
Evaluation of the Molecular Mechanism of Chlorogenic Acid in the Treatment of Pulmonary Arterial Hypertension Based on Analysis Network Pharmacology and Molecular Docking


 and
and
Abstract
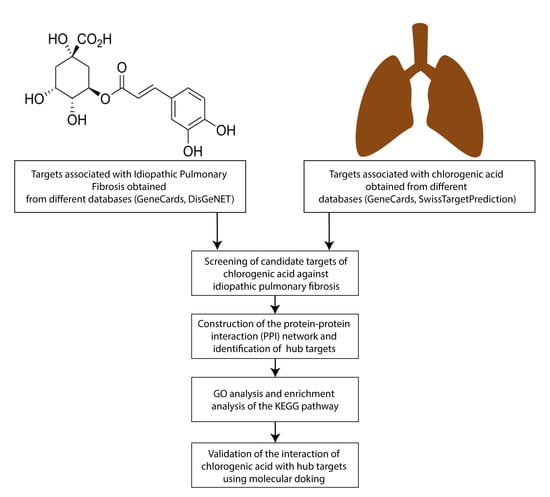
1. Introduction
2. Materials and Methods
2.1. Obtaining CGA-Related Target Genes
2.2. Obtaining PAH-Related Target Genes
2.3. Obtaining Potential Common PAH and CGA Targets
2.4. Protein-Protein Interaction (PPI) Network Construction and Detection of Hub Targets
2.5. Gene Ontology (GO) Enrichment and KEGG Pathway Enrichment Analysis
2.6. Molecular Docking
3. Results
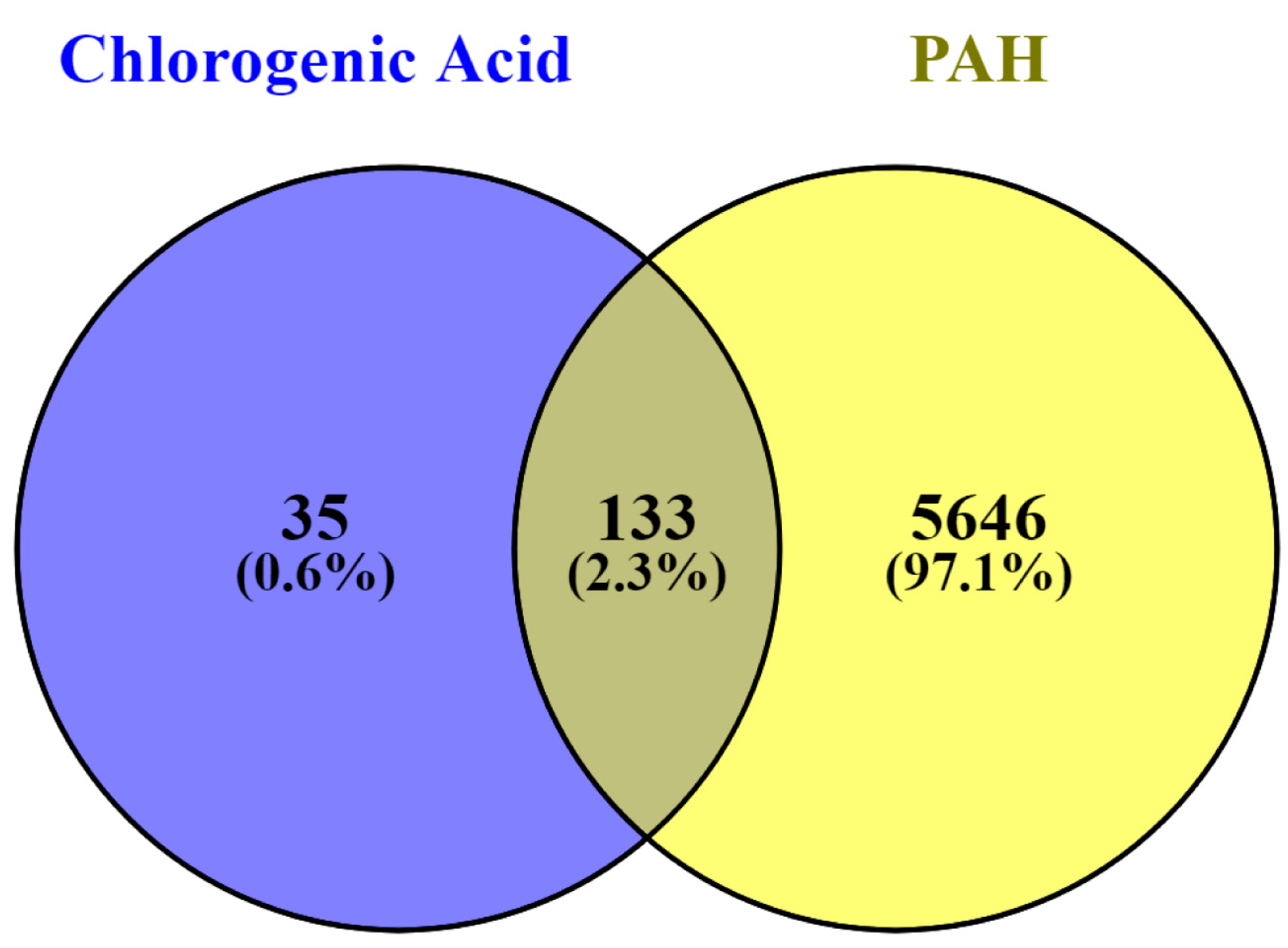
3.1. Screening of Potential PAH Targets for CGA
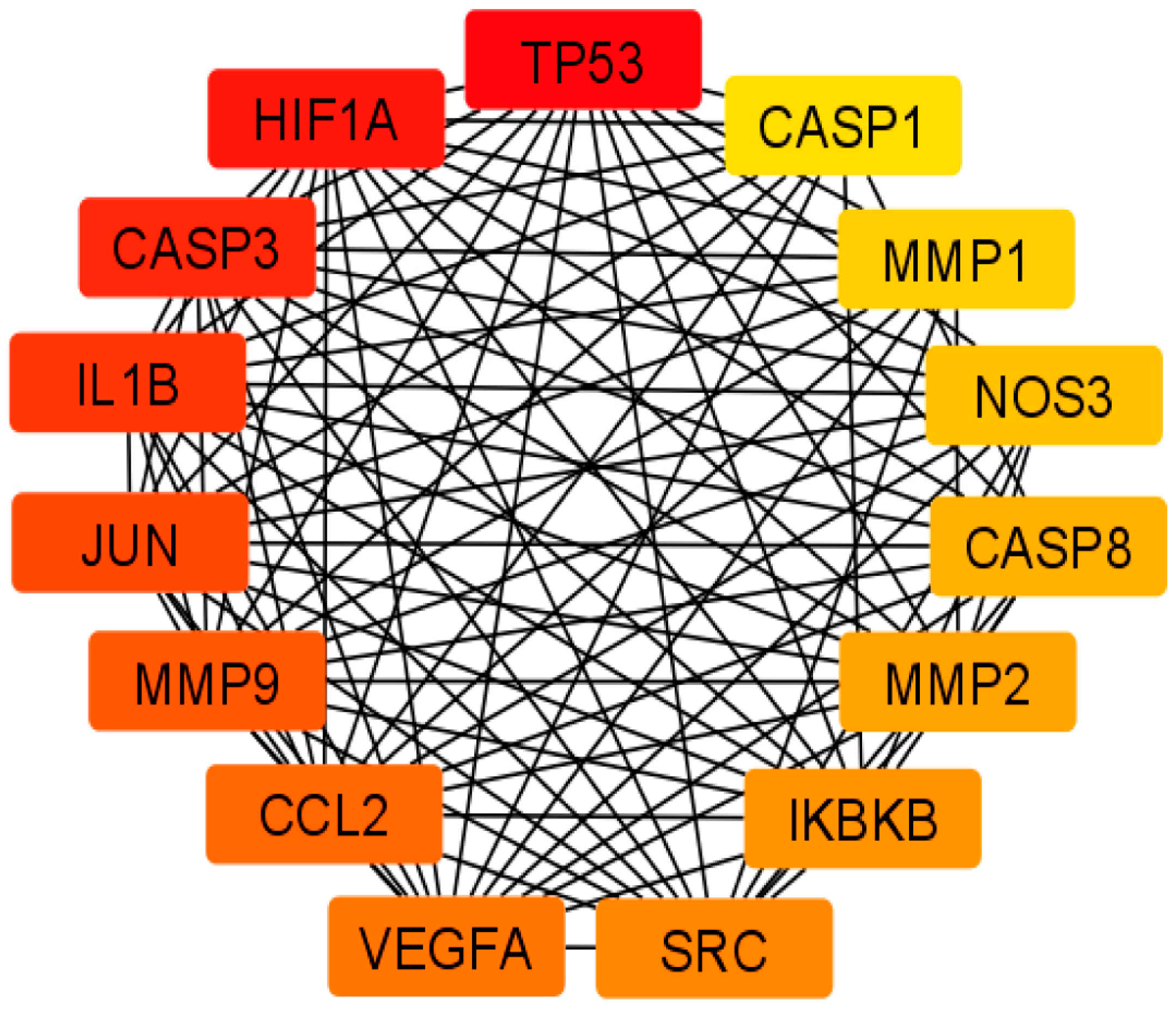
3.2. PPI Network Construction and Hub Target Analysis
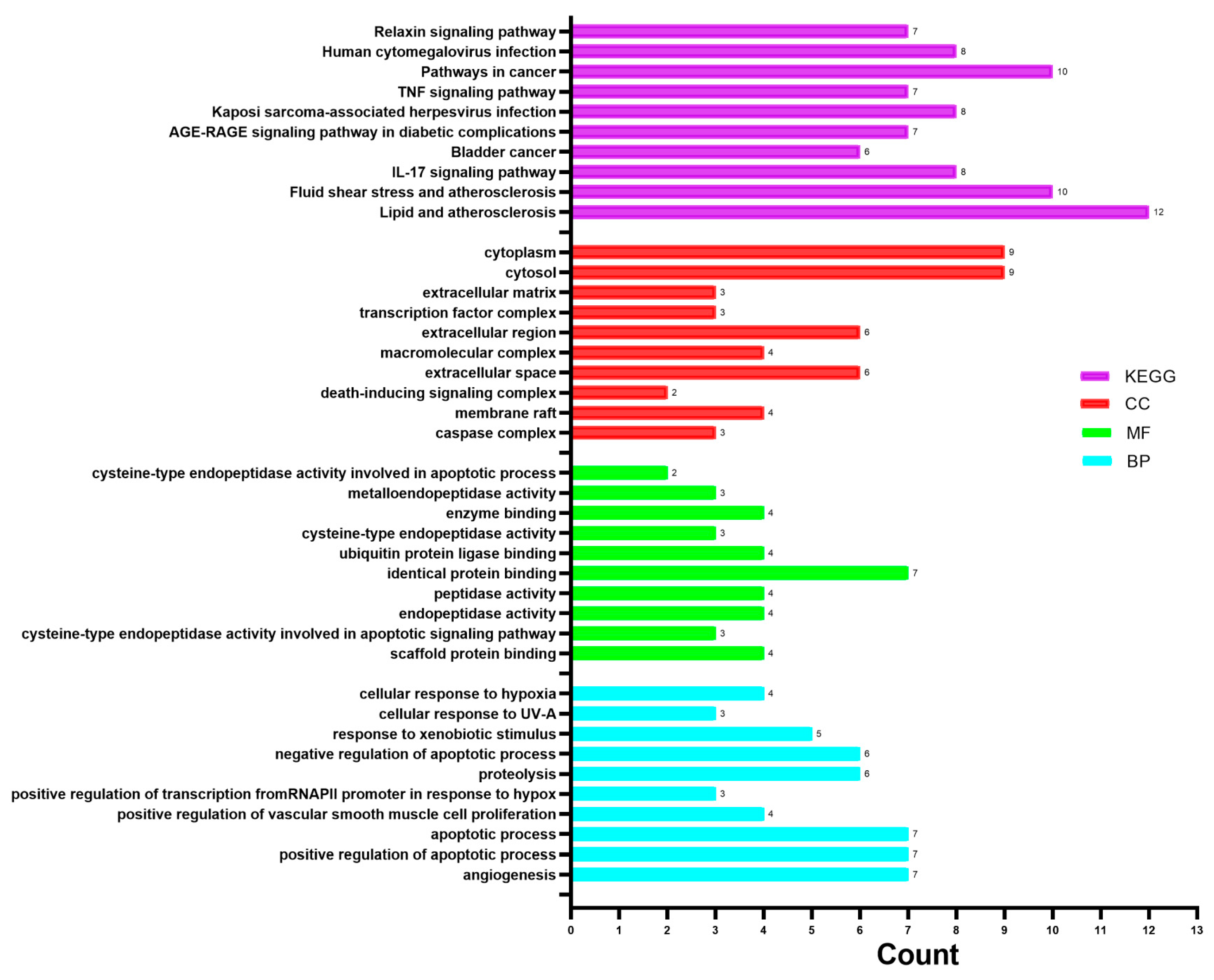
3.3. GO and KEGG Pathway Enrichment Analyses
3.4. Molecular Docking of Hub Targets
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zou, W.; Wang, X.; Sun, R.; Hu, J.; Ye, D.; Bai, G.; Liu, S.; Hong, W.; Guo, M.; Ran, P. PM2.5 Induces Airway Remodeling in Chronic Obstructive Pulmonary Diseases via the Wnt5a/β-Catenin Pathway. Int. J. Chronic Obstr. Pulm. Dis. 2021, 16, 3285–3295. [Google Scholar] [CrossRef]
- Quarck, R.; Wynants, M.; Verbeken, E.; Meyns, B.; Delcroix, M. Contribution of inflammation and impaired angiogenesis to the pathobiology of chronic thromboembolic pulmonary hypertension. Eur. Respir. J. 2015, 46, 431–443. [Google Scholar] [CrossRef]
- Hemnes, A.R.; Luther, J.M.; Rhodes, C.J.; Burgess, J.P.; Carlson, J.; Fan, R.; Fessel, J.P.; Fortune, N.; Gerszten, R.E.; Halliday, S.J.; et al. Human PAH is characterized by a pattern of lipid-related insulin resistance. JCI Insight 2019, 4, e123611. [Google Scholar] [CrossRef]
- Thenappan, T.; Chan, S.Y.; Weir, E.K. Role of extracellular matrix in the pathogenesis of pulmonary arterial hypertension. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1322–H1331. [Google Scholar] [CrossRef]
- Zeng, Z.; Wang, X.; Cui, L.; Wang, H.; Guo, J.; Chen, Y. Natural Products for the Treatment of Pulmonary Hypertension: Mechanism, Progress, and Future Opportunities. Curr. Issues Mol. Biol. 2023, 45, 2351–2371. [Google Scholar] [CrossRef]
- Moriyama, H.; Endo, J. Pathophysiological Involvement of Mast Cells and the Lipid Mediators in Pulmonary Vascular Remodeling. Int. J. Mol. Sci. 2023, 24, 6619. [Google Scholar] [CrossRef]
- Yang, Y.; Yin, L.; Zhu, M.; Song, S.; Sun, C.; Han, X.; Xu, Y.; Zhao, Y.; Qi, Y.; Xu, L.; et al. Protective effects of dioscin on vascular remodeling in pulmonary arterial hypertension via adjusting GRB2/ERK/PI3K-AKT signal. Biomed. Pharmacother. = Biomed. Pharmacother. 2021, 133, 111056. [Google Scholar] [CrossRef]
- Chang, K.Y.; Duval, S.; Badesch, D.B.; Bull, T.M.; Chakinala, M.M.; De Marco, T.; Frantz, R.P.; Hemnes, A.; Mathai, S.C.; Rosenzweig, E.B.; et al. Mortality in Pulmonary Arterial Hypertension in the Modern Era: Early Insights From the Pulmonary Hypertension Association Registry. J. Am. Heart Assoc. 2022, 11, e024969. [Google Scholar] [CrossRef]
- Nwafor, E.-O.; Lu, P.; Zhang, Y.; Liu, R.; Peng, H.; Xing, B.; Liu, Y.; Li, Z.; Zhang, K.; Zhang, Y.; et al. Chlorogenic acid: Potential source of natural drugs for the therapeutics of fibrosis and cancer. Transl. Oncol. 2022, 15, 101294. [Google Scholar] [CrossRef]
- Karpinska, J.; Świsłocka, R.; Lewandowski, W. A mystery of a cup of coffee; an insight look by chemist. BioFactors (Oxf. Engl.) 2017, 43, 621–632. [Google Scholar] [CrossRef]
- Murai, T.; Matsuda, S. The Chemopreventive Effects of Chlorogenic Acids, Phenolic Compounds in Coffee, against Inflammation, Cancer, and Neurological Diseases. Molecules 2023, 28, 2381. [Google Scholar] [CrossRef]
