
Non-Animal Technologies to Study and Target the Tumour Vasculature and Angiogenesis


Abstract
1. Introduction
1.1. Tumour-Associated Microvasculature
1.2. Therapeutic Implications
2. Non-Animal Technologies
2.1. Static Cultures
2.2. Ex Vivo Culture of Tumour Explants
2.3. Dynamic Cultures
3. Modelling Tumour Vasculature and Angiogenesis
3.1. EC Cultures and PSCs
3.2. In Vitro Tools to Study Vascular Permeability and Trans-Endothelial Cell Migration
3.3. In Vitro Tools to Study Angiogenesis
3.4. Vascularised Organoids and 3D Dynamic Cultures
3.5. Microphysiological Systems to Perfuse Engineered Microvasculature
3.6. Spontaneous Tumour Models in Companion Animals
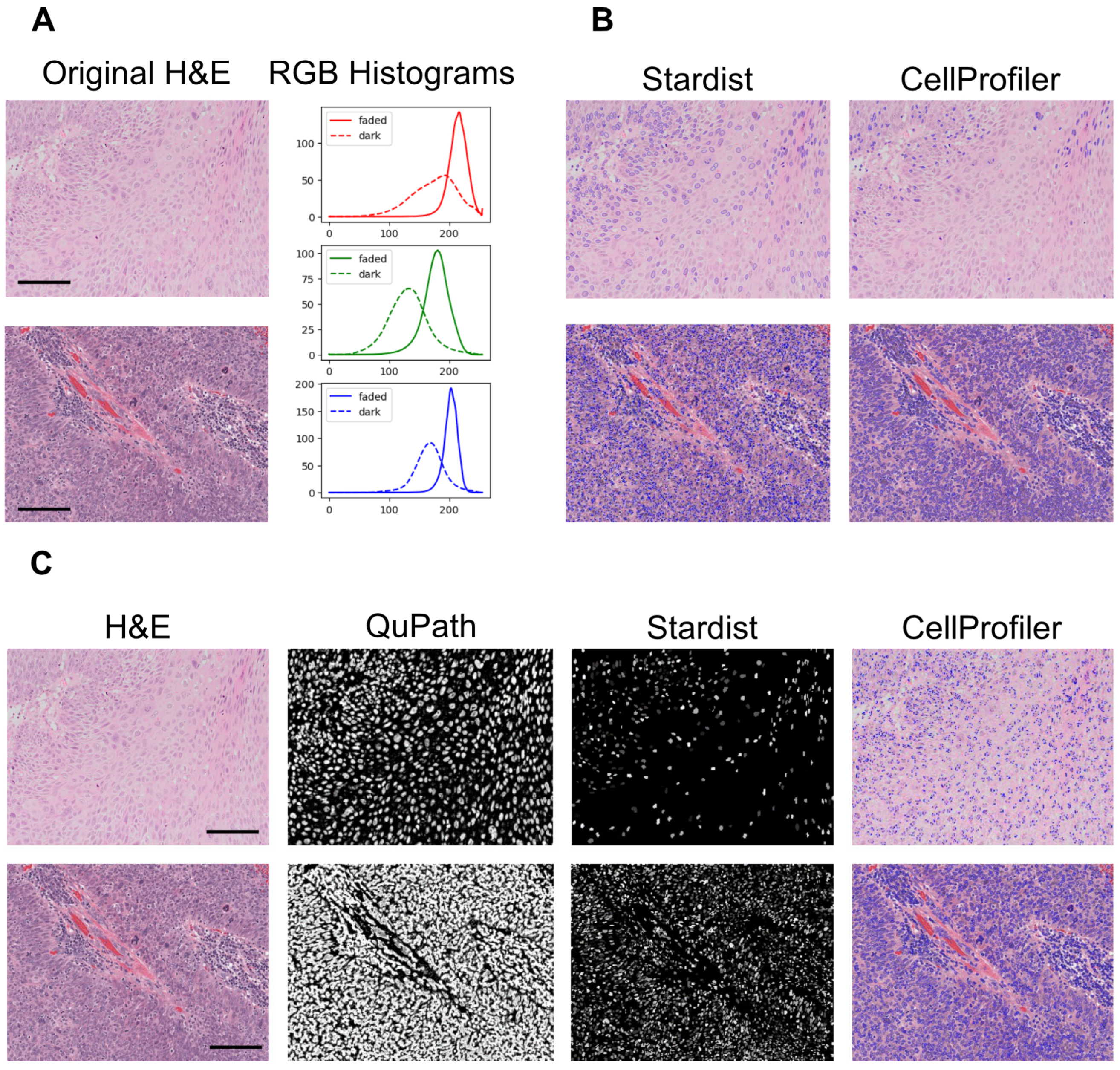
3.7. Computational Pathology and Artificial Intelligence
3.8. Mechanistic Modelling
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Cell Types | Others | ||
| Mφ | Macrophages | MVD | Microvessel density |
| ASC | Adult stem cells | OMICS | Omics technologies (genomics, proteomics, etc.) |
| CAF | Cancer-associated fibroblasts | OVAA | Organotypic vasculogenesis/angiogenesis assay |
| PCS | Pericytes | RCCS | Rotary Cell Culture System |
| EC | Endothelial cell | RGB | Red, Green, Blue |
| CSC | Cancer stem cells | SCAC | Spontaneous companion animal cancers |
| PSC-EC | Pluripotent stem cell-derived endothelial cell | TAA | Tumour-associated angiogenesis |
| PSC | Pluripotent stem cells | TAMV | Tumour-associated microvasculature |
| TAM | Tumour-associated Mφ | ||
| Drugs | TME | Tumour microenvironment | |
| SF | Sorafenib | MPS | Microphysiological system |
| MCS | Monte Carlo Steps | ||
| Proteins | HPV | Human papillomavirus | |
| PGF | Placental growth factor | AAT | Anti-angiogenic therapy |
| VEGF-A | Vascular endothelial growth factor A | AI | Artificial intelligence |
| EGFR | Epidermal growth factor receptor | CC3D | CompuCell3D (simulation software) |
| HIF | Hypoxia-inducible factor | CNN | Convolutional Neural Network |
| VEGF | Vascular endothelial growth factor | VNT | Vascular normalization therapy |
| COX-2 | Cyclooxygenase-2 | ECM | Extracellular matrix |
| RFP | Red fluorescent protein | FDA | Food and Drug Administration |
| H&E | Hematoxylin and eosin | ||
| Cell markers | |||
| CD34+ | Cluster of Differentiation 34 positive | ||
| CD31 | Platelet endothelial cell adhesion molecule | ||
| CD14+ | Cluster of Differentiation 14 positive |
References
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed]
- de Visser, K.E.; Joyce, J.A. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell 2023, 41, 374–403. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Marchesi, F.; Garlanda, C. Macrophages as tools and targets in cancer therapy. Nat. Rev. Drug Discov. 2022, 21, 799–820. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; McAndrews, K.M.; Kalluri, R. Clinical and therapeutic relevance of cancer-associated fibroblasts. Nat. Rev. Clin. Oncol. 2021, 18, 792–804. [Google Scholar] [CrossRef]
- Arner, E.N.; Rathmell, J.C. Metabolic programming and immune suppression in the tumor microenvironment. Cancer Cell 2023, 41, 421–433. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- van Seijen, M.; Lips, E.H.; Thompson, A.M.; Nik-Zainal, S.; Futreal, A.; Hwang, E.S.; Verschuur, E.; Lane, J.; Jonkers, J.; Rea, D.W.; et al. Ductal carcinoma in situ: To treat or not to treat, that is the question. Br. J. Cancer 2019, 121, 285–292. [Google Scholar] [CrossRef]
- Knoblauch, M.; Kühn, F.; von Ehrlich-Treuenstätt, V.; Werner, J.; Renz, B.W. Diagnostic and Therapeutic Management of Early Colorectal Cancer. Visc. Med. 2023, 39, 10–16. [Google Scholar] [CrossRef]
- Folkman, J.; Merler, E.; Abernathy, C.; Williams, G. Isolation of a tumor factor responsible for angiogenesis. J. Exp. Med. 1971, 133, 275–288. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [CrossRef] [PubMed]
- Leroi, N.; Lallemand, F.; Coucke, P.; Noel, A.; Martinive, P. Impacts of Ionizing Radiation on the Different Compartments of the Tumor Microenvironment. Front. Pharmacol. 2016, 7, 78. [Google Scholar] [CrossRef] [PubMed]
- Minchinton, A.I.; Tannock, I.F. Drug penetration in solid tumours. Nat. Rev. Cancer 2006, 6, 583–592. [Google Scholar] [CrossRef]
- Deyell, M.; Garris, C.S.; Laughney, A.M. Cancer metastasis as a non-healing wound. Br. J. Cancer 2021, 124, 1491–1502. [Google Scholar] [CrossRef]
- Whiteside, T.L. Tumor-Derived Exosomes and Their Role in Cancer Progression. Adv. Clin. Chem. 2016, 74, 103–141. [Google Scholar] [CrossRef]
- Patras, L.; Shaashua, L.; Matei, I.; Lyden, D. Immune determinants of the pre-metastatic niche. Cancer Cell 2023, 41, 546–572. [Google Scholar] [CrossRef]
- Fidler, I.J.; Nicolson, G.L. Organ selectivity for implantation survival and growth of B16 melanoma variant tumor lines. J. Natl. Cancer Inst. 1976, 57, 1199–1202. [Google Scholar] [CrossRef]
