHLIP). Originally identified by Engelman and coworkers, the prototype pHLIP sequence was derived from the C-helices of bacteriorhodopsin [105]. This peptide uniquely responds to acidosis at cancer cell surfaces by using protonatable residues to transit to α-helical structure, facilitating its insertion across the plasma membrane. This feature is reflected in the sequence AEQNPIY-WARADWLFTTPLLLLDLALLVDAD-EGT. It has a polar residue-rich segment in the N-terminus, a central transmembrane segment containing protonatable residues (shown in bold), and a C-terminal flanking region. Under physiological pH conditions, the negatively charged, deprotonated residues hinder membrane insertion and promote less-structured or disordered peptide conformations. Conversely, at lower pH, protonation of these residues increases the peptide’s overall hydrophobicity. This restores the helical conformation of the central segment and allows the peptide to partition across cell membranes. Upon insertion, the C-terminus of the peptide is positioned in the cytoplasm while the N-domain remains in the extracellular space. Sequence modifications have led to various pHLIP derivatives, such as Variant 3 (Var3: ADDQN-PWRAYLDLLFPTDTLLLDLLW), which exhibits significantly enhanced cancer-cell insertion efficiency [106]. The pH-responsive folding and membrane insertion properties of pHLIP offer a robust approach to cancer-cell targeting and payload delivery. A recent review summarized the applications of pHLIP technology in cancer treatment [107]. Several pHLIP-related therapeutic agents are advancing in clinical trials (Table 1). For instance, pHLIP-exatecan (CBX-12, NCT04902872, NCT05691517), which carries the topoisomerase inhibitor, is in clinical trials for treating human ovarian cancer and advanced solid tumors [66]. In addition, a pHLIP conjugated to a near-infrared fluorescent dye (indocyanine green, ICG) has been evaluated in a Phase I/II first-in-human clinical study (NCT05130801) [67]. This is expected to facilitate fluorescence-guided surgery in breast cancer patients undergoing breast-conserving surgery. Furthermore, an 18F-labeled Var3 construct is in a Phase I clinical trial as a diagnostic/imaging agent in breast cancer patients (NCT04054986) [68].
4. Conclusions and Perspective
In the field of cancer therapy, both peptide-based molecular binders and responsive targeting materials play crucial roles in facilitating cargo delivery and release [108,109,110], immune checkpoint inhibition [111], immune-effector cell recruitment [112], and organelle destruction [113]. Significant progress has been made in the search for high-affinity, protein-specific cyclic binders, many of which have advanced to clinical trials as cancer therapeutics. Unlike single-target binders, multicycle structures offer a broader scope for manipulating binding specificity, promising multi-targeting capabilities essential for advanced cancer treatment [45,114]. Moreover, recent advances in computing power and modelling frameworks have revolutionized the exploration of the chemical space of peptide-based binders, utilizing diverse molecular backbones beyond natural peptide chains. This has the potential to expand the binder library beyond the limitations of current biological display techniques [115,116]. Compared to binder-based therapeutics, stimulus-responsive targeting materials are still in their infancy and have a long way to go before reaching clinical trials (Table 2). Challenges persist in multiple aspects. For instance, many endogenous stimuli overlap with the natural physiological processes of cells. They exhibit heterogeneity in normal and pathological contexts, resulting in on-target, off-cancer effects. To address this, the next frontier is to develop next-generation materials with built-in responsiveness to multiple physiological properties or combinations of pathological cues and exogenous stimuli (i.e., light, magnetic, and acoustic energy). Unlike size-defined binders, peptide assemblies such as intracellular fibers exhibit polydispersity in diameter and length, potentially compromising targeting efficacy and reproducibility. There is an urgent need to incorporate molecular codes (i.e., frustration elements) that restrict self-assembly propensity into the design of future targeting assemblies [117,118]. Recent breakthroughs in artificial intelligence, coupled with the rapid accumulation of data on supramolecular structures (i.e., cryo-EM structural reconstruction), greatly improve the computational design of self-assembling peptides [119,120]. These advances hold the promise of creating targeting materials that integrate full molecular code (i.e., composition, sequence, and chemical moiety) within peptides in unprecedented ways. We anticipate that the combination of these areas of progress will enable continuous evolution of peptide-based cyclic binders and dynamic targeting materials to address current challenges in targeted cancer therapy.