- Ding, Q.; Xu, Y.M.; Lau, A.T.Y. The Epigenetic Effects of Coffee. Molecules 2023, 28, 1770. [Google Scholar] [CrossRef]
- Natella, F.; Nardini, M.; Giannetti, I.; Dattilo, C.; Scaccini, C. Coffee drinking influences plasma antioxidant capacity in humans. J. Agric. Food Chem. 2002, 50, 6211–6216. [Google Scholar] [CrossRef]
- Hoelzl, C.; Knasmüller, S.; Wagner, K.H.; Elbling, L.; Huber, W.; Kager, N.; Ferk, F.; Ehrlich, V.; Nersesyan, A.; Neubauer, O.; et al. Instant coffee with high chlorogenic acid levels protects humans against oxidative damage of macromolecules. Mol. Nutr. Food Res. 2010, 54, 1722–1733. [Google Scholar] [CrossRef]
- Zeng, A.; Liang, X.; Zhu, S.; Liu, C.; Wang, S.; Zhang, Q.; Zhao, J.; Song, L. Chlorogenic acid induces apoptosis, inhibits metastasis and improves antitumor immunity in breast cancer via the NF-κB signaling pathway. Oncol. Rep. 2021, 45, 717–727. [Google Scholar] [CrossRef]
- Loader, T.B.; Taylor, C.G.; Zahradka, P.; Jones, P.J. Chlorogenic acid from coffee beans: Evaluating the evidence for a blood pressure-regulating health claim. Nutr. Rev. 2017, 75, 114–133. [Google Scholar] [CrossRef]
- Larki-Harchegani, A.; Fayazbakhsh, F.; Nourian, A.; Nili-Ahmadabadi, A. Chlorogenic acid protective effects on paraquat-induced pulmonary oxidative damage and fibrosis in rats. J. Biochem. Mol. Toxicol. 2023, 37, e23352. [Google Scholar] [CrossRef]
- Wang, L.; Du, H.; Chen, P. Chlorogenic acid inhibits the proliferation of human lung cancer A549 cell lines by targeting annexin A2 in vitro and in vivo. Biomed. Pharmacother. Biomed. Pharmacother. 2020, 131, 110673. [Google Scholar] [CrossRef]
- Wang, Y.C.; Dong, J.; Nie, J.; Zhu, J.X.; Wang, H.; Chen, Q.; Chen, J.Y.; Xia, J.M.; Shuai, W. Amelioration of bleomycin-induced pulmonary fibrosis by chlorogenic acid through endoplasmic reticulum stress inhibition. Apoptosis Int. J. Program. Cell Death 2017, 22, 1147–1156. [Google Scholar] [CrossRef]
- Kong, D.; Ding, Y.; Liu, J.; Liu, R.; Zhang, J.; Zhou, Q.; Long, Z.; Peng, J.; Li, L.; Bai, H.; et al. Chlorogenic acid prevents paraquat-induced apoptosis via Sirt1-mediated regulation of redox and mitochondrial function. Free Radic. Res. 2019, 53, 680–693. [Google Scholar] [CrossRef]
- Mao, X.; Xie, X.; Ma, J.; Wei, Y.; Huang, Z.; Wang, T.; Zhu, J.; Wang, Y.; Zhao, H.; Hua, J. Chlorogenic Acid Inhibited Epithelial-Mesenchymal Transition to Treat Pulmonary Fibrosis through Modulating Autophagy. Biol. Pharm. Bull. 2023, 46, 929–938. [Google Scholar] [CrossRef]
- Li, Q.Y.; Zhu, Y.F.; Zhang, M.; Chen, L.; Zhang, Z.; Du, Y.L.; Ren, G.Q.; Tang, J.M.; Zhong, M.K.; Shi, X.J. Chlorogenic acid inhibits hypoxia-induced pulmonary artery smooth muscle cells proliferation via c-Src and Shc/Grb2/ERK2 signaling pathway. Eur. J. Pharmacol. 2015, 751, 81–88. [Google Scholar] [CrossRef]
- Hopkins, A.L. Network pharmacology: The next paradigm in drug discovery. Nat. Chem. Biol. 2008, 4, 682–690. [Google Scholar] [CrossRef]
- Min, S.; Lee, B.; Yoon, S. Deep learning in bioinformatics. Brief. Bioinform. 2016, 18, 851–869. [Google Scholar] [CrossRef]
- Maier, J.K.; Labute, P. Assessment of fully automated antibody homology modeling protocols in molecular operating environment. Proteins 2014, 82, 1599–1610. [Google Scholar] [CrossRef]
- Goodsell, D.S.; Sanner, M.F.; Olson, A.J.; Forli, S. The AutoDock suite at 30. Protein Sci. A Publ. Protein Soc. 2021, 30, 31–43. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2023 update. Nucleic Acids Res. 2022, 51, D1373–D1380. [Google Scholar] [CrossRef]
- Gfeller, D.; Grosdidier, A.; Wirth, M.; Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: A web server for target prediction of bioactive small molecules. Nucleic Acids Res. 2014, 42, W32–W38. [Google Scholar] [CrossRef]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 2016, 54, 1.30.1–1.30.33. [Google Scholar] [CrossRef]