- Wicks, E.E.; Semenza, G.L. Hypoxia-inducible factors: Cancer progression and clinical translation. J. Clin. Investig. 2022, 132, e159839. [Google Scholar] [CrossRef]
- Mole, D.R.; Ratcliffe, P.J. Cellular oxygen sensing in health and disease. Pediatr. Nephrol. 2008, 23, 681–694. [Google Scholar] [CrossRef]
- Kalucka, J.; de Rooij, L.P.M.H.; Goveia, J.; Rohlenova, K.; Dumas, S.J.; Meta, E.; Conchinha, N.V.; Taverna, F.; Teuwen, L.-A.; Veys, K.; et al. Single-Cell Transcriptome Atlas of Murine Endothelial Cells. Cell 2020, 180, 764–779.e20. [Google Scholar] [CrossRef] [PubMed]
- Eelen, G.; Treps, L.; Li, X.; Carmeliet, P. Basic and Therapeutic Aspects of Angiogenesis Updated. Circ. Res. 2020, 127, 310–329. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Sun, X.; Carmeliet, P. Hallmarks of Endothelial Cell Metabolism in Health and Disease. Cell Metab. 2019, 30, 414–433. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.M.; Kobayashi, H.; Rafii, S. Instructive role of the vascular niche in promoting tumour growth and tissue repair by angiocrine factors. Nat. Rev. Cancer 2010, 10, 138–146. [Google Scholar] [CrossRef]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.-X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef]
- Whiteside, T.L.; Demaria, S.; Rodriguez-Ruiz, M.E.; Zarour, H.M.; Melero, I. Emerging Opportunities and Challenges in Cancer Immunotherapy. Clin. Cancer Res. 2016, 22, 1845–1855. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Jaillon, S.; Garlanda, C.; Allavena, P. Tumor-associated myeloid cells: Diversity and therapeutic targeting. Cell. Mol. Immunol. 2021, 18, 566–578. [Google Scholar] [CrossRef]
- Kang, Y.; Pantel, K. Tumor cell dissemination: Emerging biological insights from animal models and cancer patients. Cancer Cell 2013, 23, 573–581. [Google Scholar] [CrossRef]
- Coste, A.; Karagiannis, G.S.; Wang, Y.; Xue, E.A.; Lin, Y.; Skobe, M.; Jones, J.G.; Oktay, M.H.; Condeelis, J.S.; Entenberg, D. Hematogenous Dissemination of Breast Cancer Cells From Lymph Nodes Is Mediated by Tumor MicroEnvironment of Metastasis Doorways. Front. Oncol. 2020, 10, 571100. [Google Scholar] [CrossRef]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef]
- Massagué, J.; Ganesh, K. Metastasis-Initiating Cells and Ecosystems. Cancer Discov. 2021, 11, 971–994. [Google Scholar] [CrossRef] [PubMed]
- Potente, M.; Gerhardt, H.; Carmeliet, P. Basic and therapeutic aspects of angiogenesis. Cell 2011, 146, 873–887. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, K.; Massagué, J. Targeting metastatic cancer. Nat. Med. 2021, 27, 34–44. [Google Scholar] [CrossRef]
- Fan, P.; Zhang, N.; Candi, E.; Agostini, M.; Piacentini, M.; TOR Centre; Shi, Y.; Huang, Y.; Melino, G. Alleviating hypoxia to improve cancer immunotherapy. Oncogene 2023, 42, 3591–3604. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Antiangiogenesis strategies revisited: From starving tumors to alleviating hypoxia. Cancer Cell 2014, 26, 605–622. [Google Scholar] [CrossRef]
- Huang, Y.; Kim, B.Y.S.; Chan, C.K.; Hahn, S.M.; Weissman, I.L.; Jiang, W. Improving immune-vascular crosstalk for cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 195–203. [Google Scholar] [CrossRef]
- Zitvogel, L.; Pitt, J.M.; Daillère, R.; Smyth, M.J.; Kroemer, G. Mouse models in oncoimmunology. Nat. Rev. Cancer 2016, 16, 759–773. [Google Scholar] [CrossRef]
- Bédard, P.; Gauvin, S.; Ferland, K.; Caneparo, C.; Pellerin, È.; Chabaud, S.; Bolduc, S. Innovative Human Three-Dimensional Tissue-Engineered Models as an Alternative to Animal Testing. Bioengineering 2020, 7, 115. [Google Scholar] [CrossRef]
- Moutinho, S. Researchers and regulators plan for a future without lab animals. Nat. Med. 2023, 29, 2151–2154. [Google Scholar] [CrossRef]
- Pampaloni, F.; Reynaud, E.G.; Stelzer, E.H.K. The third dimension bridges the gap between cell culture and live tissue. Nat. Rev. Mol. Cell Biol. 2007, 8, 839–845. [Google Scholar] [CrossRef]
- Kapałczyńska, M.; Kolenda, T.; Przybyła, W.; Zajączkowska, M.; Teresiak, A.; Filas, V.; Ibbs, M.; Bliźniak, R.; Łuczewski, Ł.; Lamperska, K. 2D and 3D cell cultures—A comparison of different types of cancer cell cultures. Arch. Med. Sci. 2018, 14, 910–919. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, J.; Castriconi, R.; Scaglione, S. Editorial: Recent 3D Tumor Models for Testing Immune-Mediated Therapies. Front. Immunol. 2021, 12, 798493. [Google Scholar] [CrossRef] [PubMed]
- Allemang, A.; Lester, C.; Roth, T.; Pfuhler, S.; Peuschel, H.; Kosemund, K.; Mahony, C.; Bergeland, T.; O’Keeffe, L. Assessing the genotoxicity and carcinogenicity of 2-chloroethanol through structure activity relationships and in vitro testing approaches. Food Chem. Toxicol. 2022, 168, 113290. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.Y.; Kenny, P.A.; Lee, E.H.; Bissell, M.J. Three-dimensional culture models of normal and malignant breast epithelial cells. Nat. Methods 2007, 4, 359–365. [Google Scholar] [CrossRef]