Author Contributions
X.C.: literature review, manuscript preparation, and writing—original draft. D.W.: literature review, manuscript preparation, and writing—original draft. Y.-B.J.: review and editing. T.J.: writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Natural Science Foundation of China (grants 22074128, 22241503, 92356308) and Fundamental Research Funds for the Central Universities (grants 20720210013, 20720220005).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Zugazagoitia, J.; Guedes, C.; Ponce, S.; Ferrer, I.; Molina-Pinelo, S.; Paz-Ares, L. Current challenges in cancer treatment. Clin. Ther. 2016, 38, 1551–1566. [Google Scholar] [CrossRef] [PubMed]
- Norsworthy, K.J.; Ko, C.-W.; Lee, J.E.; Liu, J.; John, C.S.; Przepiorka, D.; Farrell, A.T.; Pazdur, R. FDA approval summary: Mylotarg for treatment of patients with relapsed or refractory CD33-positive acute myeloid leukemia. Oncologist 2018, 23, 1103–1108. [Google Scholar] [CrossRef] [PubMed]
- Shinde-Jadhav, S.; Mansure, J.J.; Rayes, R.F.; Marcq, G.; Ayoub, M.; Skowronski, R.; Kool, R.; Bourdeau, F.; Brimo, F.; Spicer, J.; et al. Role of neutrophil extracellular traps in radiation resistance of invasive bladder cancer. Nat. Commun. 2021, 12, 2776. [Google Scholar] [CrossRef]
- Thundimadathil, J. Cancer treatment using peptides: Current therapies and future prospects. J. Amino Acids 2012, 2012, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Demaria, O.; Cornen, S.; Daëron, M.; Morel, Y.; Medzhitov, R.; Vivier, E. Harnessing innate immunity in cancer therapy. Nature 2019, 574, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Paterson, C.; Denlinger, N.; Yang, Y.P. Recent advances and challenges in cancer immunotherapy. Cancers 2022, 14, 3972. [Google Scholar] [CrossRef] [PubMed]
- Strebhardt, K.; Ullrich, A. Paul ehrlich’s magic bullet concept: 100 years of progress. Nat. Rev. Cancer 2008, 8, 473–480. [Google Scholar] [CrossRef]
- Hansel, T.T.; Kropshofer, H.; Singer, T.; Mitchell, J.A.; George, A.J.T. The safety and side effects of monoclonal antibodies. Nat. Rev. Drug Discov. 2010, 9, 325–338. [Google Scholar] [CrossRef]
- Yang, W.; Mixich, L.; Boonstra, E.; Cabral, H. Polymer-based mRNA delivery strategies for advanced therapies. Adv. Healthc. Mater. 2023, 12, e2202688. [Google Scholar] [CrossRef]
- Allen, T.M. Ligand-targeted therapeutics in anticancer therapy. Nat. Rev. Cancer 2002, 2, 750–763. [Google Scholar] [CrossRef]
- Qin, M.; Xia, H.; Xu, W.; Chen, B.; Wang, Y. The spatiotemporal journey of nanomedicines in solid tumors on their therapeutic efficacy. Adv. Drug Deliv. Rev. 2023, 203, 115137. [Google Scholar] [CrossRef] [PubMed]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Robertson, D.L.; Joyce, G.F. Selection in vitro of an RNA enzyme that specifically cleaves single-stranded DNA. Nature 1990, 344, 467–468. [Google Scholar] [CrossRef] [PubMed]
- Ellington, A.D.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.H.; Rossi, J. Aptamers as targeted therapeutics: Current potential and challenges. Nat. Rev. Drug Discov. 2017, 16, 181–202. [Google Scholar] [CrossRef] [PubMed]
- Kulabhusan, P.K.; Hussain, B.; Yüce, M. Current perspectives on aptamers as diagnostic tools and therapeutic agents. Pharmaceutics 2020, 12, 646. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.T.; Sun, W.D.; Fu, T.; Liu, X.S.; Chen, P.; Qiu, L.P.; Qu, F.L.; Tan, W.H. Aptamer-based targeted delivery of functional nucleic acids. J. Am. Chem. Soc. 2023, 145, 7677–7691. [Google Scholar] [CrossRef] [PubMed]
