1. Introduction
Microtissue cell culture models are increasingly being used, as their three-dimensional (3D) organization appears to better replicate those of tissues, compared to two-dimensional (2D) cultures, and because recent technological advances facilitate more efficient microtissue culture [
1,
2,
3,
4]. How to best combine multiple microtissues to scale or to make larger tissues or organs remain an area of intense research. Early work by Kelm et al. used microtissues as building blocks and formed larger tissues by amalgamating the smaller microtissues [
5]. Similarly, microtissues formed from different cell types have been layered on top of each other in an attempt to generate more complex or physically large macrotissues [
6]. Most recently, 3D printing has been used to impose a macrostructure on microtissues [
7]. The lack of vasculature restricts the number of microtissues that can be assembled into a larger tissue before nutrient diffusion limitations compromise tissue function or viability. Because of this limitation, there may be many instances where the elegance and utility of microtissues may be best exploited by maintaining each microtissue as a discrete unit and by simply designing systems that enable the efficient handling or manipulation of multiple microtissue units. In this paper, we focused on the development of a method to manipulate arrays of microtissues.
When bone marrow stromal cells (BMSCs, also known as “bone marrow-derived mesenchymal stem cells”) are assembled into microtissues [
8,
9,
10], they can be stimulated with growth factors to form cartilage-like tissue in vitro [
11]. While the tissue may appear cartilage-like, BMSCs have a propensity to engage intrinsic hypertrophic signaling pathways [
10], and these tissues evolve to form bone and bone marrow when implanted in vivo [
10,
12,
13,
14]. The microtissues mineralize at the surface, followed by the incremental replacement of the core cartilage-like template by a bone marrow-like structure [
10,
12,
13,
14], ultimately yielding micro-ossicles. Because these micro-ossicles are formed from and contain human stromal cells, some researchers purposefully use these tissues to study human cell biology or to model disease [
10,
12,
13,
14]. Studies have shown that these humanized bone marrow organs are capable of supporting (1) the superior engraftment of healthy human hematopoietic stem/progenitor cells (HSPCs) when compared with murine bone marrow [
15,
16]; (2) the engraftment of malignant human hematopoietic xenografts that are not readily propagated in murine bone marrow [
15,
16,
17,
18]; and (3) that these ossicles support human prostate cancer cell populations more so than murine bone marrow [
19,
20]. Cumulatively, these data suggest that humanized micro-ossicles offer a better tool to study human cell biology or disease than mouse marrow.
Serafini et al. formed microtissues from 3 × 10
5 BMSCs each, cultured in chondrogenic induction medium supplemented with transforming growth factor β (TGF-β1 or TGF-β3) [
12]. Microtissues were cultured for 3 weeks and then implanted into mice. Each animal was implanted with >16 discrete microtissues, and at 8 weeks, microtissues had mineralized and contained marrow structures within the core, effectively yielding numerous replicate micro-ossicles. In this case, the authors followed a commonly described BMSC chondrogenic induction protocol, but reported on the formation of bone marrow ossicles [
12]. In an alternative approach, Scotti et al. seeded 5 × 10
5 BMSC onto a transwell membrane and then cultured the cells in chondrogenic medium supplemented with TGF-β1 for three weeks, followed by a two-week culture in a hypertrophic medium (without TGFβ1, but with beta-glycerophosphate and thyroxine) [
21]. Scotti et al. formed larger ossicles [
21], rather than multiple repeat micro-ossicles as described by Serafini et al. [
12]. Thus, the two main approaches described in the literature are the formation of multiple micro-ossicles, or the formation of 1–2 larger tissues [
19,
20,
21].
The advantage of large ossicles is that there is more continuous volume for remodeling into bone marrow, and some studies exploit this larger volume to directly inject cells into these larger tissues [
16,
22,
23,
24]. The disadvantage of using a singular large ossicle is possible delays in vascularization, low replicate numbers, and possibly necessitating increasing the number of animals in a study. While it is possible to generate bone marrow in mice via either endochondral [
25] or intramembranous differentiation programs [
26], generating marrow through endochondral ossification would best replicate the bulk of skeletal development [
25]. A microtissue could be seen as a more suitable substrate for this process, compared to seeding cells at a low density on a large scaffold, as the aggregation of cells during microtissue assembly mimics mesenchymal condensation [
25]. Microtissues are well suited to in vitro culture and manipulation. The number of cells within a single microtissue is generally tailored to limit the maximum diameter and to ensure that there is adequate metabolite diffusion through the tissue. For chondrogenic culture, BMSCs are frequently pelleted into tissues of 2–5 × 10
5 cells each [
27]; by working in this cell range, it is possible to reliably generate a cartilage-like template that is remodeled into bone marrow when implanted into mice [
12,
14], which in theory, should enable the replication of endochondral ossification [
25]. Note that much work remains to be performed on this subject to fully elucidate the differentiation processes that yield micro-ossicles [
12]. The disadvantage of using replicate micro-ossicles is the difficulty in handling, which is further complicated by microtissues shifting under the loose skin (facia) of the mouse. We address this microtissue/micro-ossicle handling challenge in this paper.
To improve the utility of humanized micro-ossicles, we developed a micro-array system that can be initiated in vitro and transplanted in an efficient and organized manner in vivo. We used a microwell platform to deposit cells onto a nylon mesh. Each microwell deposited a specific number of cells on a nylon carrier, yielding an array of discrete microtissues. The nylon mesh functioned as a carrier for the microtissue array, making the array easy to handle. When transplanted, the nylon mesh minimally impacts microtissue vascularization due to its porosity, and because nylon does not degrade during in vivo incubation, the microtissue array remains intact. To evaluate this approach, we used the established microtissue cell number described by Serafini et al. (3 × 10
5 BMSC each [
12]) coupled with the optimized medium formulation described by Scotti et al. (three-week chondrogenic medium, followed by a two-week culture in a hypertrophic medium [
21]). Microtissue arrays were primed in culture, then implanted subcutaneously into NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice. These microtissues were permitted to remodel into micro-ossicles over 8 weeks, and the animals were then conditioned with 2.5 Gy irradiation prior to transplantation with 10
5 human umbilical cord blood-derived CD34
+ cells. Human hematopoietic cell engraftment in micro-ossicles and mouse bone marrow was characterized at 8 weeks after CD34
+ cell infusion using flow cytometry.