- Consortium, T.U. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2022, 51, D523–D531. [Google Scholar] [CrossRef]
- Zaru, R.; Orchard, S. UniProt Tools: BLAST, Align, Peptide Search, and ID Mapping. Curr. Protoc. 2023, 3, e697. [Google Scholar] [CrossRef]
- Piñero, J.; Ramírez-Anguita, J.M.; Saüch-Pitarch, J.; Ronzano, F.; Centeno, E.; Sanz, F.; Furlong, L.I. The DisGeNET knowledge platform for disease genomics: 2019 update. Nucleic Acids Res. 2019, 48, D845–D855. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, X.; Gan, J.; Chen, S.; Xiao, Z.-X.; Cao, Y. CB-Dock2: Improved protein–ligand blind docking by integrating cavity detection, docking and homologous template fitting. Nucleic Acids Res. 2022, 50, W159–W164. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Lim, C.S.; Kiriakidis, S.; Sandison, A.; Paleolog, E.M.; Davies, A.H. Hypoxia-inducible factor pathway and diseases of the vascular wall. J. Vasc. Surg. 2013, 58, 219–230. [Google Scholar] [CrossRef]
- Jiang, G.; Li, T.; Qiu, Y.; Rui, Y.; Chen, W.; Lou, Y. RNA interference for HIF-1alpha inhibits foam cells formation in vitro. Eur. J. Pharmacol. 2007, 562, 183–190. [Google Scholar] [CrossRef]
- Hänze, J.; Weissmann, N.; Grimminger, F.; Seeger, W.; Rose, F. Cellular and molecular mechanisms of hypoxia-inducible factor driven vascular remodeling. Thromb. Haemost. 2007, 97, 774–787. [Google Scholar]
- Liu, J.; Wang, W.; Wang, L.; Chen, S.; Tian, B.; Huang, K.; Corrigan, C.J.; Ying, S.; Wang, W.; Wang, C. IL-33 Initiates Vascular Remodelling in Hypoxic Pulmonary Hypertension by up-Regulating HIF-1α and VEGF Expression in Vascular Endothelial Cells. EBioMedicine 2018, 33, 196–210. [Google Scholar] [CrossRef]
- Terefe, E.M.; Ghosh, A. Molecular Docking, Validation, Dynamics Simulations, and Pharmacokinetic Prediction of Phytochemicals Isolated From Croton dichogamus Against the HIV-1 Reverse Transcriptase. Bioinform. Biol. Insights 2022, 16, 11779322221125605. [Google Scholar] [CrossRef]
- Maron, B.A.; Abman, S.H.; Elliott, C.G.; Frantz, R.P.; Hopper, R.K.; Horn, E.M.; Nicolls, M.R.; Shlobin, O.A.; Shah, S.J.; Kovacs, G.; et al. Pulmonary Arterial Hypertension: Diagnosis, Treatment, and Novel Advances. Am. J. Respir. Crit. Care Med. 2021, 203, 1472–1487. [Google Scholar] [CrossRef]
- Bhogal, S.; Khraisha, O.; Al Madani, M.; Treece, J.; Baumrucker, S.J.; Paul, T.K. Sildenafil for Pulmonary Arterial Hypertension. Am. J. Ther. 2019, 26, e520–e526. [Google Scholar] [CrossRef]
- Lei, W.; He, Y.; Shui, X.; Li, G.; Yan, G.; Zhang, Y.; Huang, S.; Chen, C.; Ding, Y. Expression and analyses of the HIF-1 pathway in the lungs of humans with pulmonary arterial hypertension. Mol. Med. Rep. 2016, 14, 4383–4390. [Google Scholar] [CrossRef]
- He, M.; Cui, T.; Cai, Q.; Wang, H.; Kong, H.; Xie, W. Iptakalim ameliorates hypoxia-impaired human endothelial colony-forming cells proliferation, migration, and angiogenesis via Akt/eNOS pathways. Pulm. Circ. 2019, 9, 2045894019875417. [Google Scholar] [CrossRef]
- Melton, E.; Qiu, H. Interleukin-1β in Multifactorial Hypertension: Inflammation, Vascular Smooth Muscle Cell and Extracellular Matrix Remodeling, and Non-Coding RNA Regulation. Int. J. Mol. Sci. 2021, 22, 8639. [Google Scholar] [CrossRef]
- Weber, A.; Wasiliew, P.; Kracht, M. Interleukin-1 (IL-1) pathway. Sci. Signal. 2010, 3, cm1. [Google Scholar] [CrossRef]
- Soon, E.; Holmes, A.M.; Treacy, C.M.; Doughty, N.J.; Southgate, L.; Machado, R.D.; Trembath, R.C.; Jennings, S.; Barker, L.; Nicklin, P.; et al. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation 2010, 122, 920–927. [Google Scholar] [CrossRef]