- Roskelley, C.D.; Desprez, P.Y.; Bissell, M.J. Extracellular matrix-dependent tissue-specific gene expression in mammary epithelial cells requires both physical and biochemical signal transduction. Proc. Natl. Acad. Sci. USA 1994, 91, 12378–12382. [Google Scholar] [CrossRef]
- Chang, T.T.; Hughes-Fulford, M. Monolayer and spheroid culture of human liver hepatocellular carcinoma cell line cells demonstrate distinct global gene expression patterns and functional phenotypes. Tissue Eng. Part A 2009, 15, 559–567. [Google Scholar] [CrossRef]
- Riedl, A.; Schlederer, M.; Pudelko, K.; Stadler, M.; Walter, S.; Unterleuthner, D.; Unger, C.; Kramer, N.; Hengstschläger, M.; Kenner, L.; et al. Comparison of cancer cells in 2D vs 3D culture reveals differences in AKT-mTOR-S6K signaling and drug responses. J. Cell Sci. 2017, 130, 203–218. [Google Scholar] [CrossRef]
- van Renterghem, A.W.J.; van de Haar, J.; Voest, E.E. Functional precision oncology using patient-derived assays: Bridging genotype and phenotype. Nat. Rev. Clin. Oncol. 2023, 20, 305–317. [Google Scholar] [CrossRef]
- Weiswald, L.-B.; Bellet, D.; Dangles-Marie, V. Spherical cancer models in tumor biology. Neoplasia 2015, 17, 1–15. [Google Scholar] [CrossRef]
- Bray, L.J.; Hutmacher, D.W.; Bock, N. Addressing Patient Specificity in the Engineering of Tumor Models. Front. Bioeng. Biotechnol. 2019, 7, 217. [Google Scholar] [CrossRef]
- Gunti, S.; Hoke, A.T.K.; Vu, K.P.; London, N.R. Organoid and Spheroid Tumor Models: Techniques and Applications. Cancers 2021, 13, 874. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-H.; Kim, T.-H. Recent Advances in Multicellular Tumor Spheroid Generation for Drug Screening. Biosensors 2021, 11, 445. [Google Scholar] [CrossRef] [PubMed]
- Nunes, A.S.; Barros, A.S.; Costa, E.C.; Moreira, A.F.; Correia, I.J. 3D tumor spheroids as in vitro models to mimic in vivo human solid tumors resistance to therapeutic drugs. Biotechnol. Bioeng. 2019, 116, 206–226. [Google Scholar] [CrossRef] [PubMed]
- Carletti, E.; Motta, A.; Migliaresi, C. Scaffolds for tissue engineering and 3D cell culture. Methods Mol. Biol. 2011, 695, 17–39. [Google Scholar] [CrossRef]
- Drost, J.; Clevers, H. Organoids in cancer research. Nat. Rev. Cancer 2018, 18, 407–418. [Google Scholar] [CrossRef]
- Datta, P.; Dey, M.; Ataie, Z.; Unutmaz, D.; Ozbolat, I.T. 3D bioprinting for reconstituting the cancer microenvironment. npj Precis. Oncol. 2020, 4, 18. [Google Scholar] [CrossRef]
- Augustine, R.; Kalva, S.N.; Ahmad, R.; Zahid, A.A.; Hasan, S.; Nayeem, A.; McClements, L.; Hasan, A. 3D Bioprinted cancer models: Revolutionizing personalized cancer therapy. Transl. Oncol. 2021, 14, 101015. [Google Scholar] [CrossRef]
- Powley, I.R.; Patel, M.; Miles, G.; Pringle, H.; Howells, L.; Thomas, A.; Kettleborough, C.; Bryans, J.; Hammonds, T.; MacFarlane, M.; et al. Patient-derived explants (PDEs) as a powerful preclinical platform for anti-cancer drug and biomarker discovery. Br. J. Cancer 2020, 122, 735–744. [Google Scholar] [CrossRef]
- Kirby, A.J.; Lavrador, J.P.; Bodi, I.; Vergani, F.; Bhangoo, R.; Ashkan, K.; Finnerty, G.T. Multicellular “hotspots” harbor high-grade potential in lower-grade gliomas. Neuro-Oncol. Adv. 2021, 3, vdab026. [Google Scholar] [CrossRef]
- Navran, S. The application of low shear modeled microgravity to 3-D cell biology and tissue engineering. Biotechnol. Annu. Rev. 2008, 14, 275–296. [Google Scholar] [CrossRef]
- Selden, C.; Fuller, B. Role of Bioreactor Technology in Tissue Engineering for Clinical Use and Therapeutic Target Design. Bioengineering 2018, 5, 32. [Google Scholar] [CrossRef] [PubMed]
- Muraro, M.G.; Muenst, S.; Mele, V.; Quagliata, L.; Iezzi, G.; Tzankov, A.; Weber, W.P.; Spagnoli, G.C.; Soysal, S.D. Ex-vivo assessment of drug response on breast cancer primary tissue with preserved microenvironments. Oncoimmunology 2017, 6, e1331798. [Google Scholar] [CrossRef] [PubMed]
- Manfredonia, C.; Muraro, M.G.; Hirt, C.; Mele, V.; Governa, V.; Papadimitropoulos, A.; Däster, S.; Soysal, S.D.; Droeser, R.A.; Mechera, R.; et al. Maintenance of Primary Human Colorectal Cancer Microenvironment Using a Perfusion Bioreactor-Based 3D Culture System. Adv. Biosyst. 2019, 3, e1800300. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.P.; Gaspar, V.M.; Mano, J.F. Design of spherically structured 3D in vitro tumor models -Advances and prospects. Acta Biomater. 2018, 75, 11–34. [Google Scholar] [CrossRef]
- Belloni, D.; Heltai, S.; Ponzoni, M.; Villa, A.; Vergani, B.; Pecciarini, L.; Marcatti, M.; Girlanda, S.; Tonon, G.; Ciceri, F.; et al. Modeling multiple myeloma-bone marrow interactions and response to drugs in a 3D surrogate microenvironment. Haematologica 2018, 103, 707–716. [Google Scholar] [CrossRef]
- Grimm, D.; Wehland, M.; Pietsch, J.; Aleshcheva, G.; Wise, P.; van Loon, J.; Ulbrich, C.; Magnusson, N.E.; Infanger, M.; Bauer, J. Growing tissues in real and simulated microgravity: New methods for tissue engineering. Tissue Eng. Part B Rev. 2014, 20, 555–566. [Google Scholar] [CrossRef]