- Diao, L.; Meibohm, B. Pharmacokinetics and pharmacokinetic-pharmacodynamic correlations of therapeutic peptides. Clin. Pharmacokinet. 2013, 52, 855–868. [Google Scholar] [CrossRef]
- Luo, X.; Wu, Y.; Zhang, X.; Tang, M.; Ju, F.; Qin, Z.; Duns, G.J.; Zhang, W.D.; Qin, J.J.; Luan, X. Peptide-based strategies for overcoming multidrug-resistance in cancer therapy. Chin. Chem. Lett. 2024, 109724, in press. [Google Scholar] [CrossRef]
- Samec, T.; Boulos, J.; Gilmore, S.; Hazelton, A.; Alexander-Bryant, A. Peptide-based delivery of therapeutics in cancer treatment. Mater. Today Bio. 2022, 14, 100248. [Google Scholar] [CrossRef]
- Li, C.M.; Haratipour, P.; Lingeman, R.G.; Perry, J.J.P.; Gu, L.; Hickey, R.J.; Malkas, L.H. Novel peptide therapeutic approaches for cancer treatment. Cells 2021, 10, 2908. [Google Scholar] [CrossRef] [PubMed]
- Ayo, A.; Laakkonen, P. Peptide-based strategies for targeted tumor treatment and imaging. Pharmaceutics 2021, 13, 481. [Google Scholar] [CrossRef] [PubMed]
- Nhàn, N.T.T.; Yamada, T.; Yamada, K.H. Peptide-based agents for cancer treatment: Current applications and future directions. Int. J. Mol. Sci. 2023, 24, 12931. [Google Scholar] [CrossRef] [PubMed]
- Jalil, A.T.; Abdulhadi, M.A.; Al-Ameer, L.R.; Taher, W.M.; Abdulameer, S.J.; Abosaooda, M.; Fadhil, A.A. Peptide-based therapeutics in cancer therapy. Mol. Biotechnol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Chagri, S.; Ng, D.Y.W.; Weil, T. Designing bioresponsive nanomaterials for intracellular self-assembly. Nat. Rev. Chem. 2022, 6, 320–338. [Google Scholar] [CrossRef] [PubMed]
- Mu, R.Q.; Zhu, D.Z.; Abdulmalik, S.; Wijekoon, S.; Wei, G.; Kumbar, S.G. Stimuli-responsive peptide assemblies: Design, self-assembly, modulation, and biomedical applications. Bioact. Mater. 2024, 35, 181–207. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Li, Q.Q.; Wu, Y.; Li, X.Y.; Zhou, Y.; Wang, Z.; Liang, H.; Ding, F.Q.; Hong, S.; Steinmetz, N.F.; et al. Molecularly stimuli-responsive self-assembled peptide nanoparticles for targeted imaging and therapy. ACS Nano 2023, 17, 8004–8025. [Google Scholar] [CrossRef]
- Fernald, K.; Kurokawa, M. Evading apoptosis in cancer. Trends Cell Biol. 2013, 23, 620–633. [Google Scholar] [CrossRef]
- Zitvogel, L.; Tesniere, A.; Kroemer, G. Cancer despite immunosurveillance: Immunoselection and immunosubversion. Nat. Rev. Immunol. 2006, 6, 715–727. [Google Scholar] [CrossRef]
- Min, H.Y.; Lee, H.Y. Molecular targeted therapy for anticancer treatment. Exp. Mol. Med. 2022, 54, 1670–1694. [Google Scholar] [CrossRef]
- Allen, G.M.; Lim, W.A. Rethinking cancer targeting strategies in the era of smart cell therapeutics. Nat. Rev. Cancer 2022, 22, 693–702. [Google Scholar] [CrossRef]
- Wang, D.R.; Wu, X.L.; Sun, Y.L. Therapeutic targets and biomarkers of tumor immunotherapy: Response versus non-response. Signal Transduct. Targted Ther. 2022, 7, 331. [Google Scholar] [CrossRef]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Pierschbacher, M.D.; Ruoslahti, E. Cell attachment activity of fibronectin can be duplicated by small synthetic fragments of the molecule. Nature 1984, 309, 30–33. [Google Scholar] [CrossRef]
- Pytela, R.; Pierschbacher, M.D.; Ruoslahti, E. Identification and isolation of a 140-kd cell-surface glycoprotein with properties expected of a fibronectin receptor. Cell 1985, 40, 191–198. [Google Scholar] [CrossRef]
- Battistini, L.; Bugatti, K.; Sartori, A.; Curti, C.; Zanardi, F. RGD peptide-drug conjugates as effective dual targeting platforms: Recent advances. Eur. J. Org. Chem. 2021, 2021, 2506–2528. [Google Scholar] [CrossRef]