- Bui, C.B.; Kolodziej, M.; Lamanna, E.; Elgass, K.; Sehgal, A.; Rudloff, I.; Schwenke, D.O.; Tsuchimochi, H.; Kroon, M.; Cho, S.X.; et al. Interleukin-1 Receptor Antagonist Protects Newborn Mice Against Pulmonary Hypertension. Front. Immunol. 2019, 10, 1480. [Google Scholar] [CrossRef]
- Voelkel, N.F.; Tuder, R.M.; Bridges, J.; Arend, W.P. Interleukin-1 receptor antagonist treatment reduces pulmonary hypertension generated in rats by monocrotaline. Am. J. Respir. Cell Mol. Biol. 1994, 11, 664–675. [Google Scholar] [CrossRef]
- Trankle, C.R.; Canada, J.M.; Kadariya, D.; Markley, R.; De Chazal, H.M.; Pinson, J.; Fox, A.; Van Tassell, B.W.; Abbate, A.; Grinnan, D. IL-1 Blockade Reduces Inflammation in Pulmonary Arterial Hypertension and Right Ventricular Failure: A Single-Arm, Open-Label, Phase IB/II Pilot Study. Am. J. Respir. Crit. Care Med. 2019, 199, 381–384. [Google Scholar] [CrossRef]
- Campos, M.; Schiopu, E. Pulmonary Arterial Hypertension in Adult-Onset Still’s Disease: Rapid Response to Anakinra. Case Rep. Rheumatol. 2012, 2012, 537613. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, S.; Wu, L.; Liu, Y.; Du, J.; Luo, Z.; Xu, J.; Guo, L.; Liu, Y. Epigenetic regulation of chemokine (CC-motif) ligand 2 in inflammatory diseases. Cell Prolif. 2023, 56, e13428. [Google Scholar] [CrossRef]
- Zhang, H.; Yang, K.; Chen, F.; Liu, Q.; Ni, J.; Cao, W.; Hua, Y.; He, F.; Liu, Z.; Li, L.; et al. Role of the CCL2-CCR2 axis in cardiovascular disease: Pathogenesis and clinical implications. Front. Immunol. 2022, 13, 975367. [Google Scholar] [CrossRef]
- Raghu, G.; Martinez, F.J.; Brown, K.K.; Costabel, U.; Cottin, V.; Wells, A.U.; Lancaster, L.; Gibson, K.F.; Haddad, T.; Agarwal, P.; et al. CC-chemokine ligand 2 inhibition in idiopathic pulmonary fibrosis: A phase 2 trial of carlumab. Eur. Respir. J. 2015, 46, 1740–1750. [Google Scholar] [CrossRef]
- Zhang, J.J.; Liu, W.; Xing, G.Z.; Xiang, L.; Zheng, W.M.; Ma, Z.L. Role of CC-chemokine ligand 2 in gynecological cancer. Cancer Cell Int. 2022, 22, 361. [Google Scholar] [CrossRef]
- O’Connor, T.; Borsig, L.; Heikenwalder, M. CCL2-CCR2 Signaling in Disease Pathogenesis. Endocr. Metab. Immune Disord. Drug Targets 2015, 15, 105–118. [Google Scholar] [CrossRef]
- Sanchez, O.; Marcos, E.; Perros, F.; Fadel, E.; Tu, L.; Humbert, M.; Dartevelle, P.; Simonneau, G.; Adnot, S.; Eddahibi, S. Role of endothelium-derived CC chemokine ligand 2 in idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2007, 176, 1041–1047. [Google Scholar] [CrossRef]
- Abid, S.; Marcos, E.; Parpaleix, A.; Amsellem, V.; Breau, M.; Houssaini, A.; Vienney, N.; Lefevre, M.; Derumeaux, G.; Evans, S.; et al. CCR2/CCR5-mediated macrophage-smooth muscle cell crosstalk in pulmonary hypertension. Eur. Respir. J. 2019, 54, 1802308. [Google Scholar] [CrossRef]
- Ochoa, S.V.; Otero, L.; Aristizabal-Pachon, A.F.; Hinostroza, F.; Carvacho, I.; Torres, Y.P. Hypoxic Regulation of the Large-Conductance, Calcium and Voltage-Activated Potassium Channel, BK. Front. Physiol. 2021, 12, 780206. [Google Scholar] [CrossRef]
- Breault, N.M.; Wu, D.; Dasgupta, A.; Chen, K.H.; Archer, S.L. Acquired disorders of mitochondrial metabolism and dynamics in pulmonary arterial hypertension. Front. Cell Dev. Biol. 2023, 11, 1105565. [Google Scholar] [CrossRef]
- Welsh, D.J.; Peacock, A.J. Cellular responses to hypoxia in the pulmonary circulation. High Alt. Med. Biol. 2013, 14, 111–116. [Google Scholar] [CrossRef]
- Sato, H.; Sato, M.; Kanai, H.; Uchiyama, T.; Iso, T.; Ohyama, Y.; Sakamoto, H.; Tamura, J.; Nagai, R.; Kurabayashi, M. Mitochondrial reactive oxygen species and c-Src play a critical role in hypoxic response in vascular smooth muscle cells. Cardiovasc. Res. 2005, 67, 714–722. [Google Scholar] [CrossRef]