- Guzzeloni, V.; Veschini, L.; Pedica, F.; Ferrero, E.; Ferrarini, M. 3D Models as a Tool to Assess the Anti-Tumor Efficacy of Therapeutic Antibodies: Advantages and Limitations. Antibodies 2022, 11, 46. [Google Scholar] [CrossRef]
- Ferrarini, M.; Steimberg, N.; Boniotti, J.; Berenzi, A.; Belloni, D.; Mazzoleni, G.; Ferrero, E. 3D-Dynamic Culture Models of Multiple Myeloma. Methods Mol. Biol. 2017, 1612, 177–190. [Google Scholar] [CrossRef]
- Ferrero, E.; Villa, A.; Stefanoni, D.; Nemkov, T.; D’Alessandro, A.; Tengesdal, I.; Belloni, D.; Molteni, R.; Vergani, B.; De Luca, G.; et al. Immunometabolic activation of macrophages leads to cytokine production in the pathogenesis of KRAS-mutated histiocytosis. Rheumatology 2022, 61, e93–e96. [Google Scholar] [CrossRef]
- Holton, A.B.; Sinatra, F.L.; Kreahling, J.; Conway, A.J.; Landis, D.A.; Altiok, S. Microfluidic Biopsy Trapping Device for the Real-Time Monitoring of Tumor Microenvironment. PLoS ONE 2017, 12, e0169797. [Google Scholar] [CrossRef]
- Marei, I.; Abu Samaan, T.; Al-Quradaghi, M.A.; Farah, A.A.; Mahmud, S.H.; Ding, H.; Triggle, C.R. 3D Tissue-Engineered Vascular Drug Screening Platforms: Promise and Considerations. Front. Cardiovasc. Med. 2022, 9, 847554. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Alitalo, K.; Annex, B.H.; Augustin, H.G.; Beam, C.; Berk, B.C.; Byzova, T.; Carmeliet, P.; Chilian, W.; Cooke, J.P.; et al. State-of-the-Art Methods for Evaluation of Angiogenesis and Tissue Vascularization: A Scientific Statement from the American Heart Association. Circ. Res. 2015, 116, 99–132. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.E.; Jung, C.; Yoon, Y.-S. Human Induced Pluripotent Stem Cell-Derived Vascular Cells: Recent Progress and Future Directions. J. Cardiovasc. Dev. Dis. 2021, 8, 148. [Google Scholar] [CrossRef] [PubMed]
- Palpant, N.J.; Pabon, L.; Friedman, C.E.; Roberts, M.; Hadland, B.; Zaunbrecher, R.J.; Bernstein, I.; Zheng, Y.; Murry, C.E. Generating high-purity cardiac and endothelial derivatives from patterned mesoderm using human pluripotent stem cells. Nat. Protoc. 2016, 12, 15–31. [Google Scholar] [CrossRef]
- Kumar, M.; Toprakhisar, B.; Van Haele, M.; Antoranz, A.; Boon, R.; Chesnais, F.; De Smedt, J.; Tricot, T.; Idoype, T.I.; Canella, M.; et al. A fully defined matrix to support a pluripotent stem cell derived multi-cell-liver steatohepatitis and fibrosis model. Biomaterials 2021, 276, 121006. [Google Scholar] [CrossRef]
- Ferrero, E.; Bondanza, A.; Leone, B.E.; Manici, S.; Poggi, A.; Zocchi, M.R. CD14+ CD34+ peripheral blood mononuclear cells migrate across endothelium and give rise to immunostimulatory dendritic cells. J. Immunol. 1998, 160, 2675–2683. [Google Scholar] [CrossRef]
- Langheim, S.; Dreas, L.; Veschini, L.; Maisano, F.; Foglieni, C.; Ferrarello, S.; Sinagra, G.; Zingone, B.; Alfieri, O.; Ferrero, E.; et al. Increased expression and secretion of resistin in epicardial adipose tissue of patients with acute coronary syndrome. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H746–H753. [Google Scholar] [CrossRef]
- Chesnais, F.; Hue, J.; Roy, E.; Branco, M.; Stokes, R.; Pellon, A.; Le Caillec, J.; Elbahtety, E.; Battilocchi, M.; Danovi, D.; et al. High-content image analysis to study phenotypic heterogeneity in endothelial cell monolayers. J. Cell Sci. 2022, 135, jcs259104. [Google Scholar] [CrossRef]
- Veschini, L.; Sailem, H.; Malani, D.; Pietiäinen, V.; Stojiljkovic, A.; Wiseman, E.; Danovi, D. High-Content Imaging to Phenotype Human Primary and iPSC-Derived Cells. Methods Mol. Biol. 2021, 2185, 423–445. [Google Scholar] [CrossRef]
- Kerns, S.J.; Belgur, C.; Petropolis, D.; Kanellias, M.; Barrile, R.; Sam, J.; Weinzierl, T.; Fauti, T.; Freimoser-Grundschober, A.; Eckmann, J.; et al. Human immunocompetent Organ-on-Chip platforms allow safety profiling of tumor-targeted T-cell bispecific antibodies. eLife 2021, 10, e67106. [Google Scholar] [CrossRef]
- Ewart, L.; Apostolou, A.; Briggs, S.A.; Carman, C.V.; Chaff, J.T.; Heng, A.R.; Jadalannagari, S.; Janardhanan, J.; Jang, K.-J.; Joshipura, S.R.; et al. Performance assessment and economic analysis of a human Liver-Chip for predictive toxicology. Commun. Med. 2022, 2, 154. [Google Scholar] [CrossRef] [PubMed]
- Pediaditakis, I.; Kodella, K.R.; Manatakis, D.V.; Le, C.Y.; Barthakur, S.; Sorets, A.; Gravanis, A.; Ewart, L.; Rubin, L.L.; Manolakos, E.S.; et al. A microengineered Brain-Chip to model neuroinflammation in humans. iScience 2022, 25, 104813. [Google Scholar] [CrossRef] [PubMed]
- Ronaldson-Bouchard, K.; Baldassarri, I.; Tavakol, D.N.; Graney, P.L.; Samaritano, M.; Cimetta, E.; Vunjak-Novakovic, G. Engineering complexity in human tissue models of cancer. Adv. Drug Deliv. Rev. 2022, 184, 114181. [Google Scholar] [CrossRef] [PubMed]
