1. Introduction
Over the last decades, we have seen a decrease in the number of new medicines introduced to the market. This is primarily due to the high costs of research and the length of the various stages of new substance testing [
1]. Only 48 drugs have been approved for use by the Food and Drug Administration (FDA) in 2019 [
2] and another 53 in 2020 [
3]. Moreover, 2018 is considered to be one of the record-breaking years, where this number reached a value of only 62 [
2]. Despite the constant development of medicine, the number of new medicines and the indications for which they can be applied is still insufficient to provide effective treatment for any condition. In order to improve the effectiveness and quality of pharmacotherapy, researchers are increasingly emphasizing so-called repurposing (or repositioning, reprofiling, or re-tasking), i.e., searching for new indications for drugs already in use [
4]. The method requires activities within two areas of drug development: in silico studies, based on drug-drug and drug-target interactions, and experimental methods, including in vitro and in vivo assessments. Connecting these two areas provides an opportunity to reduce the costs associated with introducing a new treatment method and significantly accelerate the process. Repositioning is developing for a multitude of reasons. Firstly, the drug, which has already been authorized for marketing, has undergone all safety tests, has passed all phases of clinical trials, and therefore the risk of causing adverse effects is reduced significantly. Frequently, the most suitable form of administration is already known, which significantly speeds up the whole process of introduction of the drug medication. Certainly, the costs of phase I and II clinical trials are reduced. Additionally, this method offers the possibility of discovering new therapeutic targets for investigational drugs. All the above arguments prove that repurposing brings many benefits for both patients and pharmaceutical companies [
5].
The well-known example of such a drug is sildenafil, which was originally tested for the treatment of angina pectoris, but turned out to have more measurable effects when used as an erectile dysfunction drug. Another example is minoxidil, which was originally designed to treat ulcers but has been applied as a hair growth drug [
6]. The utilization of this method also found application in March 2020, when the COVID-19 pandemic caused by the SARS-CoV-2 virus was announced by the World Health Organization (WHO). Among the drugs used
off-label were CQ (
N′-(7-chloroquinolin-4-yl)-
N,
N-diethylpentane-1,4-diamine, trade name: Aralen; Arechin) and HCQ (trade name: Plaquenil). Both CQ and HCQ were approved by the FDA for the use of the abovementioned virus in emergency situations [
7]. These pharmacotherapeutics have long been of considerable interest to the scientific community due to the phenomenon of hormesis observed during therapy with these drugs [
8]. A multitude of scientific studies have confirmed that both these drugs are highly effective in controlling the infection caused by SARS-CoV-2, reducing the development of pneumonia and thus shortening the duration and intensity of the disease [
7]. However, on 15 June 2020, the FDA decided to withdraw these drugs from emergency use authorization (EUA) for Covid-19 therapy due to the risk of cardiotoxicity [
3]. This news caused a stir among patients and scientists alike. The main source of controversy was the FDA’s ambiguous and shifting stance on the use of CQ and HCQ. In addition, the mechanism of the observed prolongation of the QT interval of the action potential following administration of CQ and HCQ has not been clearly defined. Nevertheless, some researchers suggest that one of the possible causes of this phenomenon may be the influence of these drugs on the activity of cardiac ion channels [
9,
10].
Ion Channels
Ion channels are protein molecules dispersed in the cell membrane, whose role is to transport ions through a lipid bilayer. Special attention of scientists is focused on
voltage-gated ion channels (VGIC), which have been among the most common molecular drug targets [
11], due to the fact that the risk of cardiotoxicity has been a frequent reason for withdrawal of drugs from the market. In 2005, studies were undertaken to explain the causes of cardiotoxicity. This extensive research, as the source of the two most commonly reported disorders, i.e., torsade de pointes and QT elongation of action potentials, has shown blocking of the potassium heart channel, which is encoded by the human ether-related gene (hERG gene). This allowed for the inclusion of new drugs in the basic safety assessment process of hERG blocking studies which consequently significantly limited drug withdrawal for this reason [
12]. Nevertheless, the new guidelines on drug cardiotoxicity studies proved that not only hERG but other VGIC channels are the site of drug action, and disturbances of VGIC-drug interaction may be a probable cause of cardiotoxicity development. The most important ion channels proposed in the comprehensive in vitro pro-arrhythmia test (CiPA) include: K
V11.1 (hERG), Na
V1.5-late and Ca
V1.2, K
V4.3, K
VLQT1/mink and K
ir2.1. Their influence on the repolarization and depolarization of the heart action potential (AP) has been confirmed and proven in numerous tests covering the physiological state and pathological state of the human organism [
13]. However, the first three channels are considered the most crucial in assessing the risk of cardiotoxicity.