- Kapp, T.G.; Rechenmacher, F.; Neubauer, S.; Maltsev, O.V.; Cavalcanti-Adam, E.A.; Zarka, R.; Reuning, U.; Notni, J.; Wester, H.J.; Mas-Moruno, C.; et al. A comprehensive evaluation of the activity and selectivity profile of ligands for RGD-binding integrins. Sci. Rep. 2017, 7, 39805. [Google Scholar] [CrossRef] [PubMed]
- Javid, H.; Oryani, M.A.; Rezagholinejad, N.; Esparham, A.; Tajaldini, M.; Karimi-Shahri, M. RGD peptide in cancer targeting: Benefits, challenges, solutions, and possible integrin-RGD interactions. Cancer Med-Us 2024, 13, e6800. [Google Scholar] [CrossRef]
- Smith, G.P. Filamentous fusion phage-novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317. [Google Scholar] [CrossRef]
- Roberts, R.W.; Szostak, J.W. RNA-peptide fusions for the in vitro selection of peptides and proteins. Proc. Natl. Acad. Sci. USA 1997, 94, 12297–12302. [Google Scholar] [CrossRef]
- Swain, S.M.; Shastry, M.; Hamilton, E. Targeting HER2-positive breast cancer: Advances and future directions. Nat. Rev. Drug Discov. 2023, 22, 101–126. [Google Scholar] [CrossRef] [PubMed]
- Karasseva, N.G.; Glinsky, V.V.; Chen, N.X.; Komatireddy, R.; Quinn, T.P. Identification and characterization of peptides that bind human ErbB-2 selected from a bacteriophage display library. J. Protein Chem. 2002, 21, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Shadidi, M.; Sioud, M. Identification of novel carrier peptides for the specific delivery of therapeutics into cancer cells. FASEB J. 2002, 17, 256–258. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, A.A.; Yin, Y.Z.; Suga, H. Macrocyclic peptides as drug candidates: Recent progress and remaining challenges. J. Am. Chem. Soc. 2019, 141, 4167–4181. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.J.; Nielsen, A.L.; Heinis, C. Cyclic peptides for drug development. Angew. Chem. Int. Ed. 2024, 63, e202308251. [Google Scholar] [CrossRef] [PubMed]
- Dechantsreiter, M.A.; Planker, E.; Mathä, B.; Lohof, E.; Hölzemann, G.; Jonczyk, A.; Goodman, S.L.; Kessler, H. N-methylated cyclic RGD peptides as highly active and selective αβ integrin antagonists. J. Med. Chem. 1999, 42, 3033–3040. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.P.; Wang, Q.; Liu, Y.C.; Xie, Y. Molecular basis for the targeted binding of RGD-containing peptide to integrin αvβ3. Biomaterials 2014, 35, 1667–1675. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.L.; Leroux, J.C.; Gauthier, M.A. Twin disulfides for orthogonal disulfide pairing and the directed folding of multicyclic peptides. Nat. Chem. 2012, 4, 1045–1050. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.M.; Fan, S.H.; Xiao, S.L.; Li, J.J.; Zhang, S.L.; Wu, Y.P.; Kong, C.L.; Zhuang, J.; Liu, H.T.; Zhao, Y.B.; et al. Disulfide-directed multicyclic peptide libraries for the discovery of peptide ligands and drugs. J. Am. Chem. Soc. 2023, 145, 1964–1972. [Google Scholar] [CrossRef]
- White, C.J.; Yudin, A.K. Contemporary strategies for peptide macrocyclization. Nat. Chem. 2011, 3, 509–524. [Google Scholar] [CrossRef]
- Deyle, K.; Kong, X.D.; Heinis, C. Phage selection of cyclic peptides for application in research and drug development. Acc. Chem. Res. 2017, 50, 1866–1874. [Google Scholar] [CrossRef]
- Heinis, C.; Rutherford, T.; Freund, S.; Winter, G. Phage-encoded combinatorial chemical libraries based on bicyclic peptides. Nat. Chem. Biol. 2009, 5, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, Y.; Shoji, I.; Miyagawa, S.; Kawakami, T.; Katoh, T.; Goto, Y.; Suga, H. Natural product-like macrocyclic-methyl-peptide inhibitors against a ubiquitin ligase uncovered from a ribosome-expressed de novo library. Chem. Biol. 2011, 18, 1562–1570. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.C.; Wiedmann, M.M.; Suga, H. RNA display methods for the discovery of bioactive macrocycles. Chem. Rev. 2019, 119, 10360–10391. [Google Scholar] [CrossRef]