- Ronaldson-Bouchard, K.; Teles, D.; Yeager, K.; Tavakol, D.N.; Zhao, Y.; Chramiec, A.; Tagore, S.; Summers, M.; Stylianos, S.; Tamargo, M.; et al. A multi-organ chip with matured tissue niches linked by vascular flow. Nat. Biomed. Eng. 2022, 6, 351–371. [Google Scholar] [CrossRef]
- Nakatsu, M.N.; Davis, J.; Hughes, C.C.W. Optimized fibrin gel bead assay for the study of angiogenesis. J. Vis. Exp. 2007, 186. [Google Scholar] [CrossRef]
- Jauhiainen, S.; Ilmonen, H.; Vuola, P.; Rasinkangas, H.; Pulkkinen, H.H.; Keränen, S.; Kiema, M.; Liikkanen, J.J.; Laham-Karam, N.; Laidinen, S.; et al. ErbB signaling is a potential therapeutic target for vascular lesions with fibrous component. eLife 2023, 12, e82543. [Google Scholar] [CrossRef]
- Francis, C.R.; Kincross, H.; Kushner, E.J. Rab35 governs apicobasal polarity through regulation of actin dynamics during sprouting angiogenesis. Nat. Commun. 2022, 13, 5276. [Google Scholar] [CrossRef]
- Hetheridge, C.; Mavria, G.; Mellor, H. Uses of the in vitro endothelial-fibroblast organotypic co-culture assay in angiogenesis research. Biochem. Soc. Trans. 2011, 39, 1597–1600. [Google Scholar] [CrossRef]
- Strobel, H.A.; Moss, S.M.; Hoying, J.B. Vascularized Tissue Organoids. Bioengineering 2023, 10, 124. [Google Scholar] [CrossRef]
- Yu, J. Vascularized Organoids: A More Complete Model. Int. J. Stem Cells 2021, 14, 127–137. [Google Scholar] [CrossRef]
- Orlova, V.V.; van den Hil, F.E.; Petrus-Reurer, S.; Drabsch, Y.; Ten Dijke, P.; Mummery, C.L. Generation, expansion and functional analysis of endothelial cells and pericytes derived from human pluripotent stem cells. Nat. Protoc. 2014, 9, 1514–1531. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; D’Souza, S.S.; Moskvin, O.V.; Toh, H.; Wang, B.; Zhang, J.; Swanson, S.; Guo, L.-W.; Thomson, J.A.; Slukvin, I.I. Specification and Diversification of Pericytes and Smooth Muscle Cells from Mesenchymoangioblasts. Cell Rep. 2017, 19, 1902–1916. [Google Scholar] [CrossRef] [PubMed]
- Ferrarini, M.; Steimberg, N.; Ponzoni, M.; Belloni, D.; Berenzi, A.; Girlanda, S.; Caligaris-Cappio, F.; Mazzoleni, G.; Ferrero, E. Ex-vivo dynamic 3-D culture of human tissues in the RCCSTM bioreactor allows the study of Multiple Myeloma biology and response to therapy. PLoS ONE 2013, 8, e71613. [Google Scholar] [CrossRef]
- Osaki, T.; Sivathanu, V.; Kamm, R.D. Vascularized microfluidic organ-chips for drug screening, disease models and tissue engineering. Curr. Opin. Biotechnol. 2018, 52, 116–123. [Google Scholar] [CrossRef]
- Chen, M.B.; Whisler, J.A.; Fröse, J.; Yu, C.; Shin, Y.; Kamm, R.D. On-chip human microvasculature assay for visualization and quantification of tumor cell extravasation dynamics. Nat. Protoc. 2017, 12, 865–880. [Google Scholar] [CrossRef]
- Yamamoto, K.; Tanimura, K.; Watanabe, M.; Sano, H.; Uwamori, H.; Mabuchi, Y.; Matsuzaki, Y.; Chung, S.; Kamm, R.D.; Tanishita, K.; et al. Construction of Continuous Capillary Networks Stabilized by Pericyte-like Perivascular Cells. Tissue Eng. Part A 2019, 25, 499–510. [Google Scholar] [CrossRef]
- Boussommier-Calleja, A.; Atiyas, Y.; Haase, K.; Headley, M.; Lewis, C.; Kamm, R.D. The effects of monocytes on tumor cell extravasation in a 3D vascularized microfluidic model. Biomaterials 2019, 198, 180–193. [Google Scholar] [CrossRef]
- Sobrino, A.; Phan, D.T.T.; Datta, R.; Wang, X.; Hachey, S.J.; Romero-López, M.; Gratton, E.; Lee, A.P.; George, S.C.; Hughes, C.C.W. 3D microtumors in vitro supported by perfused vascular networks. Sci. Rep. 2016, 6, 31589. [Google Scholar] [CrossRef]
- Hachey, S.J.; Sobrino, A.; Lee, J.G.; Jafari, M.D.; Klempner, S.J.; Puttock, E.J.; Edwards, R.A.; Lowengrub, J.S.; Waterman, M.L.; Zell, J.A.; et al. A human vascularized microtumor model of patient-derived colorectal cancer recapitulates clinical disease. Transl. Res. J. Lab. Clin. Med. 2023, 255, 97–108. [Google Scholar] [CrossRef]
- Chesnais, F.; Joel, J.; Hue, J.; Shakib, S.; Di Silvio, L.; Grigoriadis, A.E.; Coward, T.; Veschini, L. Continuously perfusable, customisable, and matrix-free vasculature on a chip platform. Lab. Chip 2023, 23, 761–772. [Google Scholar] [CrossRef]
- Razavirad, A.; Rismanchi, S.; Mortazavi, P.; Muhammadnejad, A. Canine Mammary Tumors as a Potential Model for Human Breast Cancer in Comparative Oncology. Vet. Med. Int. 2024, 2024, 9319651. [Google Scholar] [CrossRef] [PubMed]
- Palma, S.D.; McConnell, A.; Verganti, S.; Starkey, M. Review on Canine Oral Melanoma: An Undervalued Authentic Genetic Model of Human Oral Melanoma? Vet. Pathol. 2021, 58, 881–889. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.; van der Weyden, L.; Schott, C.R.; Foote, A.; Constantino-Casas, F.; Smith, S.; Dobson, J.M.; Murchison, E.P.; Wu, H.; Yeh, I.; et al. Cross-species genomic landscape comparison of human mucosal melanoma with canine oral and equine melanoma. Nat. Commun. 2019, 10, 353. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.H.; Cho, J.-Y. Comparative oncology: Overcoming human cancer through companion animal studies. Exp. Mol. Med. 2023, 55, 725–734. [Google Scholar] [CrossRef]