2. Materials and Methods
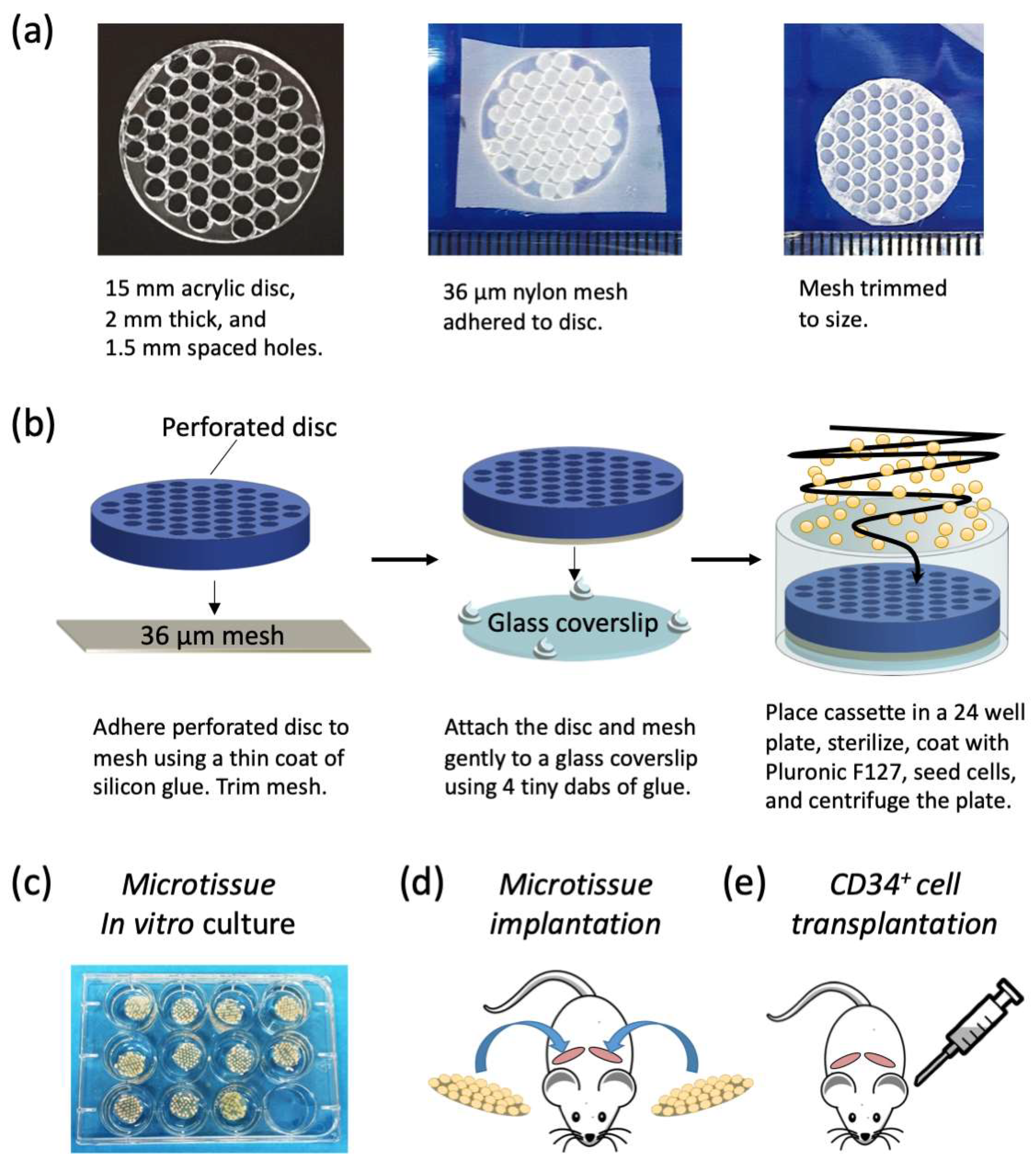
Microtissue array cassette manufacture: We manufactured a custom cell-seeding cassette that consisted of a perforated acrylic disc that facilitated the seeding of cells onto a nylon mesh in an organized array. Perforated acrylic discs were fabricated by Microsurfaces Pty Ltd. (Flemington, VIC, Australia). Discs were 15 mm in diameter and 2 mm thick with 51 circular perforations laser cut 1.5 mm on center (
Figure 1a). A schematic of the microtissue array cassette fabrication and cell seeding is shown in
Figure 1b. Nylon mesh (Amazon.com, part number CMN-0035) with 36 μm square openings was bonded to the perforated acrylic disc by applying a thin coat of silicone glue (Selleys, Aquarium safe, New Zealand), with care taken not to fill the perforations. Excess mesh was trimmed from the perforated acrylic disc, and the disc, with mesh side down, was attached to a glass cover slip with a 15 mm diameter using four tiny dabs of silicone glue at the periphery. Cassettes were sterilized in 70% ethanol for one hour and then rinsed repeatedly with sterile PBS. Sterile cassettes were then placed at the bottom of 24 well plates (Nunc, ThermoFisher, Waltham, MA, USA). To minimize cell attachment to the cassettes, and to encourage cell aggregation, the cassettes and wells were soaked in sterile 5% Pluronic-F127 (Sigma-Aldrich, St. Louis, MO, USA) for 10 min [
28] and then rinsed thrice with PBS before cell seeding.