- Han, X.J.; Zhang, W.F.; Wang, Q.; Li, M.; Zhang, C.B.; Yang, Z.J.; Tan, R.J.; Gan, L.J.; Zhang, L.L.; Lan, X.M.; et al. HIF-1α promotes the proliferation and migration of pulmonary arterial smooth muscle cells via activation of Cx43. J. Cell. Mol. Med. 2021, 25, 10663–10673. [Google Scholar] [CrossRef]
- Zhang, R.; Wu, Y.; Zhao, M.; Liu, C.; Zhou, L.; Shen, S.; Liao, S.; Yang, K.; Li, Q.; Wan, H. Role of HIF-1alpha in the regulation ACE and ACE2 expression in hypoxic human pulmonary artery smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L631–L640. [Google Scholar] [CrossRef]
- Pullamsetti, S.S.; Mamazhakypov, A.; Weissmann, N.; Seeger, W.; Savai, R. Hypoxia-inducible factor signaling in pulmonary hypertension. J. Clin. Investig. 2020, 130, 5638–5651. [Google Scholar] [CrossRef]
- Guignabert, C.; Tu, L.; Girerd, B.; Ricard, N.; Huertas, A.; Montani, D.; Humbert, M. New molecular targets of pulmonary vascular remodeling in pulmonary arterial hypertension: Importance of endothelial communication. Chest 2015, 147, 529–537. [Google Scholar] [CrossRef]
- Papaioannou, A.I.; Zakynthinos, E.; Kostikas, K.; Kiropoulos, T.; Koutsokera, A.; Ziogas, A.; Koutroumpas, A.; Sakkas, L.; Gourgoulianis, K.I.; Daniil, Z.D. Serum VEGF levels are related to the presence of pulmonary arterial hypertension in systemic sclerosis. BMC Pulm. Med. 2009, 9, 18. [Google Scholar] [CrossRef]
- Wang, L.; Wang, Y.; Lei, Z. Chrysin ameliorates ANTU-induced pulmonary edema and pulmonary arterial hypertension via modulation of VEGF and eNOs. J. Biochem. Mol. Toxicol. 2019, 33, e22332. [Google Scholar] [CrossRef]
- Pokall, S.; Maldonado, A.R.; Klanke, C.A.; Katayama, S.; Morris, L.M.; Vuletin, J.F.; Lim, F.Y.; Crombleholme, T.M. Compensatory lung growth in NOS3 knockout mice suggests synthase isoform redundancy. Eur. J. Pediatr. Surg. 2012, 22, 148–156. [Google Scholar] [CrossRef]
- Vyas-Read, S.; Shaul, P.W.; Yuhanna, I.S.; Willis, B.C. Nitric oxide attenuates epithelial-mesenchymal transition in alveolar epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L212–L221. [Google Scholar] [CrossRef]
- Varshney, R.; Ali, Q.; Wu, C.; Sun, Z. Monocrotaline-Induced Pulmonary Hypertension Involves Downregulation of Antiaging Protein Klotho and eNOS Activity. Hypertension 2016, 68, 1255–1263. [Google Scholar] [CrossRef]
- Jian, J.; Xia, L. miR-1226-3p Promotes eNOS Expression of Pulmonary Arterial Endothelial Cells to Mitigate Hypertension in Rats via Targeting Profilin-1. BioMed Res. Int. 2021, 2021, 1724722. [Google Scholar] [CrossRef]
- Somani, A.; Nair, S.L.; Milbauer, L.C.; Zhu, G.; Sajja, S.; Solovey, A.; Chen, Y.; Hebbel, R.P. Blood outgrowth endothelial cells overexpressing eNOS mitigate pulmonary hypertension in rats: A unique carrier cell enabling autologous cell-based gene therapy. Transl. Res. J. Lab. Clin. Med. 2019, 210, 1–7. [Google Scholar] [CrossRef]
- Thomas, S.M.; Brugge, J.S. Cellular functions regulated by Src family kinases. Annu. Rev. Cell Dev. Biol. 1997, 13, 513–609. [Google Scholar] [CrossRef]
- Miller, B.; Sewell-Loftin, M.K. Mechanoregulation of Vascular Endothelial Growth Factor Receptor 2 in Angiogenesis. Front. Cardiovasc. Med. 2021, 8, 804934. [Google Scholar] [CrossRef]
- Paulin, R.; Meloche, J.; Jacob, M.H.; Bisserier, M.; Courboulin, A.; Bonnet, S. Dehydroepiandrosterone inhibits the Src/STAT3 constitutive activation in pulmonary arterial hypertension. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1798–H1809. [Google Scholar] [CrossRef]
- Wang, J.L.; Li, L.; Hu, M.B.; Wu, B.; Fan, W.X.; Peng, W.; Wei, D.N.; Wu, C.J. In silico drug design of inhibitor of nuclear factor kappa B kinase subunit beta inhibitors from 2-acylamino-3-aminothienopyridines based on quantitative structure-activity relationships and molecular docking. Comput. Biol. Chem. 2019, 78, 297–305. [Google Scholar] [CrossRef]