- Onaciu, A.; Munteanu, R.; Munteanu, V.C.; Gulei, D.; Raduly, L.; Feder, R.-I.; Pirlog, R.; Atanasov, A.G.; Korban, S.S.; Irimie, A.; et al. Spontaneous and Induced Animal Models for Cancer Research. Diagnostics 2020, 10, 660. [Google Scholar] [CrossRef]
- Mariano, L.C.; Warnakulasuriya, S.; Straif, K.; Monteiro, L. Secondhand smoke exposure and oral cancer risk: A systematic review and meta-analysis. Tob. Control 2022, 31, 597–607. [Google Scholar] [CrossRef]
- Zaccone, R.; Renzi, A.; Chalfon, C.; Lenzi, J.; Bellei, E.; Marconato, L.; Ros, E.; Rigillo, A.; Bettini, G.; Faroni, E.; et al. Environmental risk factors for the development of oral squamous cell carcinoma in cats. J. Vet. Intern. Med. 2022, 36, 1398–1408. [Google Scholar] [CrossRef]
- Sarver, A.L.; Makielski, K.M.; DePauw, T.A.; Schulte, A.J.; Modiano, J.F. Increased risk of cancer in dogs and humans: A consequence of recent extension of lifespan beyond evolutionarily-determined limitations? Aging Cancer 2022, 3, 3–19. [Google Scholar] [CrossRef]
- Carvalho, M.I.; Guimarães, M.J.; Pires, I.; Prada, J.; Silva-Carvalho, R.; Lopes, C.; Queiroga, F.L. EGFR and microvessel density in canine malignant mammary tumours. Res. Vet. Sci. 2013, 95, 1094–1099. [Google Scholar] [CrossRef]
- Queiroga, F.L.; Pires, I.; Parente, M.; Gregório, H.; Lopes, C.S. COX-2 over-expression correlates with VEGF and tumour angiogenesis in canine mammary cancer. Vet. J. 2011, 189, 77–82. [Google Scholar] [CrossRef]
- Laufer-Amorim, R.; Fonseca-Alves, C.E.; Villacis, R.A.R.; Linde, S.A.D.; Carvalho, M.; Larsen, S.J.; Marchi, F.A.; Rogatto, S.R. Comprehensive Genomic Profiling of Androgen-Receptor-Negative Canine Prostate Cancer. Int. J. Mol. Sci. 2019, 20, 1555. [Google Scholar] [CrossRef] [PubMed]
- Nordby, Y.; Andersen, S.; Richardsen, E.; Ness, N.; Al-Saad, S.; Melbø-Jørgensen, C.; Patel, H.R.H.; Dønnem, T.; Busund, L.-T.; Bremnes, R.M. Stromal expression of VEGF-A and VEGFR-2 in prostate tissue is associated with biochemical and clinical recurrence after radical prostatectomy. Prostate 2015, 75, 1682–1693. [Google Scholar] [CrossRef] [PubMed]
- Warszawik-Hendzel, O.; Słowińska, M.; Olszewska, M.; Rudnicka, L. Melanoma of the oral cavity: Pathogenesis, dermoscopy, clinical features, staging and management. J. Dermatol. Case Rep. 2014, 8, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Bergman, P.J. Canine oral melanoma. Clin. Tech. Small Anim. Pract. 2007, 22, 55–60. [Google Scholar] [CrossRef]
- Patel, V.; Di Silvio, L.; Kwok, J.; Burns, M.; Henley Smith, R.; Thavaraj, S.; Veschini, L. The impact of intensity-modulated radiation treatment on dento-alveolar microvasculature in pharyngeal cancer implant patients. J. Oral Rehabil. 2020, 47, 1411–1421. [Google Scholar] [CrossRef]
- Nafe, R.; Schlote, W. Histomorphometry of brain tumours. Neuropathol. Appl. Neurobiol. 2004, 30, 315–328. [Google Scholar] [CrossRef]
- Mahmood, H.; Shaban, M.; Indave, B.I.; Santos-Silva, A.R.; Rajpoot, N.; Khurram, S.A. Use of artificial intelligence in diagnosis of head and neck precancerous and cancerous lesions: A systematic review. Oral Oncol. 2020, 110, 104885. [Google Scholar] [CrossRef]
- Veschini, L.; Crippa, L.; Dondossola, E.; Doglioni, C.; Corti, A.; Ferrero, E. The vasostatin-1 fragment of chromogranin A preserves a quiescent phenotype in hypoxia-driven endothelial cells and regulates tumor neovascularization. FASEB J. 2011, 25, 3906–3914. [Google Scholar] [CrossRef]
- Madusanka, N.; Jayalath, P.; Fernando, D.; Yasakethu, L.; Lee, B.-I. Impact of H&E Stain Normalization on Deep Learning Models in Cancer Image Classification: Performance, Complexity, and Trade-Offs. Cancers 2023, 15, 4144. [Google Scholar] [CrossRef]
- Hue, J.; Valinciute, Z.; Thavaraj, S.; Veschini, L. Multifactorial estimation of clinical outcome in HPV-associated oropharyngeal squamous cell carcinoma via automated image analysis of routine diagnostic H&E slides and neural network modelling. Oral Oncol. 2023, 141, 106399. [Google Scholar] [CrossRef]
- Graham, S.; Vu, Q.D.; Jahanifar, M.; Weigert, M.; Schmidt, U.; Zhang, W.; Zhang, J.; Yang, S.; Xiang, J.; Wang, X.; et al. CoNIC Challenge: Pushing the frontiers of nuclear detection, segmentation, classification and counting. Med. Image Anal. 2024, 92, 103047. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.; Vu, Q.D.; Raza, S.E.A.; Azam, A.; Tsang, Y.W.; Kwak, J.T.; Rajpoot, N. Hover-Net: Simultaneous segmentation and classification of nuclei in multi-tissue histology images. Med. Image Anal. 2019, 58, 101563. [Google Scholar] [CrossRef] [PubMed]
- Cabral, B.P.; Braga, L.A.M.; Syed-Abdul, S.; Mota, F.B. Future of Artificial Intelligence Applications in Cancer Care: A Global Cross-Sectional Survey of Researchers. Curr. Oncol. 2023, 30, 3432–3446. [Google Scholar] [CrossRef] [PubMed]