BMSC isolation and expansion: BMSCs were isolated and enriched from the bone marrow of healthy human adults, who each provided informed and voluntary written consent. Bone marrow aspirates (20 mL) were collected from the iliac crest for research purposes. Ethics approval for aspirate collection was granted by the Mater Health Services Human Research Ethics Committee and the Queensland University of Technology Human Ethics Committee (Ethics number: 1000000938), and all procedures were in accordance with the National Health and Medical Research Council of Australia guidelines. BMSC isolation, culture, and characterization were performed as previously described [
9,
29]. In brief, mononuclear cells were isolated from 20 mL of bone marrow aspirates using Ficoll-Paque density gradient centrifugation. BMSCs were enriched using plastic adherence overnight in a 20% O
2 and 5% CO
2 atmosphere at 37 °C in expansion medium formulated from low glucose DMEM (ThermoFisher, Waltham, MA, USA), 10% fetal bovine serum (FBS; ThermoFisher), 10 ng/mL of fibroblast growth factor-1 (FGF-1; Peprotech, Israel), 5 μg/mL of porcine heparin sodium salt (Sigma-Aldrich), and 100 U/mL of penicillin/streptomycin (PenStrep; ThermoFisher). On the following day, non-adherent cells were discarded, and the medium was refreshed. Subsequent BMSC expansion was performed in a 2% O
2 and 5% CO
2 atmosphere at 37 °C. We supplemented expansion medium with 10 ng/mL FGF-1 and used a hypoxic atmosphere when culturing BMSC [
9,
10,
28,
30] to maintain consistency with our previous studies. FGF-1 and FGF-2 signal through the same receptor [
31], and hypoxia aids in the maintenance of multilineage differentiation capacity [
32,
33,
34]. Cells were passaged at 80% confluence using 0.25% trypsin/EDTA (ThermoFisher), and new flasks were re-seeded at ~1500 cells per cm
2. Using flow cytometry, BMSCs were characterized for their expression of CD44, CD90, CD73, CD105, and CD146; their lack of CD45, CD34, and HLA-DR; and their potential for in vitro tri-lineage differentiation capacity, as described previously [
28,
29]. BMSCs used for the mouse study described here were from a 23-year-old male (donor 2) and were at passage 3, and this donor’s flow cytometry marker profile and osteogenic and adipogenic differentiation potential were previously detailed [
29]. The chondrogenic differentiation potential of this BMSC population is detailed here.
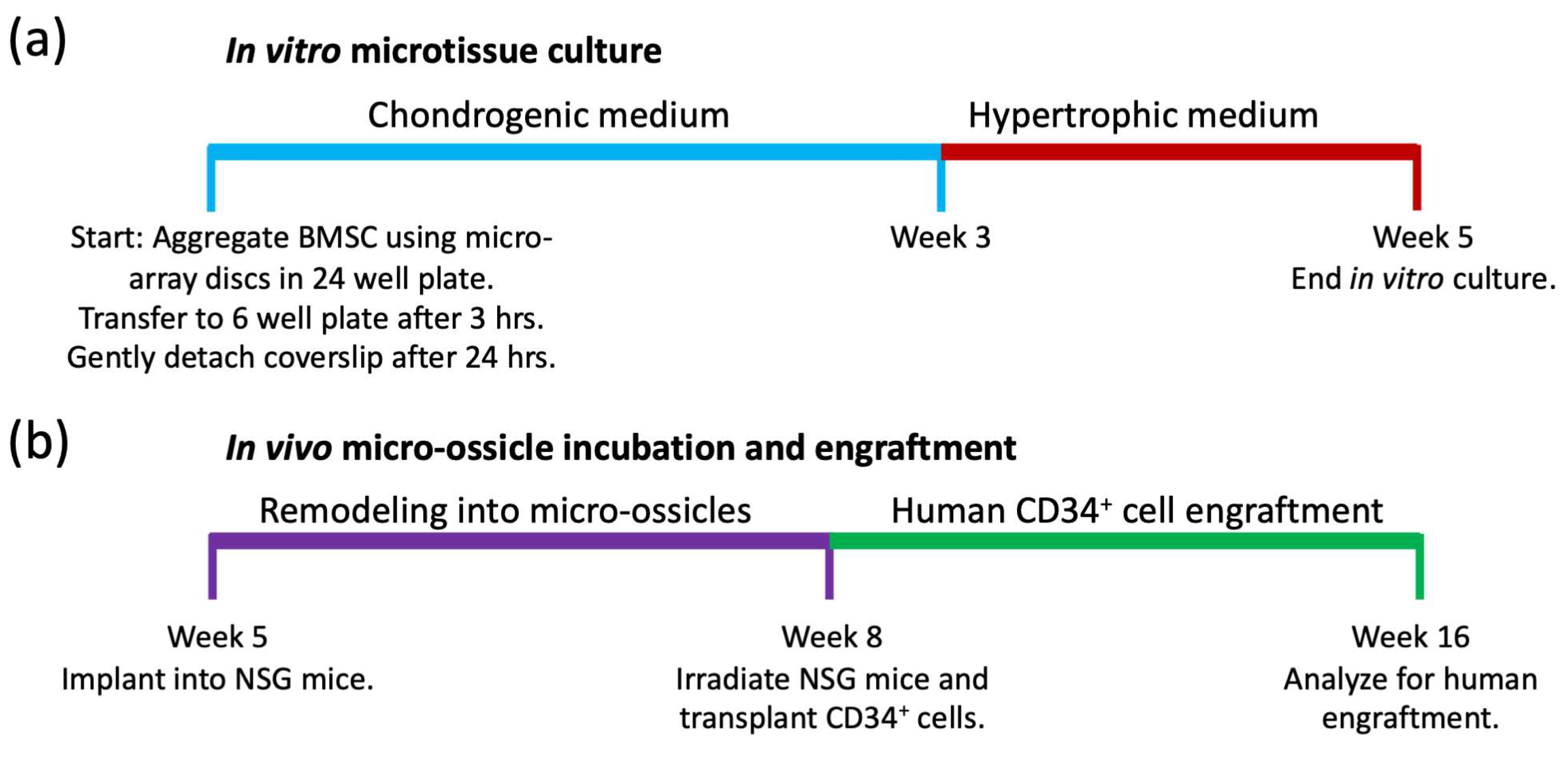
Microtissue array seeding and culture: Microtissue culture timelines are summarized schematically in
Figure 2a. BMSCs were seeded into microtissue array discs contained in 24 well plates by adding 15 × 10
6 cells in 1 mL of chondrogenic induction medium, and the plates were centrifuged for 1 min at 100×
g. This yielded approximately 3 × 10
5 BMSCs per microwell. Chondrogenic induction medium was formulated from high glucose-DMEM (HG-DMEM) containing GlutaMax and 100 μM of sodium pyruvate (ThermoFisher), supplemented with 1X ITS-X, 100 U/mL of penicillin/streptomycin (both from ThermoFisher), 10 ng/mL of TGF-β1 (PeproTech), 100 nM of dexamethasone, 200 μM of ascorbic acid 2-phosphate, and 40 μg/mL of L-proline (all from Sigma-Aldrich). Cells were permitted to aggregate for 3 h, and then the microtissue array cassettes were transferred to 6 well plates, with media volumes of 5 mL per well. After 24 h, the glass coverslips were gently detached from the cassette to allow media access to all sides of the microtissues. Chondrogenic medium was replaced daily for three weeks, and then microtissues were cultured in hypertrophic medium [