- Sripathi, S.R.; Hu, M.W.; Turaga, R.C.; Mikeasky, R.; Satyanarayana, G.; Cheng, J.; Duan, Y.; Maruotti, J.; Wahlin, K.J.; Berlinicke, C.A.; et al. IKKβ Inhibition Attenuates Epithelial Mesenchymal Transition of Human Stem Cell-Derived Retinal Pigment Epithelium. Cells 2023, 12, 1155. [Google Scholar] [CrossRef]
- Zhou, P.; Hua, F.; Wang, X.; Huang, J.L. Therapeutic potential of IKK-β inhibitors from natural phenolics for inflammation in cardiovascular diseases. Inflammopharmacology 2020, 28, 19–37. [Google Scholar] [CrossRef]
- Price, L.C.; Caramori, G.; Perros, F.; Meng, C.; Gambaryan, N.; Dorfmuller, P.; Montani, D.; Casolari, P.; Zhu, J.; Dimopoulos, K.; et al. Nuclear factor κ-B is activated in the pulmonary vessels of patients with end-stage idiopathic pulmonary arterial hypertension. PLoS ONE 2013, 8, e75415. [Google Scholar] [CrossRef]
- Mumby, S.; Gambaryan, N.; Meng, C.; Perros, F.; Humbert, M.; Wort, S.J.; Adcock, I.M. Bromodomain and extra-terminal protein mimic JQ1 decreases inflammation in human vascular endothelial cells: Implications for pulmonary arterial hypertension. Respirology 2017, 22, 157–164. [Google Scholar] [CrossRef]
- Price, L.C.; Shao, D.; Meng, C.; Perros, F.; Garfield, B.E.; Zhu, J.; Montani, D.; Dorfmuller, P.; Humbert, M.; Adcock, I.M.; et al. Dexamethasone induces apoptosis in pulmonary arterial smooth muscle cells. Respir. Res. 2015, 16, 114. [Google Scholar] [CrossRef]
- Vaillancourt, M.; Chia, P.; Sarji, S.; Nguyen, J.; Hoftman, N.; Ruffenach, G.; Eghbali, M.; Mahajan, A.; Umar, S. Autonomic nervous system involvement in pulmonary arterial hypertension. Respir. Res. 2017, 18, 201. [Google Scholar] [CrossRef]
- Arvidsson, M.; Ahmed, A.; Bouzina, H.; Rådegran, G. Matrix metalloproteinase 7 in diagnosis and differentiation of pulmonary arterial hypertension. Pulm. Circ. 2019, 9, 2045894019895414. [Google Scholar] [CrossRef]
- Schäfer, M.; Ivy, D.D.; Nguyen, K.; Boncella, K.; Frank, B.S.; Morgan, G.J.; Miller-Reed, K.; Truong, U.; Colvin, K.; Yeager, M.E. Metalloproteinases and their inhibitors are associated with pulmonary arterial stiffness and ventricular function in pediatric pulmonary hypertension. Am. J. Physiol. Heart Circ. Physiol. 2021, 321, H242–H252. [Google Scholar] [CrossRef]
- George, J.; D’Armiento, J. Transgenic expression of human matrix metalloproteinase-9 augments monocrotaline-induced pulmonary arterial hypertension in mice. J. Hypertens. 2011, 29, 299–308. [Google Scholar] [CrossRef]
- Qin, Y.; Zhou, A.; Ben, X.; Shen, J.; Liang, Y.; Li, F. All-trans retinoic acid in pulmonary vascular structural remodeling in rats with pulmonary hypertension induced by monocrotaline. Chin. Med. J. 2001, 114, 462–465. [Google Scholar]
- Benisty, J.I.; Folkman, J.; Zurakowski, D.; Louis, G.; Rich, S.; Langleben, D.; Moses, M.A. Matrix metalloproteinases in the urine of patients with pulmonary arterial hypertension. Chest 2005, 128, 572s. [Google Scholar] [CrossRef]
- Bai, P.; Lyu, L.; Yu, T.; Zuo, C.; Fu, J.; He, Y.; Wan, Q.; Wan, N.; Jia, D.; Lyu, A. Macrophage-Derived Legumain Promotes Pulmonary Hypertension by Activating the MMP (Matrix Metalloproteinase)-2/TGF (Transforming Growth Factor)-β1 Signaling. Arterioscler. Thromb. Vasc. Biol. 2019, 39, e130–e145. [Google Scholar] [CrossRef]
- Kumar, R.; Mickael, C.; Kassa, B.; Gebreab, L.; Robinson, J.C.; Koyanagi, D.E.; Sanders, L.; Barthel, L.; Meadows, C.; Fox, D.; et al. TGF-β activation by bone marrow-derived thrombospondin-1 causes Schistosoma- and hypoxia-induced pulmonary hypertension. Nat. Commun. 2017, 8, 15494. [Google Scholar] [CrossRef]