- Timakova, A.; Ananev, V.; Fayzullin, A.; Makarov, V.; Ivanova, E.; Shekhter, A.; Timashev, P. Artificial Intelligence Assists in the Detection of Blood Vessels in Whole Slide Images: Practical Benefits for Oncological Pathology. Biomolecules 2023, 13, 1327. [Google Scholar] [CrossRef]
- Gniadek, T.; Kang, J.; Theparee, T.; Krive, J. Framework for Classifying Explainable Artificial Intelligence (XAI) Algorithms in Clinical Medicine. Online J. Public Health Inform. 2023, 15, e50934. [Google Scholar] [CrossRef]
- Ali, S.; Akhlaq, F.; Imran, A.S.; Kastrati, Z.; Daudpota, S.M.; Moosa, M. The enlightening role of explainable artificial intelligence in medical & healthcare domains: A systematic literature review. Comput. Biol. Med. 2023, 166, 107555. [Google Scholar] [CrossRef]
- Wang, X.; Chen, F.; Guo, N.; Gu, Z.; Lin, H.; Xiang, X.; Shi, Y.; Han, B. Application of physiologically based pharmacokinetics modeling in the research of small-molecule targeted anti-cancer drugs. Cancer Chemother. Pharmacol. 2023, 92, 253–270. [Google Scholar] [CrossRef]
- Calzone, L.; Noël, V.; Barillot, E.; Kroemer, G.; Stoll, G. Modeling signaling pathways in biology with MaBoSS: From one single cell to a dynamic population of heterogeneous interacting cells. Comput. Struct. Biotechnol. J. 2022, 20, 5661–5671. [Google Scholar] [CrossRef]
- Choi, K.; Medley, J.K.; König, M.; Stocking, K.; Smith, L.; Gu, S.; Sauro, H.M. Tellurium: An extensible python-based modeling environment for systems and synthetic biology. Biosystems 2018, 171, 74–79. [Google Scholar] [CrossRef]
- Graner, F.; Glazier, J.A. Simulation of biological cell sorting using a two-dimensional extended Potts model. Phys. Rev. Lett. 1992, 69, 2013–2016. [Google Scholar] [CrossRef]
- Starruß, J.; de Back, W.; Brusch, L.; Deutsch, A. Morpheus: A user-friendly modeling environment for multiscale and multicellular systems biology. Bioinformatics 2014, 30, 1331–1332. [Google Scholar] [CrossRef]
- Swat, M.H.; Thomas, G.L.; Belmonte, J.M.; Shirinifard, A.; Hmeljak, D.; Glazier, J.A. Multi-scale modeling of tissues using CompuCell3D. Methods Cell Biol. 2012, 110, 325–366. [Google Scholar] [CrossRef]
- Shirinifard, A.; Gens, J.S.; Zaitlen, B.L.; Popławski, N.J.; Swat, M.; Glazier, J.A. 3D multi-cell simulation of tumor growth and angiogenesis. PLoS ONE 2009, 4, e7190. [Google Scholar] [CrossRef]
- Merks, R.M.H.; Glazier, J.A. Dynamic mechanisms of blood vessel growth. Nonlinearity 2006, 19, C1–C10. [Google Scholar] [CrossRef]
- Merks, R.M.H.; Perryn, E.D.; Shirinifard, A.; Glazier, J.A. Contact-Inhibited Chemotaxis in De Novo and Sprouting Blood-Vessel Growth. PLOS Comput. Biol. 2008, 4, e1000163. [Google Scholar] [CrossRef]
- Niarakis, A.; Laubenbacher, R.; An, G.; Ilan, Y.; Fisher, J.; Flobak, Å.; Reiche, K.; Rodríguez Martínez, M.; Geris, L.; Ladeira, L.; et al. Immune digital twins for complex human pathologies: Applications, limitations, and challenges. npj Syst. Biol. Appl. 2024, 10, 141. [Google Scholar] [CrossRef]
- Yang, H.M. Mathematical modeling of solid cancer growth with angiogenesis. Theor. Biol. Med. Model. 2012, 9, 2. [Google Scholar] [CrossRef]
- Moffett, A.S.; Deng, Y.; Levine, H. Modeling the Role of Immune Cell Conversion in the Tumor-Immune Microenvironment. Bull. Math. Biol. 2023, 85, 93. [Google Scholar] [CrossRef]
- Uatay, A.; Gall, L.; Irons, L.; Tewari, S.G.; Zhu, X.S.; Gibbs, M.; Kimko, H. Physiological Indirect Response Model to Omics-Powered Quantitative Systems Pharmacology Model. J. Pharm. Sci. 2023, 113, 11–21. [Google Scholar] [CrossRef]
- Pereira, M.; Pinto, J.; Arteaga, B.; Guerra, A.; Jorge, R.N.; Monteiro, F.J.; Salgado, C.L. A Comprehensive Look at In Vitro Angiogenesis Image Analysis Software. Int. J. Mol. Sci. 2023, 24, 17625. [Google Scholar] [CrossRef]
- Yan, H.H.N.; Siu, H.C.; Law, S.; Ho, S.L.; Yue, S.S.K.; Tsui, W.Y.; Chan, D.; Chan, A.S.; Ma, S.; Lam, K.O.; et al. A Comprehensive Human Gastric Cancer Organoid Biobank Captures Tumor Subtype Heterogeneity and Enables Therapeutic Screening. Cell Stem Cell 2018, 23, 882–897.e11. [Google Scholar] [CrossRef] [PubMed]
- Oyoshi, H.; Du, J.; Sakai, S.A.; Yamashita, R.; Okumura, M.; Motegi, A.; Hojo, H.; Nakamura, M.; Hirata, H.; Sunakawa, H.; et al. Comprehensive single-cell analysis demonstrates radiotherapy-induced infiltration of macrophages expressing immunosuppressive genes into tumor in esophageal squamous cell carcinoma. Sci. Adv. 2023, 9, eadh9069. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Ni, S.; Li, S.; Lv, B. Role of stemness-related genes TIMP1, PGF, and SNAI1 in the prognosis of colorectal cancer through single-cell RNA-seq. Cancer Med. 2023, 12, 11611–11623. [Google Scholar] [CrossRef] [PubMed]