21] for a further two weeks with daily medium exchanges. Hypertrophic medium was formulated from HG-DMEM supplemented with 50 nM thyroxine, 7.0 × 10
−3 M β-glycerophosphate, 10
−8 M dexamethasone, and 2.5 × 10
−4 M ascorbic acid (all from Sigma-Aldrich). Some cultures were maintained in chondrogenic induction medium for the full 5-weeks, rather than switching to hypertrophic medium as a control.
Live/Dead staining: Tissues were stained with Live/Dead viability stain as per the manufacturer’s instructions (ThermoFischer). Briefly, tissues were submerged in 2 μM of calcein AM and 4 μM of ethidium homodimer-1 in PBS and incubated in the dark at room temperature for 45 min. To generate a dead cell control, tissues were first incubated for 30 min in 70% ethanol.
MicroCT analysis: MicroCT analysis was performed to detect dense mineralized tissue formation. Fixed tissues were scanned in plastic tubes containing 70% ethanol and stuffed with a piece of biopsy pad to reduce movement. Tissues were scanned at 15 μm voxel size, 50 kV, 200 uA, no filter, at 150 ms exposure using a SkyScan 1272 (Bruker Micro-CT, Kontich, Belgium). Reconstruction was performed with InstaRecon and NRecon software (Bruker Micro-CT).
In vivo transplants in mice: Timelines for microtissue implantation into animals, remodeling into micro-ossicles, and CD34
+ cell engraftment are summarized in
Figure 2b. The University of Queensland (UQ) and the Queensland University of Technology (QUT) Animal Ethics Committees authorized the animal procedures described here. All animal procedures were carried out as described in the approved ethics protocol (AEMAR62827). All animal procedures were approved as per the National Health and Medical Research Council of Australia Guidelines, which in effect align with ARRIVE guidelines. NSG mice were purchased from the Jackson Laboratory and bred in the Animal Facility at the Translational Research Institute (TRI) in Brisbane (breeding ethics approval AEMAR62825). Male mice (6–8 weeks old) were used for experiments. Mice were anesthetized with a 15 to 30 µL solution containing 100 mg/mL of ketamine and 20 mg/mL of xylazine solution delivered via intraperitoneal (IP) injection, their backs were shaved, and their skin was sterilized with iodine and alcohol wipes. Human microtissue arrays were transplanted into subcutaneous pockets, and staples were used to close the wounds. Mice were observed daily for two weeks following surgery by the research team, with daily maintenance of cages, water, and feed by the animal facility staff. Twelve mice received full discs, while two mice received half-discs. Once the wounds healed, the staples were removed. Following 8 weeks of incubation, the 2 mice with half-discs were euthanized to evaluate the tissue histologically, while the remaining 12 mice were sub-lethally irradiated with 250 cGy using a Gamma Cell 40 Cesium source (Nordion, Ottawa, ON, Canada). Following 24 h, irradiated NSG mice were transplanted with human umbilical cord blood CD34
+ cells. CD34
+ cells were purchased from STEMCELL Technologies. Mice were anaesthetized by isoflurane inhalation. Thawed CD34
+ cells were resuspended in X-Vivo 15 media (Lonza, Basel, Switzerland), and 10
5 cells suspended in 100 μL of medium were injected into the retro-orbital sinus as described previously [
30].