- Eskandari, E.; Eaves, C.J. Paradoxical roles of caspase-3 in regulating cell survival, proliferation, and tumorigenesis. J. Cell Biol. 2022, 221, e202201159. [Google Scholar] [CrossRef]
- Udjus, C.; Cero, F.T.; Halvorsen, B.; Behmen, D.; Carlson, C.R.; Bendiksen, B.A.; Espe, E.K.S.; Sjaastad, I.; Løberg, E.M.; Yndestad, A.; et al. Caspase-1 induces smooth muscle cell growth in hypoxia-induced pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 316, L999–L1012. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Dixit, V.M. Death receptors: Signaling and modulation. Scicence 1998, 281, 1305–1308. [Google Scholar] [CrossRef]
- Salvesen, G.S. Caspase 8: Igniting the death machine. Structure 1999, 7, R225–R229. [Google Scholar] [CrossRef]
- Rong, W.; Liu, C.; Li, X.; Wan, N.; Wei, L.; Zhu, W.; Bai, P.; Li, M.; Ou, Y.; Li, F.; et al. Caspase-8 Promotes Pulmonary Hypertension by Activating Macrophage-Associated Inflammation and IL-1β (Interleukin 1β) Production. Arterioscler. Thromb. Vasc. Biol. 2022, 42, 613–631. [Google Scholar] [CrossRef]
- Lagna, G.; Nguyen, P.H.; Ni, W.; Hata, A. BMP-dependent activation of caspase-9 and caspase-8 mediates apoptosis in pulmonary artery smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L1059–L1067. [Google Scholar] [CrossRef]
- Cai, C.; Xiang, Y.; Wu, Y.; Zhu, N.; Zhao, H.; Xu, J.; Lin, W.; Zeng, C. Formononetin attenuates monocrotaline-induced pulmonary arterial hypertension via inhibiting pulmonary vascular remodeling in rats. Mol. Med. Rep. 2019, 20, 4984–4992. [Google Scholar] [CrossRef]
- Jankov, R.P.; Kantores, C.; Belcastro, R.; Yi, M.; Tanswell, A.K. Endothelin-1 inhibits apoptosis of pulmonary arterial smooth muscle in the neonatal rat. Pediatr. Res. 2006, 60, 245–251. [Google Scholar] [CrossRef]
- White, K.; Dempsie, Y.; Caruso, P.; Wallace, E.; McDonald, R.A.; Stevens, H.; Hatley, M.E.; Van Rooij, E.; Morrell, N.W.; MacLean, M.R.; et al. Endothelial apoptosis in pulmonary hypertension is controlled by a microRNA/programmed cell death 4/caspase-3 axis. Hypertension 2014, 64, 185–194. [Google Scholar] [CrossRef]
- Bhosale, P.B.; Kim, H.H.; Abusaliya, A.; Vetrivel, P.; Ha, S.E.; Park, M.Y.; Lee, H.J.; Kim, G.S. Structural and Functional Properties of Activator Protein-1 in Cancer and Inflammation. Evid.-Based Complement. Altern. Med. Ecam. 2022, 2022, 9797929. [Google Scholar] [CrossRef]
- Kappelmann, M.; Bosserhoff, A.; Kuphal, S. AP-1/c-Jun transcription factors: Regulation and function in malignant melanoma. Eur. J. Cell Biol. 2014, 93, 76–81. [Google Scholar] [CrossRef]
- Bennett, B.L. c-Jun N-terminal kinase-dependent mechanisms in respiratory disease. Eur. Respir. J. 2006, 28, 651–661. [Google Scholar] [CrossRef]
- Ortiz, L.A.; Champion, H.C.; Lasky, J.A.; Gambelli, F.; Gozal, E.; Hoyle, G.W.; Beasley, M.B.; Hyman, A.L.; Friedman, M.; Kadowitz, P.J. Enalapril protects mice from pulmonary hypertension by inhibiting TNF-mediated activation of NF-kappaB and AP-1. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 282, L1209–L1221. [Google Scholar] [CrossRef]
- Zabini, D.; Crnkovic, S.; Xu, H.; Tscherner, M.; Ghanim, B.; Klepetko, W.; Olschewski, A.; Kwapiszewska, G.; Marsh, L.M. High-mobility group box-1 induces vascular remodelling processes via c-Jun activation. J. Cell. Mol. Med. 2015, 19, 1151–1161. [Google Scholar] [CrossRef]
- Hassin, O.; Oren, M. Drugging p53 in cancer: One protein, many targets. Nat. Rev. Drug Discov. 2023, 22, 127–144. [Google Scholar] [CrossRef]
- Lacroix, M.; Riscal, R.; Arena, G.; Linares, L.K.; Le Cam, L. Metabolic functions of the tumor suppressor p53: Implications in normal physiology, metabolic disorders, and cancer. Mol. Metab. 2020, 33, 2–22. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, S.; Bogaard, H.J.; Kraskauskas, D.; Alhussaini, A.; Gomez-Arroyo, J.; Voelkel, N.F.; Ishizaki, T. p53 Gene deficiency promotes hypoxia-induced pulmonary hypertension and vascular remodeling in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 300, L753–L761. [Google Scholar] [CrossRef] [PubMed]