- Goto, Y.; Katoh, T.; Suga, H. Flexizymes for genetic code reprogramming. Nat. Protoc. 2011, 6, 779–790. [Google Scholar] [CrossRef] [PubMed]
- Worm, D.J.; Els-Heindl, S.; Beck-Sickinger, A.G. Targeting of peptide-binding receptors on cancer cells with peptide-drug conjugates. Pept. Sci. 2020, 112, e24171. [Google Scholar] [CrossRef]
- Vadevoo, S.M.P.; Gurung, S.; Lee, H.S.; Gunassekaran, G.R.; Lee, S.M.; Yoon, J.W.; Lee, Y.K.; Lee, B.Y.H. Peptides as multifunctional players in cancer therapy. Exp. Mol. Med. 2023, 55, 1099–1109. [Google Scholar] [CrossRef]
- Li, X.T.; Craven, T.W.; Levine, P.M. Cyclic peptide screening methods for preclinical drug discovery. J. Med. Chem. 2022, 65, 11913–11926. [Google Scholar] [CrossRef]
- Nakajima, H.; Kim, Y.B.; Terano, H.; Yoshida, M.; Horinouchi, S. FR901228, a potent antitumor antibiotic, is a novel histone deacetylase inhibitor. Exp. Cell Res. 1998, 241, 126–133. [Google Scholar] [CrossRef]
- Hennrich, U.; Kopka, K. Lutathera®: The First FDA- and EMA-approved radiopharmaceutical for peptide receptor radionuclide therapy. Pharmaceuticals 2019, 12, 114. [Google Scholar] [CrossRef]
- Shi, J.; Fan, D.; Dong, C.; Liu, H.; Jia, B.; Zhao, H.; Jin, X.; Liu, Z.; Li, F.; Wang, F. Anti-tumor effect of integrin targeted 177Lu-3PRGD2 and combined therapy with endostar. Theranostics 2014, 4, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Loriot, Y.; Siefker-Radtke, A.O.; Friedlander, T.W.; Necchi, A.; Wei, A.Z.; Sridhar, S.S.; Garmezy, B.; Arroyo, S.; Gartside, E.; Liu, J.; et al. A phase 2/3 study of bicycle toxin conjugate BT8009 targeting nectin-4 in patients with locally advanced or metastatic urothelial cancer (la/mUC): Duravelo-2. J. Clin. Oncol. 2024, 42, TPS4619. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, S. Cyclic peptide drugs approved in the last two decades (2001–2021). RSC Chem. Biol. 2022, 3, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Yu, L.; Miao, Y.; Liu, X.; Yu, Z.; Wei, M. Peptide-drug conjugates (PDCs): A novel trend of research and development on targeted therapy, hype or hope? Acta Pharm. Sin. B 2023, 13, 498–516. [Google Scholar] [CrossRef]
- Wang, M.D.; Lv, G.T.; An, H.W.; Zhang, N.Y.; Wang, H. In situ self-assembly of bispecific peptide for cancer immunotherapy. Angew. Chem. Int. Ed. 2022, 61, e202113649. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.; O’Sullivan Coyne, G.H.; Rubinstein, L.V.; Takebe, N.; Wright, J.J.; Wilsker, D.; Ferry-Galow, K.V.; Karlovich, C.A.; Anderson, L.; Kuhlmann, L.; et al. Pilot study of CBX-12 pharmacodynamics in patients with advanced solid tumors. J. Clin. Oncol. 2024, 42, TPS3187. [Google Scholar] [CrossRef]
- Wang, F.; Qu, L.; Ren, F.; Baghdasaryan, A.; Jiang, Y.; Hsu, R.; Liang, P.; Li, J.; Zhu, G.; Ma, Z.; et al. High-precision tumor resection down to few-cell level guided by NIR-IIb molecular fluorescence imaging. Proc. Natl. Acad. Sci. USA 2022, 119, e2123111119. [Google Scholar] [CrossRef] [PubMed]
- Van der Heide, C.D.; Dalm, S.U. Radionuclide imaging and therapy directed towards the tumor microenvironment: A multi-cancer approach for personalized medicine. Eur. J. Nucl. Med. Mol. Imaging 2022, 49, 4616–4641. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.L.; Thatte, A.S.; Mai, D.; Haley, R.M.; Gong, N.Q.; Han, X.X.; Wang, K.; Sheppard, N.C.; June, C.H.; Mitchell, M.J. Responsive biomaterials: Optimizing control of cancer immunotherapy. Nat. Rev. Mater. 2024, 9, 100–118. [Google Scholar] [CrossRef]