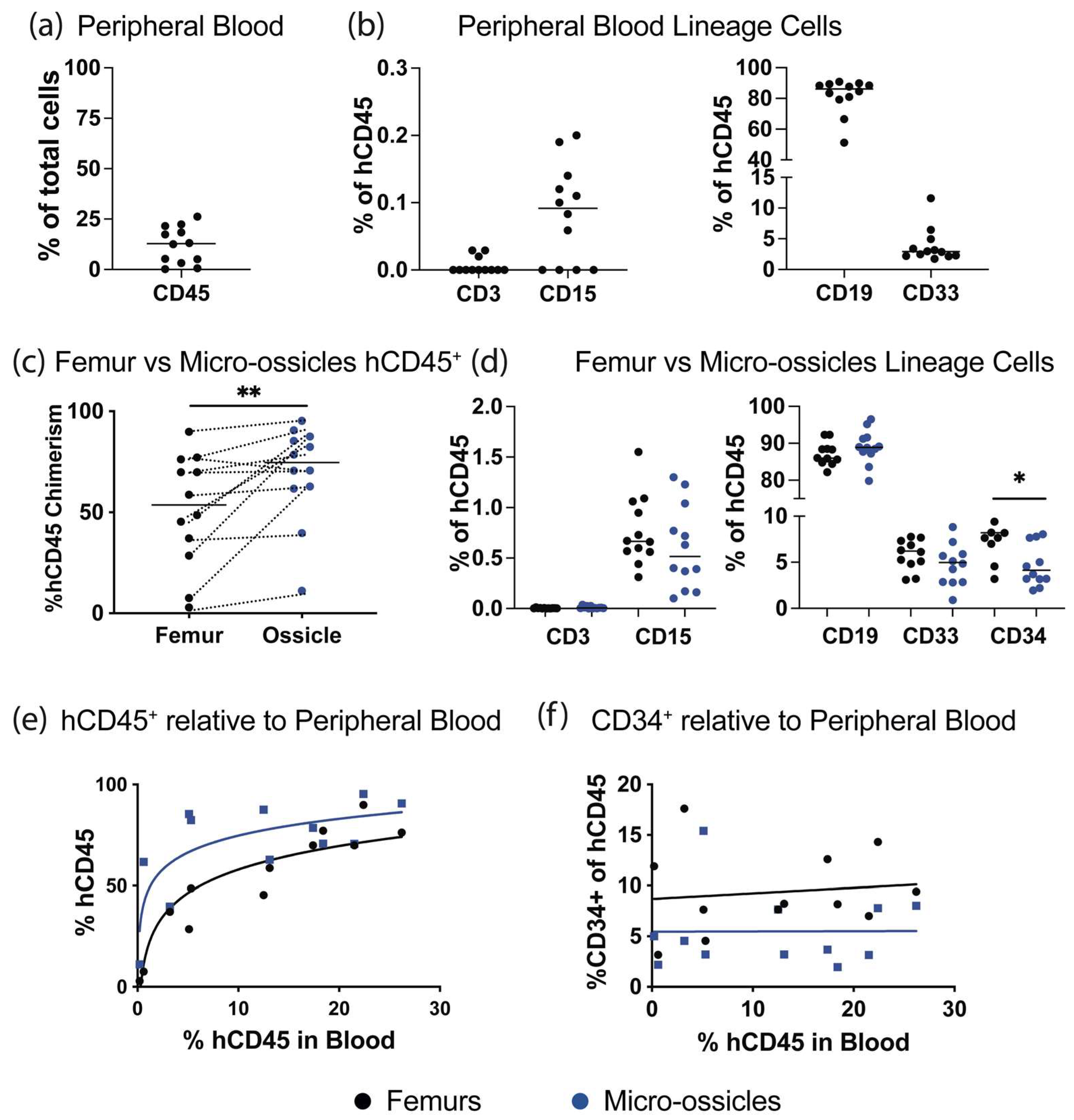
Characterization of engraftment by flow cytometry: Human CD34+ cells were permitted to engraft over 8 weeks, and then animals were euthanized and human hematopoietic cell content in the peripheral blood, micro-ossicle arrays, and mouse femurs were compared. Mice were euthanized by CO2 inhalation, and peripheral blood was collected using cardiac puncture with heparin-coated syringe needles and heparin-containing tubes. Red blood cells were eliminated using lysis buffer (eBiosciences, San Diego, CA, USA). Femurs or ossicle arrays were collected separately, crushed using a mortar and pestle, washed extensively, and passed through a 40 μm strainer to remove debris. Cells were resuspended in blocking solution containing mouse-Fc block (1 μg per sample, BD Biosciences, NJ, USA), 10% normal mouse serum (ThermoFisher), and 1 mg/mL of human IgG (Sigma-Aldrich), and they were incubated for 20 min at 4 °C. Relative human hematopoietic cell engraftment was assessed by staining the cells with anti-human CD45 (hCD45)-VioBright FITC (Miltenyi, Bergisch Gladbach, Germany), anti-human CD34-APC (Miltenyi), hCD3-APC_Cy7, hCD15-PE, hCD19-PE_Cy7, hCD33-BV421 (all from Biolegend, San Diego, CA, USA), and mouse CD45 (mCD45)-APC or mCD45-APC_Cy7 (BD Biosciences). Antibodies were incubated with cells for 30 min at 4°C. To exclude dead cells from flow cytometry analysis, cells were stained with 7-aminoactinomycin D (7-AAD; ThermoFisher). Cells were characterized on an LSRII flow cytometer (BD Biosciences), and data were analyzed using FlowJo software (BD Biosciences). Human hematopoietic engraftment was calculated as: % hCD45+ engraftment = hCD45+ cells/(hCD45+ cells + mCD45+ cells) × 100.