- Mouraret, N.; Marcos, E.; Abid, S.; Gary-Bobo, G.; Saker, M.; Houssaini, A.; Dubois-Rande, J.L.; Boyer, L.; Boczkowski, J.; Derumeaux, G.; et al. Activation of lung p53 by Nutlin-3a prevents and reverses experimental pulmonary hypertension. Circulation 2013, 127, 1664–1676. [Google Scholar] [CrossRef] [PubMed]
- Jacquin, S.; Rincheval, V.; Mignotte, B.; Richard, S.; Humbert, M.; Mercier, O.; Londoño-Vallejo, A.; Fadel, E.; Eddahibi, S. Inactivation of p53 Is Sufficient to Induce Development of Pulmonary Hypertension in Rats. PLoS ONE 2015, 10, e0131940. [Google Scholar] [CrossRef]
- Khalil Alyahya, H.; Subash-Babu, P.; Mohammad Salamatullah, A.; Hayat, K.; Albader, N.; Alkaltham, M.S.; Ahmed, M.A.; Arzoo, S.; Bourhia, M. Quantification of Chlorogenic Acid and Vanillin from Coffee Peel Extract and its Effect on α-Amylase Activity, Immunoregulation, Mitochondrial Oxidative Stress, and Tumor Suppressor Gene Expression Levels in H(2)O(2)-Induced Human Mesenchymal Stem Cells. Front. Pharmacol. 2021, 12, 760242. [Google Scholar] [CrossRef]

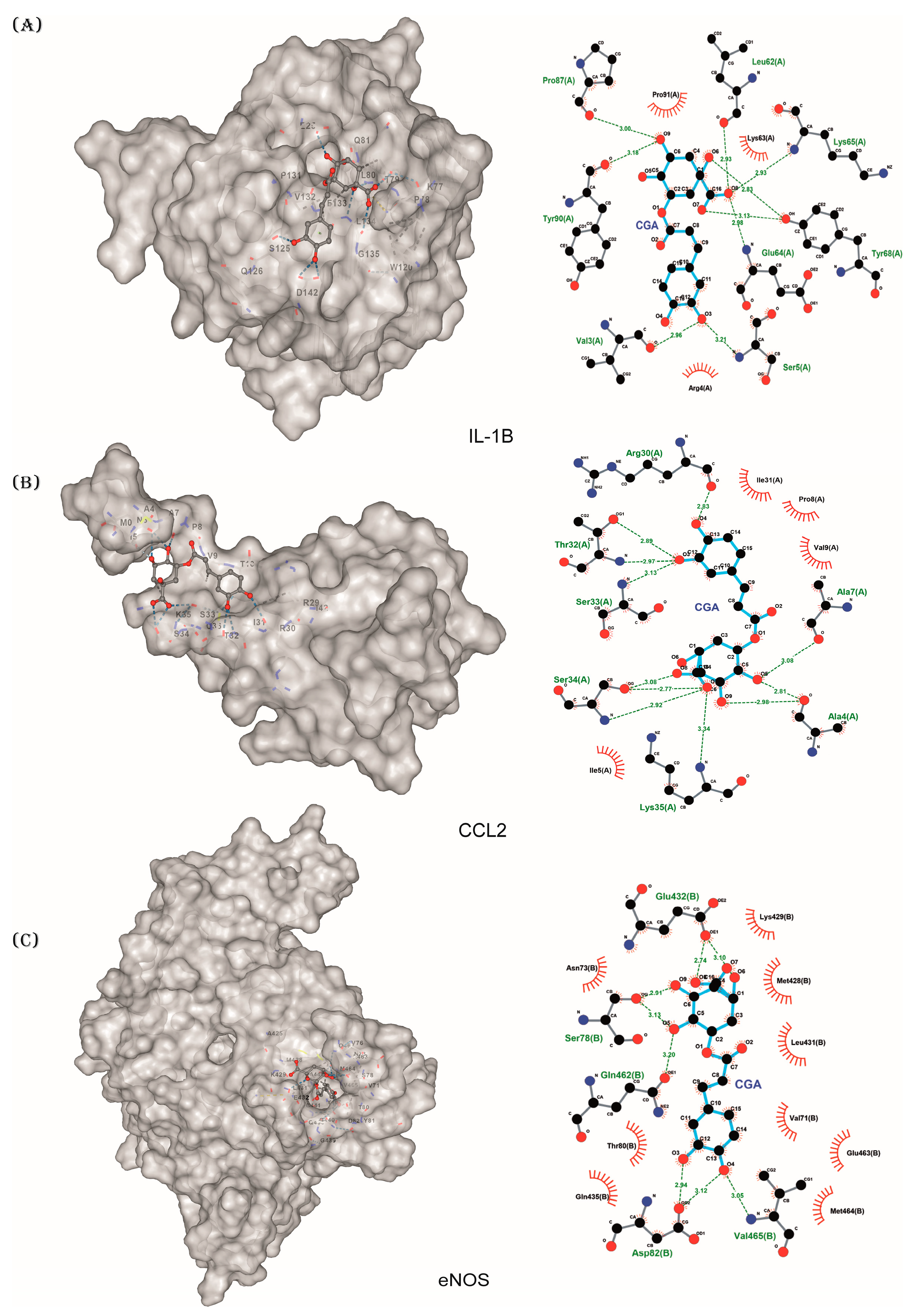
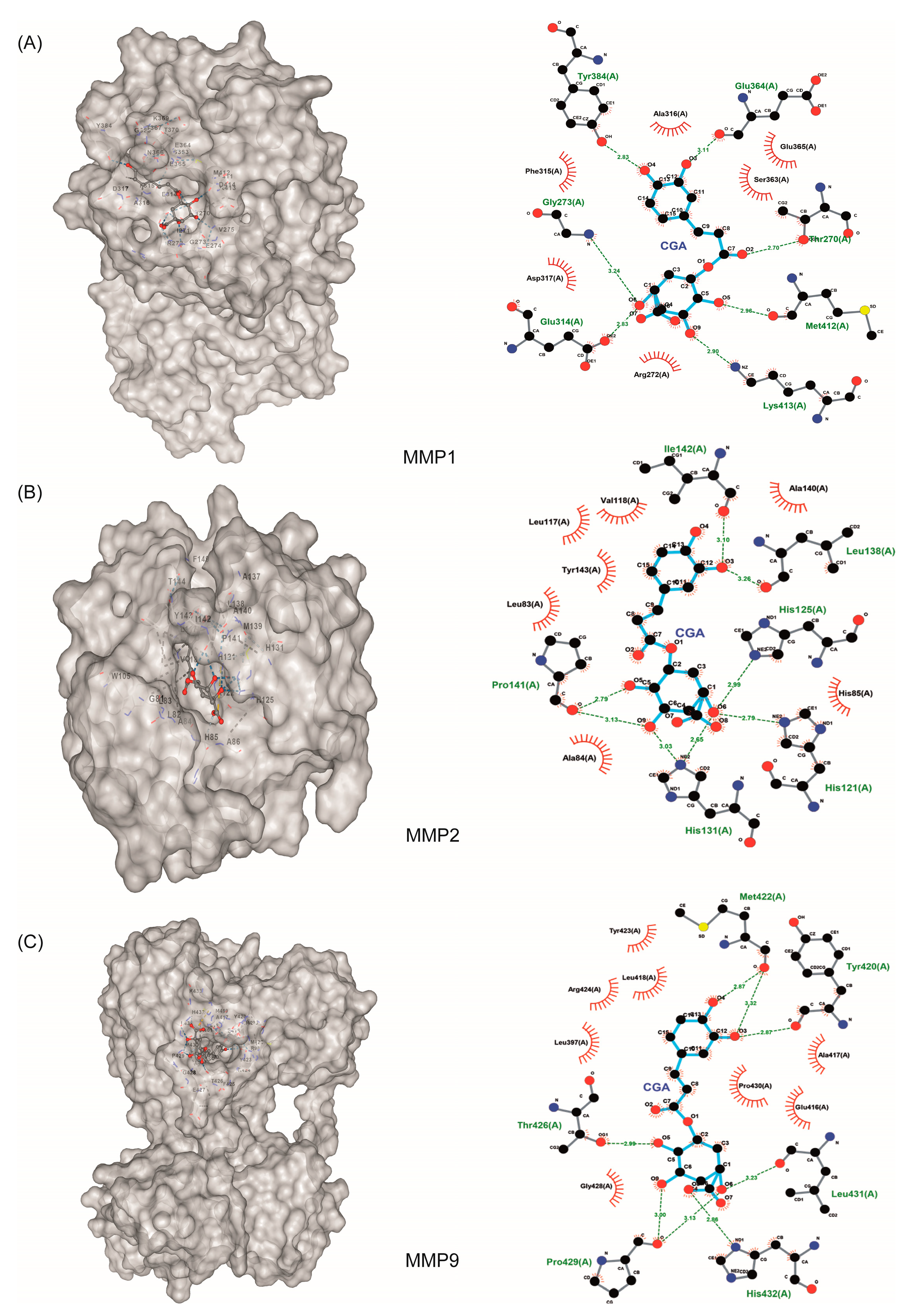

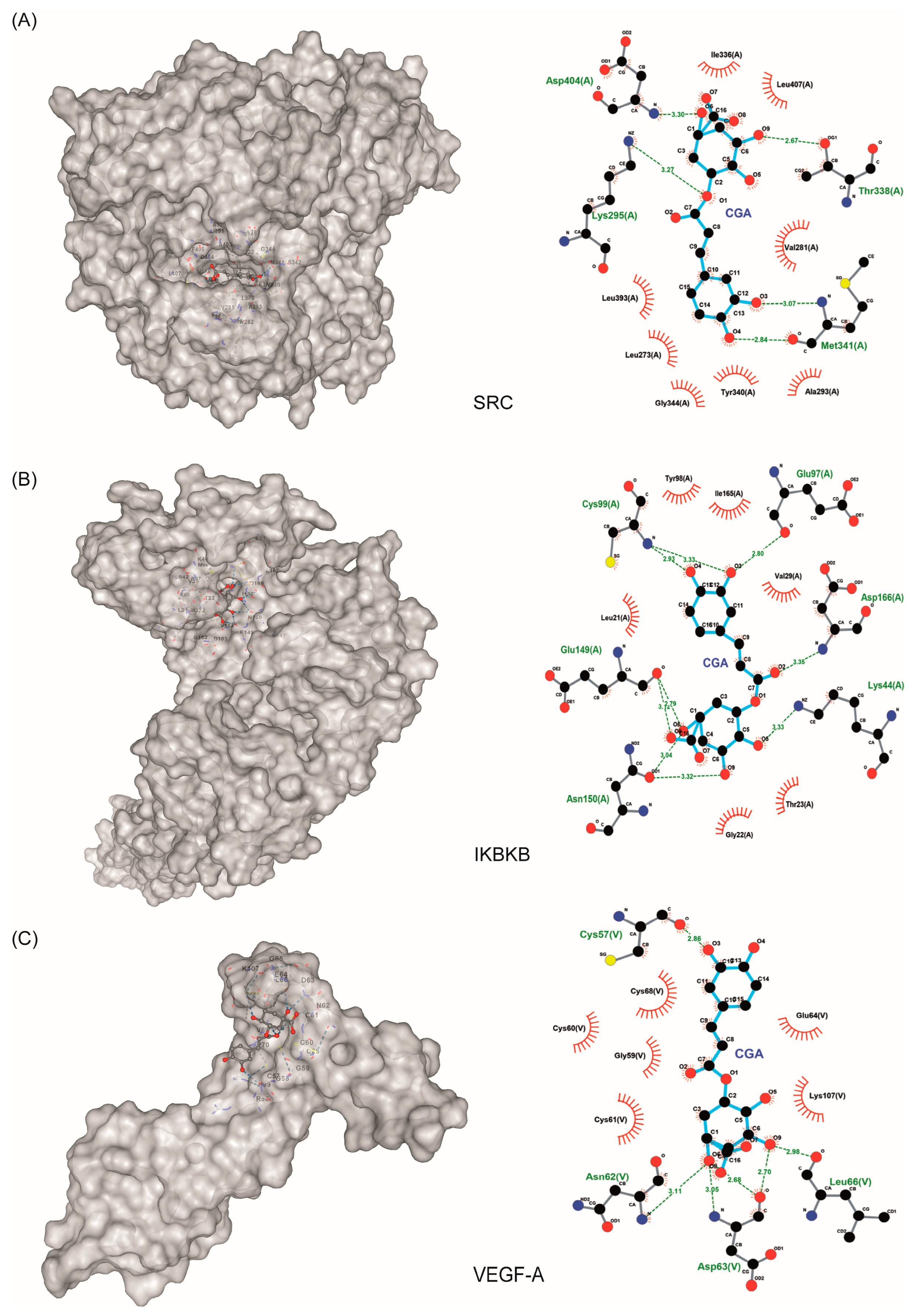


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | ID PDB | Kcal/mol | Â3 | |
|---|---|---|---|---|
| 1 | TUMOR SUPPRESSOR P53 | 1KZY | −7.1 | 115 |
| 2 | HYPOXIA INDUCIBLE FACTOR 1-ALPHA | 4H6J | −5.8 | 63 |
| 3 | CASPASE-3 | 4H6J | −7.3 | 255 |
| 4 | INTERLEUKIN-1 BETA | 2NVH | −6.6 | 182 |
| 5 | C-JUN | 1JUN | −5.6 | 12 |
| 6 | MATRIX METALLOPROTEINASE-9 | 1L6J | −7.8 | 399 |
| 7 | CHEMOKINE (C-C MOTIF) LIGAND 2 | 1DOK | −5.9 | 20 |
| 8 | VASCULAR ENDOTHELIAL GROWTH FACTOR A | 1BJ1 | −5.8 | 29 |
| 9 | PROTO-ONCOGENE TYROSINE-PROTEIN KINASE SRC | 2H8H | −8.0 | 662 |
| 10 | INHIBITOR OF NUCLEAR FACTOR KAPPA-B KINASE SUBUNIT BETA | 4KIK | −8.6 | 564 |
| 11 | MATRIX METALLOPROTEINASE-2 | 7XGJ | −9.4 | 789 |
| 12 | CASPASE-8 | 3KJN | −7.3 | 154 |
| 13 | NITRIC OXIDE SYNTHASE, ENDOTHELIAL | 4D1O | −8.8 | 329 |
| 14 | MATRIX METALLOPROTEINASE-1 | 2CLT | −7.3 | 372 |
| 15 | CASPASE-1 | 2H54 | −7.2 | 152 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santos-Álvarez, J.C.; Velázquez-Enríquez, J.M.; Baltiérrez-Hoyos, R. Evaluation of the Molecular Mechanism of Chlorogenic Acid in the Treatment of Pulmonary Arterial Hypertension Based on Analysis Network Pharmacology and Molecular Docking. J. Vasc. Dis. 2024, 3, 11-33. https://doi.org/10.3390/jvd3010002
Santos-Álvarez JC, Velázquez-Enríquez JM, Baltiérrez-Hoyos R. Evaluation of the Molecular Mechanism of Chlorogenic Acid in the Treatment of Pulmonary Arterial Hypertension Based on Analysis Network Pharmacology and Molecular Docking. Journal of Vascular Diseases. 2024; 3(1):11-33. https://doi.org/10.3390/jvd3010002
Chicago/Turabian StyleSantos-Álvarez, Jovito Cesar, Juan Manuel Velázquez-Enríquez, and Rafael Baltiérrez-Hoyos. 2024. "Evaluation of the Molecular Mechanism of Chlorogenic Acid in the Treatment of Pulmonary Arterial Hypertension Based on Analysis Network Pharmacology and Molecular Docking" Journal of Vascular Diseases 3, no. 1: 11-33. https://doi.org/10.3390/jvd3010002
APA StyleSantos-Álvarez, J. C., Velázquez-Enríquez, J. M., & Baltiérrez-Hoyos, R. (2024). Evaluation of the Molecular Mechanism of Chlorogenic Acid in the Treatment of Pulmonary Arterial Hypertension Based on Analysis Network Pharmacology and Molecular Docking. Journal of Vascular Diseases, 3(1), 11-33. https://doi.org/10.3390/jvd3010002