- Martínez-Reyes, I.; Chandel, N.S. Cancer metabolism: Looking forward. Nat. Rev. Cancer 2021, 21, 669–680. [Google Scholar] [CrossRef]
- Chao, Y.; Liu, Z. Biomaterials tools to modulate the tumour microenvironment in immunotherapy. Nat. Rev. Bioeng. 2023, 1, 125–138. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Shahriari, M.; Zahiri, M.; Abnous, K.; Taghdisi, S.M.; Ramezani, M.; Alibolandi, M. Enzyme responsive drug delivery systems in cancer treatment. J. Control Release 2019, 308, 172–189. [Google Scholar] [CrossRef]
- Lou, X.F.; Du, Y.Z.; Xu, X.L. Endogenous enzyme-responsive nanoplatforms for anti-tumor therapy. Curr. Drug Targets 2021, 22, 845–855. [Google Scholar] [CrossRef] [PubMed]
- Maggi, M.; Scotti, C. Enzymes in metabolic anticancer therapy. Adv. Exp. Med. Biol. 2019, 1148, 173–199. [Google Scholar]
- Li, M.Q.; Zhao, G.K.; Su, W.K.; Shuai, Q. Enzyme-responsive nanoparticles for anti-tumor drug delivery. Front. Chem. 2020, 8, 647. [Google Scholar] [CrossRef]
- Zhao, Y.; Guo, Y.G.; Tang, L. Engineering cancer vaccines using stimuli-responsive biomaterials. Nano Res. 2018, 11, 5355–5371. [Google Scholar] [CrossRef]
- Yang, Z.; Liang, G.; Xu, B. Enzymatic hydrogelation of small molecules. Acc. Chem. Res. 2008, 41, 315–326. [Google Scholar] [CrossRef] [PubMed]
- He, H.J.; Tan, W.Y.; Guo, J.Q.; Yi, M.H.; Shy, A.N.; Xu, B. Enzymatic noncovalent synthesis. Chem. Rev. 2020, 120, 9994–10078. [Google Scholar] [CrossRef]
- Yang, Z.M.; Gu, H.W.; Fu, D.G.; Gao, P.; Lam, J.K.; Xu, B. Enzymatic formation of supramolecular hydrogels. Adv. Mater. 2004, 16, 1440–1444. [Google Scholar] [CrossRef]
- Kuang, Y.; Shi, J.; Li, J.; Yuan, D.; Alberti, K.A.; Xu, Q.; Xu, B. Pericellular hydrogel/nanonets inhibit cancer cells. Angew. Chem. Int. Ed. 2014, 53, 8104–8107. [Google Scholar] [CrossRef]
- Chen, Y.M.; Li, W.; Wang, Z.Q.; Yu, Y.Y.; Li, J.; Ding, Y.H.; Hu, Z.W.; Liu, Q.; Yang, Z.M.; Gao, J. A transformable supramolecular bispecific cell engager for augmenting natural killer and T cell-based cancer immunotherapy. Adv. Mater. 2024, 36, e2306736. [Google Scholar] [CrossRef]
- Zhang, W.; Hu, J.J.; Liu, R.; Dai, J.; Yuan, L.Z.; Liu, Y.H.; Chen, B.C.; Gong, M.X.; Xia, F.; Lou, X.D. A peptide-conjugated probe with cleavage-induced morphological change for treatment on tumor cell membrane. Adv. Sci. 2023, 10, 2207228. [Google Scholar] [CrossRef]
- Liu, X.Y.; Zhan, W.J.; Gao, G.; Jiang, Q.C.; Zhang, X.P.; Zhang, H.B.; Sun, X.B.; Han, W.; Wu, F.G.; Liang, G.L. Apoptosis-amplified assembly of porphyrin nanofiber enhances photodynamic therapy of oral tumor. J. Am. Chem. Soc. 2023, 145, 7918–7930. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.B.; Wang, D.; Gao, Y.J.; Wang, L.; Qiao, Z.Y.; Wang, H. Autocatalytic morphology transformation platform for targeted drug accumulation. J. Am. Chem. Soc. 2019, 141, 4406–4411. [Google Scholar] [CrossRef] [PubMed]
- Cheung, E.C.; Vousden, K.H. The role of ROS in tumour development and progression. Nat. Rev. Cancer 2022, 22, 280–297. [Google Scholar] [CrossRef] [PubMed]
- Curvello, R.; Kast, V.; Ordoñez-Morán, P.; Mata, A.; Loessner, D. Biomaterial-based platforms for tumour tissue engineering. Nat. Rev. Mater. 2023, 8, 314–330. [Google Scholar] [CrossRef]
- De Angelis, B.; Depalo, N.; Petronella, F.; Quintarelli, C.; Curri, M.L.; Pani, R.; Calogero, A.; Locatelli, F.; De Sio, L. Stimuli-responsive nanoparticle-assisted immunotherapy: A new weapon against solid tumours. J. Mater. Chem. B 2020, 8, 1823–1840. [Google Scholar] [CrossRef]