Histological analysis: Tissues were fixed in 4% paraformaldehyde (PFA, Sigma- Aldrich) for 24 h. Post-implant or in vivo tissues, but not pre-implant tissues, were additionally decalcified in 10% EDTA solution until soft. Tissues were dehydrated in an ethanol series, paraffin embedded, and sectioned at 5 μm. Sections were stained with Hematoxylin and Eosin, Alcian Blue for cartilage glycosaminoglycan (GAG) matrix, or Alizarin Red S for mineralized matrix deposition as previously described [
6]. Antigen retrieval prior to immunohistochemistry or RNAScope labeling was performed by incubating slides in RNAScope (Advanced Cell Diagnostics, Hayward, CA, USA) antigen retrieval reagent at 75 °C for 1 h. To identify blood vessels, tissue sections were stained using rat anti-CD31 antibody (BD Biosciences) and rat anti-endomucin (V.7C7) primary antibody (Santa Cruz, Dallas, TX, USA) at 4 °C, overnight, as previously described [
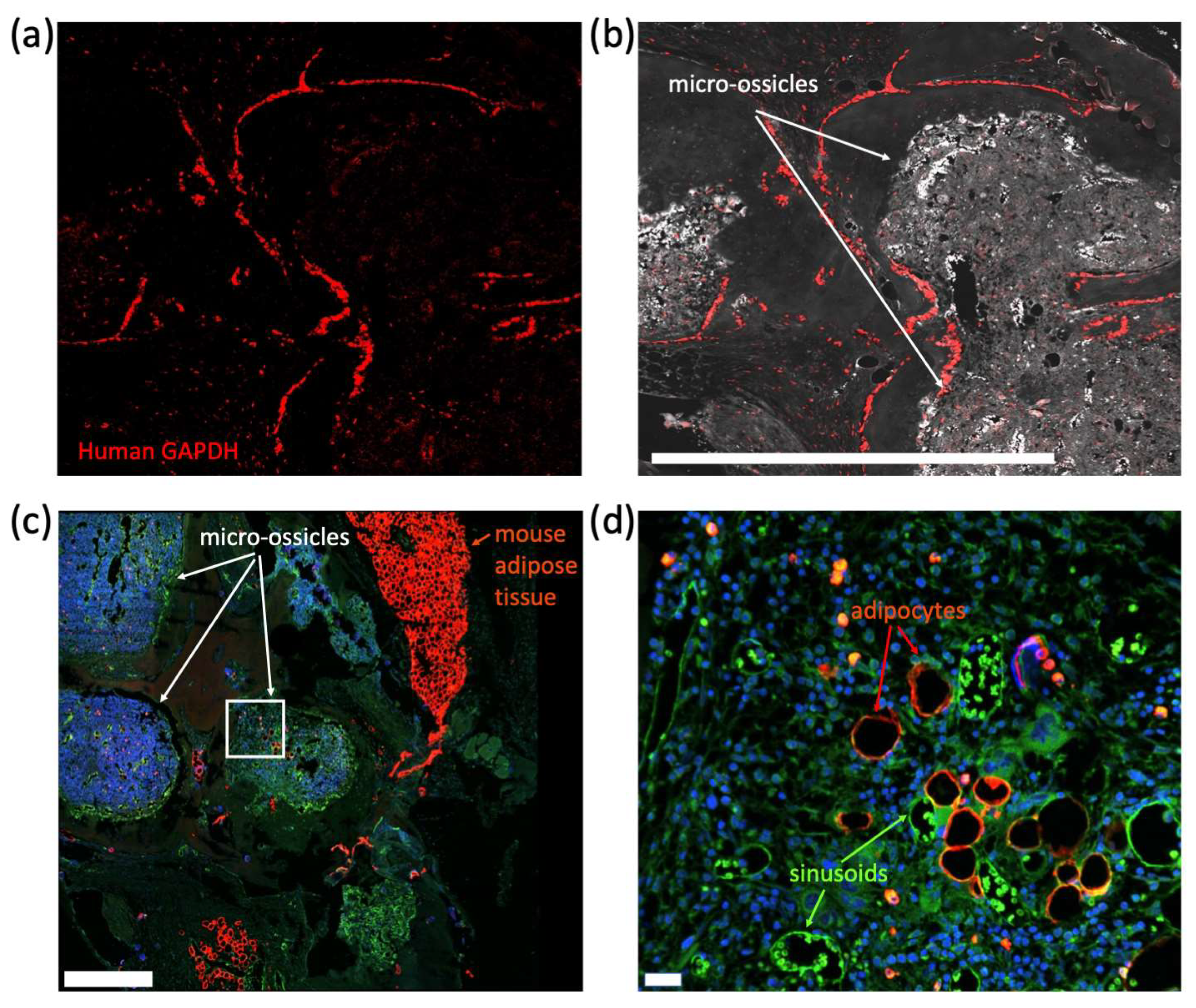
35], followed by secondary anti-rat IgG HRP-linked antibody (Cell Signaling Technology, Danvers, MA, USA) for 30 min at room temperature and then Opal 690 dye (Akoya Biosciences, Marlborough, MA, USA) for 10 min at room temperature. To identify human cells in micro-ossicle sections, tissues were stained using an RNAScope probe for human GAPDH (hGAPDH) as per the manufacturer’s instructions (Advanced Cell Diagnostics; Fluorescent Reagent Kit V2 and proprietary hGAPDH, which did not cross-react with the mice). Fluorescence was developed using Opal 690 dye. Fluorescence imaging was captured using a Dragonfly 200 Confocal microscopy system (Oxford Instruments, Abingdon, UK) with a mechanical stage and Micro-Manager software [
36] with the ImageJ plugin [
37]. Picrosirius Red Staining and polarized light were used to view collagen fibril organization in micro-ossicles. The Picrosirius Red Stain Kit (Polysciences Inc., Warrington, PA, USA) was applied to tissue sections as recommended by the manufacturer. Brightfield and polarized light images were captured using an Axio Imager 2 (ZEISS, Baden-Württemberg, Germany) equipped with a polarizing filter.