2. Methodology
It should be emphasized that the safety requirements for medicines have become more stringent over the years. At the same time, new methods of computational chemistry, based on the structure of drugs and their similarity to already known active molecules, allow early identification of potential adverse interactions.
In Silico Methods
In this work, the in silico methods were applied to answer the question as to whether CQ and HCQ can interact with voltage-gated ion channels in the heart. ProTox-II [
14] and AdmetSAR software [
15,
16] were used to calculate the probability of induction of toxicity in selected areas of activity. The mechanisms of action of the studied drugs in the cardiovascular and nervous systems are underlined. In order to compare the cardio and neurotoxicity of the investigated drugs, their full safety profiles were tested.
Using the ProTox-II software, one can determine the properties of an already known molecule structure by using its name or drawing the structure. The calculations included in this study present the results of the oral toxicity predictions. A full safety assessment report is also prepared based on the likelihood of toxicity or non-toxicity in a particular activity model.
According to statistical studies, one of the most common reasons for suspension or withdrawal of drugs from the market is the risk of cardiotoxicity and neurotoxicity [
17]. Unfortunately, one of the drawbacks of the ProTox-II program is lack of information on potential neuro- and cardiotoxic effects of calculated compounds. Therefore, in the present analyses, the AdmetSAR program was used to test the safe application of test compounds on the heart and neuronal system. It is worth emphasizing that the computational algorithms of both programs are very similar [
15]. In both algorithms, the results of toxicity probability are yielded from a comparison of the test molecule with data collected in available compound databases and literature containing 717 toxic molecules (IC
50 < 30 µMol) and 216 non-toxic particles [