- Qin, J.L.; Sun, M.; Hu, W.; Cheng, J.J.; Fan, Z.; Du, J.Z. Stimuli-responsive hydrogels for cancer immunotherapy. Polym. Chem. 2023, 14, 793–802. [Google Scholar] [CrossRef]
- Cheng, D.B.; Zhang, X.H.; Gao, Y.J.; Ji, L.; Hou, D.Y.; Wang, Z.Q.; Xu, W.H.; Qiao, Z.Y.; Wang, H. Endogenous reactive oxygen species-triggered morphology transformation for enhanced cooperative interaction with mitochondria. J. Am. Chem. Soc. 2019, 141, 7235–7239. [Google Scholar] [CrossRef]
- Estrela, J.M.; Ortega, A.; Obrador, E. Glutathione in cancer biology and therapy. Crit. Rev. Clin. Lab. Sci. 2006, 43, 143–181. [Google Scholar] [CrossRef] [PubMed]
- Niu, B.Y.; Liao, K.X.; Zhou, Y.X.; Wen, T.; Quan, G.L.; Pan, X.; Wu, C.B. Application of glutathione depletion in cancer therapy: Enhanced ROS-based therapy, ferroptosis, and chemotherapy. Biomaterials 2021, 277, 121110. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, L.; Sandhu, J.K.; Harper, M.E.; Cuperlovic-Culf, M. Role of glutathione in cancer: From mechanisms to therapies. Biomolecules 2020, 10, 1429. [Google Scholar] [CrossRef] [PubMed]
- Bansal, A.; Simon, M.C. Glutathione metabolism in cancer progression and treatment resistance. J. Cell Biol. 2018, 217, 2291–2298. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, M.M.; Liu, J.Z.; Song, Y.Q.; Hu, B.B.; Wu, C.X.; Liu, A.A.; Zhou, H.; Long, J.F.; Shi, L.Q.; et al. Self-sorting peptide assemblies in living cells for simultaneous organelle targeting. J. Am. Chem. Soc. 2022, 144, 9312–9323. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.D.; Zhang, R.; Hu, Y.X.; Wu, L.Y.; Bai, H.; Song, D.F.; Wang, Y.F.; An, R.B.; Weng, J.H.; Zhang, S.R.; et al. Controlled sequential in situ self-assembly and disassembly of a fluorogenic cisplatin prodrug for cancer theranostics. Nat. Commun. 2023, 14, 800. [Google Scholar] [CrossRef] [PubMed]
- Koltai, T. The pH paradigm in cancer. Eur. J. Clin. Nutr. 2020, 74, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Bogdanov, A.; Bogdanov, A.; Chubenko, V.; Volkov, N.; Moiseenko, F.; Moiseyenko, V. Tumor acidity: From hallmark of cancer to target of treatment. Front. Oncol. 2022, 12, 979154. [Google Scholar] [CrossRef]
- Li, Z.; Zhu, Y.; Matson, J.B. pH-responsive self-assembling peptide-based biomaterials: Designs and applications. ACS Appl. Bio Mater. 2022, 5, 4635–4651. [Google Scholar] [CrossRef]
- Yan, Y.F.; Ding, H.W. pH-responsive nanoparticles for cancer immunotherapy: A brief review. Nanomaterials 2020, 10, 1613. [Google Scholar] [CrossRef]
- Manchun, S.; Dass, C.R.; Sriamornsak, P. Targeted therapy for cancer using pH-responsive nanocarrier systems. Life Sci. 2012, 90, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Pieszka, M.; Han, S.; Volkmann, C.; Graf, R.; Lieberwirth, I.; Landfester, K.; Ng, D.Y.W.; Weil, T. Controlled supramolecular assembly inside living cells by sequential multistaged chemical reactions. J. Am. Chem. Soc. 2020, 142, 15780–15789. [Google Scholar] [CrossRef]
- Zhang, K.; Yang, P.P.; He, P.P.; Wen, S.F.; Zou, X.R.; Fan, Y.; Chen, Z.M.; Cao, H.; Yang, Z.; Yue, K.; et al. Peptide-based nanoparticles mimic fibrillogenesis of laminin in tumor vessels for precise embolization. ACS Nano 2020, 14, 7170–7180. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.R.; Zhu, Y.X.; Liu, X.Y.; Pan, G.Y.; Gao, G.; Sun, W.; Zhang, X.D.; Jiang, Y.W.; Wu, F.G. Construction of dually responsive nanotransformers with nanosphere-nanofiber-nanosphere transition for overcoming the size paradox of anticancer nanodrugs. ACS Nano 2019, 13, 11781–11792. [Google Scholar] [CrossRef]