Statistical Analysis: For engraftment comparisons, statistical analysis was performed using the Wilcoxon matched-pairs signed rank test in GraphPad Prism version 8 (Boston, MA, USA). p-values of 0.05 or less were considered statistically significant. Each mouse is represented as a separate point on the graphs to make data and analysis as transparent as possible.
4. Discussion
Mouse bone marrow is frequently used to characterize human hematopoietic stem cells [
39], to study hematopoiesis [
40], or to study cancers forming from human blood stem cells or human cancers known to metastasize to the bone marrow [
41,
42]. Despite the utility of mouse marrow, species–species mismatches between human and mouse cells make mouse bone marrow an imperfect tool [
18]. This has motivated the development of a number of so-called humanized marrows formed in mice from human cells [
15,
16,
17,
18,
19,
20]. These human bone marrow models are purported to better maintain human hemopoietic stem progenitor cell quiescence [
43], improve establishment of malignant human hematopoietic populations that are challenging to engraft into mouse marrow [
15,
16,
17,
18], and facilitate better modeling of human cancers that metastasize to the bone and marrow in human patients [
19,
20]. Some groups have formed large humanized marrows using scaffolds [
19,
20] or gels [
16], while other groups have used multiple smaller microtissues [
12,
21,
43,
44]. Because all the purported input tissues require remodeling in vivo to yield a humanized marrow, we reasoned that it may be worthwhile to view the input into the process as a tissue unit, where if more bone/marrow units are required, more input tissue units can be added. Relative to using large scaffold units, a benefit of using microtissues is that the unit size and composition can be easily standardized by controlling the number of cells used to generate each microtissue. A benefit of small diameter microtissue units is that they can be sized such that they do not exceed in vitro mass transport limitations, enabling the extended culture required to guide cell fate for optimal marrow formation [
45,
46,
47]. A weakness of the microtissue approach is that handling multiple microtissue units can become unwieldly. In an effort to facilitate the handling and scaling of microtissues for the formation of humanized bone marrow ossicles, we developed the methods described herein.
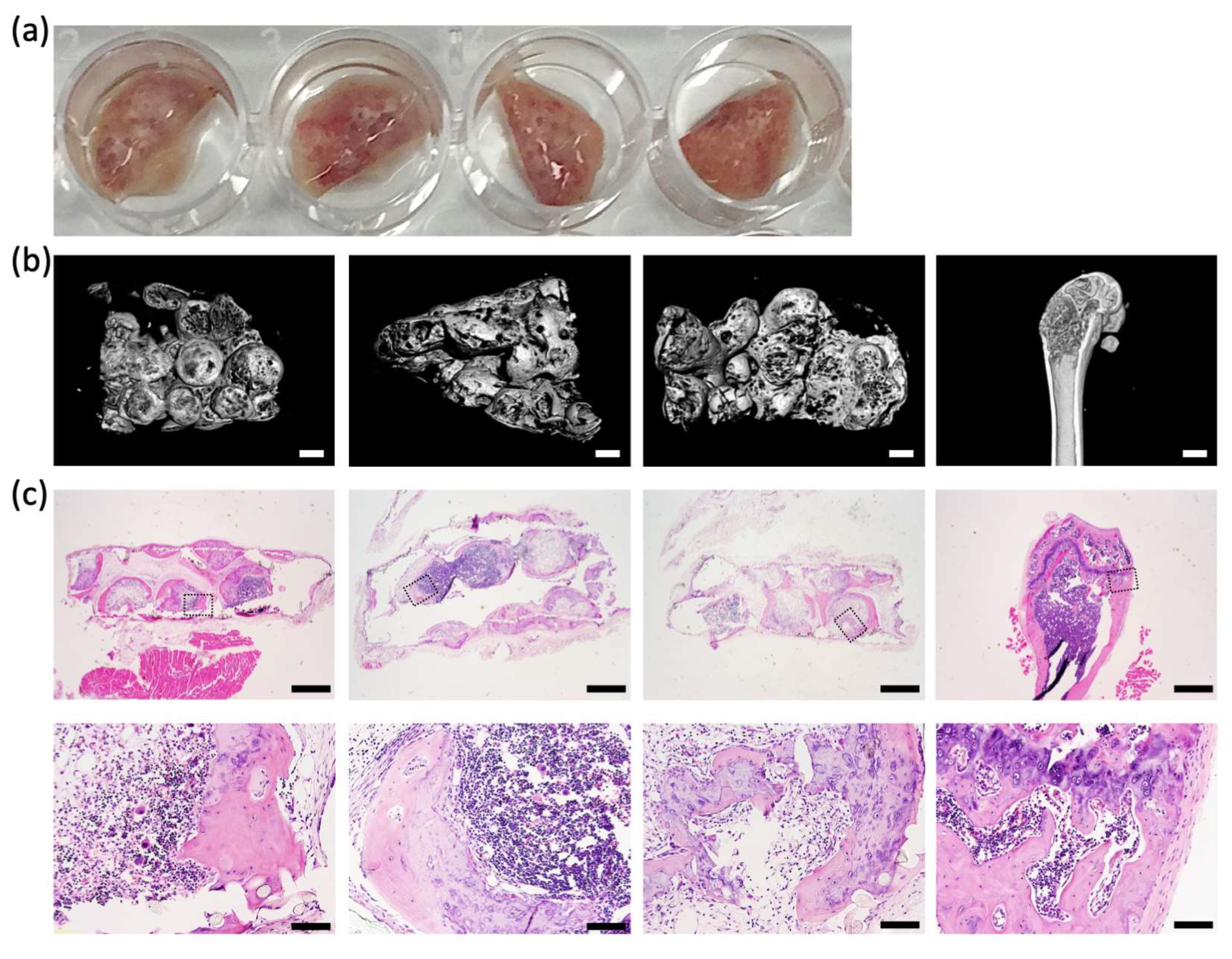
Using a microwell platform, arrays of ~50 microtissues, each assembled from 3 × 10
5 human BMSCs, were organized onto sheets of nylon mesh. Microtissue extracellular matrix formed around the nylon mesh fibers, firmly anchoring each microtissue to the mesh. The array of microtissues could be easily manipulated, and microtissue arrays were transferred to standard 6 well plates for the majority of the culture period and then implanted into NSG mice. After 8 weeks of subcutaneous incubation in mice, microtissues were remodeled into micro-ossicles containing a mineralized shell of bone-like tissue and a bone marrow core. The mineralized bone-like shell was evident in microCT images, and the bone marrow structure evident in the histological sections of micro-ossicles. The observed remodeling paralleled previous studies that reported the mineralization of microtissues in the first 3–4 weeks following implantation in mice, followed by progressive replacement of the cartilage/bone-like core with marrow stroma [
10,
12,
13,
14]. In micro-ossicles, we observed the development of mineralized tissue reminiscent of trabecular and cortical bone, similar to a previously reported study by Bianco’s team [
12].