- Laubenbacher, R.; Niarakis, A.; Helikar, T.; An, G.; Shapiro, B.; Malik-Sheriff, R.S.; Sego, T.J.; Knapp, A.; Macklin, P.; Glazier, J.A. Building digital twins of the human immune system: Toward a roadmap. npj Digit. Med. 2022, 5, 64. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NAT Category | Advantages | Limitations | Translational Challenges | Regulatory/Commercial Aspects | Value in Studying EC Biology/Angiogenesis | Value in Studying Tumour-Associated Angiogenesis |
|---|---|---|---|---|---|---|
| 2D Static Cultures | Simple, cost-effective, high-throughput | Lack of 3D architecture and TME complexity | Moderate clinical predictivity | Established for first-line screens | Used in toxicity testing, barrier function assays, leukocyte transmigration, and endothelial signalling studies. | Applied in screening VEGF signalling, endothelial permeability, and immune cell transmigration; foundational in early angiogenesis modelling. |
| 3D Spheroids and Organoids | Better mimicry of tumour architecture; patient-derived | Variable yield and standardisation; lack vasculature | Scale-up and patient-specific validation | Gaining traction in precision oncology | EC spheroids used to investigate angiogenesis in matrices or microtissues. | Support indirect exploration of angiogenic signalling under hypoxia or drug conditions |
| Ex Vivo Tumour Explants | Preserve native TME; clinically relevant | Short culture lifespan; access to specimens | Integration with drug screening pipelines | Valuable for personalised medicine; yet under-utilised | Enable investigation of native endothelial structures, vessel morphology, and angiogenic responses. | Preserve native tumour vasculature enabling direct evaluation of angiogenic features, drug effects, and EC-TME interaction in patient tissues. |
| Dynamic Bioreactors (e.g., RCCS) | Sustain viability in complex 3D tissues | Complex handling; limited throughput | Standardising protocols for clinical translation | Increasingly explored under FDA Modernization Act | Allow monitoring of endothelial and vascular behaviour over time in viable 3D cultures; Angiogenic response studies and drug testing. | Used to culture tumour explants with intact vasculature; facilitate real-time observation of angiogenic modulation under therapeutic conditions. |
| Microphysiological Systems (MPS) | Allow perfusion, mass transfer; scalable | Costly, complex fabrication and operation | Inter-device reproducibility, regulatory acceptance | Key to non-animal preclinical validation; high priority | Controlled study of EC function under flow, including vessel formation, barrier properties, and signalling. | Allow reproduction of tumour vascular environments with flow; applied in mechanistic studies, drug screening, and metastasis research. |
| Vascularised Organoids | Integrate vascular features into tissue models | Perfusion and maturation still limited | Demonstrating consistent vascularisation | Potential to fulfil unmet modelling needs | Support formation of capillary-like structures within organoids; Study EC-stroma interaction and vascular self-organisation. | Enable angiocrine and vessel remodelling studies within patient-derived or stem-cell-based tumour constructs |
| In Silico/Computational Models | Hypothesis generation and testing; multi-scale integration | Require high-quality experimental validation | Validation, regulatory uncertainty | Seen as decision-support tools; still unregulated | Enable in silico experimentation on EC dynamics, angiogenic pathways, and network behaviour. | Support modelling of tumour-induced angiogenesis, VEGF diffusion, and EC-tumour cell crosstalk at multiple scales. |
| Spontaneous Tumours in Companion Animals | Human-relevant, ethically viable, naturally occurring cancers | Logistics, sample standardisation, limited availability | Data harmonisation across species | Supports One Health approach; gaining interest | Physiologic endothelial diversity and vascular changes in spontaneous diseases; informative for natural history and treatment response studies. | Offer clinically relevant insights into tumour angiogenesis and vascular responses in natural disease; valuable for translational and comparative research. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferrero, E.; Hue, J.; Ferrarini, M.; Veschini, L. Non-Animal Technologies to Study and Target the Tumour Vasculature and Angiogenesis. Organoids 2025, 4, 12. https://doi.org/10.3390/organoids4020012
Ferrero E, Hue J, Ferrarini M, Veschini L. Non-Animal Technologies to Study and Target the Tumour Vasculature and Angiogenesis. Organoids. 2025; 4(2):12. https://doi.org/10.3390/organoids4020012
Chicago/Turabian StyleFerrero, Elisabetta, Jonas Hue, Marina Ferrarini, and Lorenzo Veschini. 2025. "Non-Animal Technologies to Study and Target the Tumour Vasculature and Angiogenesis" Organoids 4, no. 2: 12. https://doi.org/10.3390/organoids4020012
APA StyleFerrero, E., Hue, J., Ferrarini, M., & Veschini, L. (2025). Non-Animal Technologies to Study and Target the Tumour Vasculature and Angiogenesis. Organoids, 4(2), 12. https://doi.org/10.3390/organoids4020012