- Hunt, J.F.; Rath, P.; Rothschild, K.J.; Engelman, D.M. Spontaneous, pH-dependent membrane insertion of a transbilayer alpha-helix. Biochemistry 1997, 36, 15177–15192. [Google Scholar] [CrossRef] [PubMed]
- Weerakkody, D.; Moshnikova, A.; Thakur, M.S.; Moshnikova, V.; Daniels, J.; Engelman, D.M.; Andreev, O.A.; Reshetnyak, Y.K. Family of pH (low) insertion peptides for tumor targeting. Proc. Natl. Acad. Sci. USA 2013, 110, 5834–5839. [Google Scholar] [CrossRef] [PubMed]
- Reshetnyak, Y.K.; Andreev, O.A.; Engelman, D.M. Aiming the magic bullet: Targeted delivery of imaging and therapeutic agents to solid tumors by pHLIP peptides. Front. Pharmacol. 2024, 15, 1355893. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Y.; Song, G.; He, Y.; Zhang, X.; Liu, Y.; Ju, H. A DNA-azobenzene nanopump fueled by upconversion luminescence for controllable intracellular drug release. Angew. Chem. Int. Ed. 2019, 58, 18207–18211. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.; Huang, Y.; Wang, B.; Ma, L.; Karges, J.; Xiao, H. Photo-reduction with NIR light of nucleus-targeting PtIVNanoparticles for combined tumor-targeted chemotherapy and photodynamic immunotherapy. Angew. Chem. Int. Ed. 2022, 61, e202201486. [Google Scholar] [CrossRef]
- Han, K.; Lei, Q.; Wang, S.B.; Hu, J.J.; Qiu, W.X.; Zhu, J.Y.; Yin, W.N.; Luo, X.; Zhang, X.Z. Dual-stage-light-guided tumor inhibition by mitochondria-targeted photodynamic therapy. Adv. Funct. Mater. 2015, 25, 2961–2971. [Google Scholar] [CrossRef]
- Fuchs, N.; Zhang, L.; Calvo-Barreiro, L.; Kuncewicz, K.; Gabr, M. Inhibitors of immune checkpoints: Small molecule and peptide-based approaches. J. Pers. Med. 2024, 14, 68. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, C.; Wu, C.; Song, L. Natural peptides for immunological regulation in cancer therapy: Mechanism, facts and perspectives. Biomed. Pharmacother. 2023, 159, 114257. [Google Scholar] [CrossRef] [PubMed]
- Gabernet, G.; Müller, A.T.; Hiss, J.A.; Schneider, G. Membranolytic anticancer peptides. MedChemComm 2016, 7, 2232–2245. [Google Scholar] [CrossRef]
- Wang, P.; Liu, J.; Zhu, X.; Kenry; Yan, Z.; Yan, J.; Jiang, J.; Fu, M.; Ge, J.; Zhu, Q.; et al. Modular synthesis of clickable peptides via late-stage maleimidation on C (7)-H tryptophan. Nat. Commun. 2023, 14, 3973. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, G.; O’Connor, J.; Rettie, S.; Huang, Y.H.; Ramelot, T.A.; Mulligan, V.K.; Alpkilic, G.G.; Palmer, J.; Bera, A.K.; Bick, M.J.; et al. Accurate de novo design of membrane-traversing macrocycles. Cell 2022, 185, 3520–3532. [Google Scholar] [CrossRef] [PubMed]
- Salveson, P.J.; Moyer, A.P.; Said, M.Y.; Gökçe, G.; Li, X.; Kang, A.; Nguyen, H.; Bera, A.K.; Levine, P.M.; Bhardwaj, G.; et al. Expansive discovery of chemically diverse structured macrocyclic oligoamides. Science 2024, 384, 420–428. [Google Scholar] [CrossRef]
- Cheng, D.; Chen, X.; Zhang, W.; Guo, P.; Xue, W.; Xia, J.; Wu, S.; Shi, J.; Ma, D.; Zuo, X.B.; et al. Design of multicomponent peptide fibrils with ordered and programmable compositional patterns. Angew. Chem. Int. Ed. 2023, 62, e202303684. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Jia, F.; Jiang, Y.B.; Conticello, V.P.; Jiang, T. Assembly of peptide nanostructures with controllable sizes. Nano Res. 2024, 17, 151–161. [Google Scholar] [CrossRef]
- Notin, P.; Rollins, N.; Gal, Y.; Sander, C.; Marks, D. Machine learning for functional protein design. Nat. Biotechnol. 2024, 42, 216–228. [Google Scholar] [CrossRef]
- Min, J.W.; Rong, X.; Zhang, J.X.; Su, R.X.; Wang, Y.F.; Qi, W. Computational design of peptide assemblies. J. Chem. Theory Comput. 2024, 20, 532–550. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).