We conditioned mice carrying micro-ossicle arrays with sublethal irradiation and transplanted animals with human umbilical cord blood-derived CD34
+ cells. After 8 weeks, human hematopoietic cell content in the mouse marrow and micro-ossicles was compared. While overall hCD45
+ cell content was greater in the humanized micro-ossicles than in the mouse marrow, human CD34
+ cell content was greater in the mouse marrow than in the humanized micro-ossicles. We observed a similar percentage of CD34
+ cells in the mouse bone marrow as in previous studies where human hematopoietic cells were transplanted via intravenous injection (up to ~15% of all human CD45 cells in the marrow [
30]). The reduced number of human CD34
+ progenitor cells in the humanized micro-ossicles, relative to the mouse marrow, paralleled a previous study that reported fewer cycling human HSPC in humanized ossicles than in the matched mouse bone marrow [
43]. Human HSPC are known to cycle, rather than maintain quiescence, in mouse marrow [
43], resulting in elevated numbers of human HSPC in mouse marrow. Fewer CD34
+ HSPC in the humanized micro-ossicles, relative to matched mouse marrow, likely reflects the reduced cycling of the human progenitor cells in micro-ossicles. In this sense, the behavior of human HSPC in the humanized micro-ossicles better replicated physiological behavior than the human HSPC in the matched mouse marrow. Thus, as has been previously demonstrated [
43], humanized marrow appears to offer a more physiological mimic than mouse marrow.
How to best direct human BMSCs to form an ossicle in mice is unresolved. Dupard and colleagues provided a nice review of progress in the development of humanized ossicles and the range of technologies being explored [
26]. They made the interesting statement that “Overall, the current limitations of hOss primarily lie in the lack of characterization and the need for standardization.” We agree, noting that there are many in vitro priming protocols that have been used to generate microtissue precursors for micro-ossicles [
12,
21,
43,
44], as well as other protocols that have been used to prime and organize human BMSCs to form ectopic marrows in mice [
15,
16,
17,
18,
19,
20,
43]. While there are several methodologies and putative input cell populations, these have not been compared. A lack of comparative studies likely reflects the fact that tools used to generate ectopic marrows are either low-throughput or are suboptimal for side-by-side comparison, and the fact that this is a relatively new area of research. We do not claim to have provided the “best” humanized marrow model, but instead argue that the comparative analysis of methods is an important next step in the development of technologies in this field. The proposed microtissue to micro-ossicle array could be used to standardize methods and may offer a relatively efficient method to compare different priming protocols or input cell types. With a modified cell-seeding cassette, it may be possible to establish micro-ossicles from different cell compositions on the same mesh carrier. Using such an approach, it may be possible to have an array of tissues, where each of the tissues could be tailored to represent a slightly different niche composition or disease phenotype.
Previous studies demonstrated that when BMSC microtissues are implanted into mice, they are remodeled to form micro-ossicles [
12,
21,
43,
44], and these micro-ossicles can provide a superior bone marrow model, relative to mouse marrow. The microtissue or micro-ossicle unit on its own is small, requiring that multiple units be used for most studies. Herein, we outlined a simple and efficient strategy to generate multiple microtissues, anchored in an organized array on a nylon mesh that is easy to handle. The microtissue array can be easily implanted into mice and harvested as a unit, providing a viable method to scale up microtissue/micro-ossicle studies. This method could be adapted to study the engraftment of leukemic cells, the metastasis of cancer cells, or could be exploited to anchor micro-ossicles to facilitate the live animal imaging of cell homing. Finally, the concept of organizing arrays of microtissues on a mesh, or similar delivery device, could be adapted to other applications where controlled arrays of microtissues would be useful for research or therapy. For example, this concept could be adapted to deposit arrays of intestinal organoids on mesh scaffolds to generate tubular intestinal structures or arrays of hair follicle organoids for cosmetic applications.
A limitation of our study is that only a single BMSC donor was used; this was due to the relocation and closing of our laboratory in Australia. Nevertheless, the number of BMSC per tissue and the differentiation protocols used in our study parallel those previously described by Paolo Bianco [
12], which used BMSC from nine pediatric and seven adult donors. These studies [
12], and a study by Farrell and colleagues using three BMSC donors [
14], showed that chondrogenically induced pellets reliably form micro-ossicles in vivo. In addition to replicating these studies with multiple BMSC donors, there may be merit in analyzing each of the individual micro-ossicles separately, to understand the variability between micro-ossicles. The focus of our study and the data contained in this paper demonstrate the feasibility of generating arrays of microtissues, culturing these tissues extensively in vitro, and then implanting these arrays in vivo—an elegant strategy to scale previously described methods.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