18]. The specific toxicity model is created by using an algorithm called AtomPairs coded with the appropriate vector notation. It allows one to assess a drug administration safety profile, based on the chemical structure of the compounds. Full assessment of the pharmacological safety profile of the studied drugs is possible by combining results of ProTox-II and AdmetSAR. The resulting data are shown in
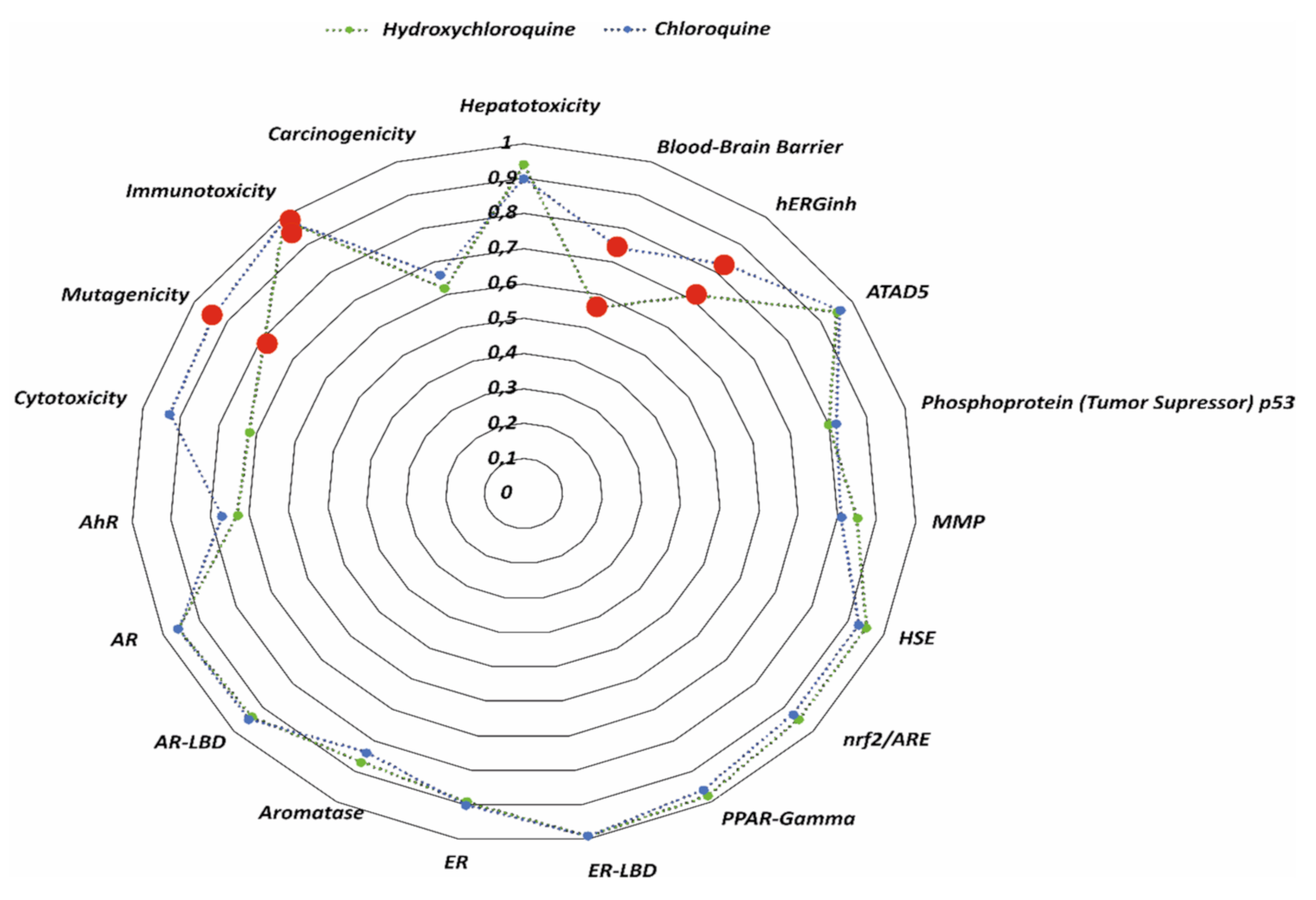
Figure 1 as juxtaposition of the toxicity profiles of studied compounds with the average values of the probability of inactivity for substances in the considered toxicity class of the model/molecule.
Both drugs have been reported to be active at the same toxicity levels/types. The pharmaceuticals exhibit immunotoxic properties with a 99% probability. The study indicates that the risk of mutagenicity for CQ is higher (94%) compared to HCQ (79%). Probability results obtained using AdmetSAR program provided information on the inhibition of the hERG gene. The probability of QT prolongation is: 83% for CQ and 71% for HCQ. Moreover, the substances are highly likely (74% for CQ and 68% for HCQ) to pass through the blood-brain barrier (BBB). Attention should also be paid to the risk of carcinogenicity—calculations show that both drugs should not participate in tumorigenesis. However, the probability of no toxicity in this area is for CQ only 66%, while for HCQ it is 62%.
3. Discussion
The in silico studies conducted within this project provide a preliminary view of the potentially toxic areas of action of CQ and HCQ. The data are particularly important in changing the pharmacotherapeutic position of drugs, i.e., discovering new or extended uses of clinically available drugs. Of course, one should remember that this type of research cannot replace animal testing, but it can significantly limit their number. By modulating cell excitability and altering action potentials, quinoline derivatives have been reported to affect excitable cells (both neuronal and cardiac). In our discussions, we decided to confirm the result of the neuro- and cardiotoxicity of the tested derivatives.
3.1. Neurotoxicity
Tests showed that quinoline derivatives easily pass through the blood-brain barrier, which was confirmed by literature data [
19]. This ability allows the tested drugs to exert effects in the nervous system. Studies have demonstrated that CQ and HCQ affect the secretion and effect of neurotransmitters such as serotonin [
20] and adenosine [
21]. These compounds can also affect opioid receptors; this is related to the possibility of drug accumulation in the central nervous system (CNS). Dysfunction of these neurotransmitters may lead to abnormal communication between cells. Additionally, inhibition of the neurotransmitter γ amino butyric acid (GABA) may lead to brain dysfunction (encephalopathy) [
22].
The action of CQ in the nervous system is also manifested by the effects of lowering and raising the convulsive threshold. According to the collected data, in low doses (1.0–5.0 mg/kg) the drug increases the seizure threshold, but in high doses (10.0–50.0 mg/kg) it has a pro-convulsant effect. According to data, this effect may be related to the influence of CQ on the opioid system [
23,
24].
Adverse effects after CQ intoxication are manifested by extrapyramidal nervous system symptoms. These include ataxia, dysphonia and involuntary upward tilt of the eyes (oculogyric dystonia). Most of the symptoms resulting from CQ inhibition are reversible, but chronic neurological disorders such as temporal lobe epilepsy and dysautonomia may develop. Most neurological symptoms disappear when QT is discontinued, but studies in animal models have reported that CQ can cause permanent brain and brainstem damage [
23].
Free passage through the blood-brain barrier also made it possible to use CQ as a neuroprotective agent. Very low concentrations of CQ can induce GM1-lipid gangliosides, which are an integral part of the nervous tissue, with proven neuroprotective effects. It has also been proven that the neuroprotective effect may result from the influence of CQ on sigma-1 receptors [
25].
3.2. Cardiotoxicity
In silico methods conducted for cardiotoxicity showed that the drugs are toxic to the heart with high probability. Cardiovascular side effects following CQ/HCQ use include cardiac arrhythmia, vasodilation and hypotension [
26]. The mechanism of cardiac dysfunction is related to prolongation of the QT interval of the action potential due to blockade of the hERG gene. Repolarization of the action potential is inhibited, which results in blocking the delayed rectifier potassium current (I
kr). Molecular modeling studies have shown that the inhibitory potency value of hERG is IC
50 = 4.56 µM in the case of CQ racemic form, while for the HCQ racemate it is approx. 3-fold lower (IC
50 = 12.8 µM) [
27].
Additionally, it has been proven that CQ and HCQ influence HCN channels regulating the activity of neurons (HCN1, 2, 4) and cardiomyocytes (HCN3) [
28,
29]. Consequently, the investigational drugs may affect both myocardial contractility and heart rhythm. It is also worth emphasizing that the literature data confirm the influence of the studied drugs on another channel proposed by CIPA, i.e., K
ir2.1 channel [
28,
30].
Previously presented molecular modeling data showed that the inhibitory potency value reflecting the CQ blocking value of IK1 is IC50 = 0.69 ± 0.09 µM. The data confirm high probability of CQ cardiotoxicity.
4. Conclusions
The combination of preliminary in silico tests with cell-line-based methods and validation of results in vitro provides a high probability that the test substance can be used by humans. However, it should be noted that these results cannot be the only way to analyze the compound in terms of safety. The decision to limit the use of CQ and HCQ seems to be correct, because the administration of these drugs may cause neurological and cardiovascular disorders. Nevertheless, it is worth conducting research on the reasonable dosage of the abovementioned preparations, because most in silico tests do not take into account such parameters as: dose of the substance, the rate of metabolism or elimination of a specific chemical, which are of key importance in the treatment process. Additional assays, such as molecular modeling, are recommended for a more accurate in silico assessment of interactions between ion channels and test drugs.
However, presented comparison of CQ and HCQ toxicity revealed that the toxicity probability values for both tested drugs were high. Additionally, CQ is more toxic than HCQ. Considering high neurotoxicity and cardiotoxicity of these drugs, it seems peculiar as to why the FDA decided to approve them for use during the COVID-19 pandemic so quickly. As the presented research proves, even simple in silico tests provide information about serious side effects of using CQ and HCQ.
Author Contributions
M.K., Ł.F. and A.N. conceived and directed the project, designed the study, collected the data, analyzed the data, interpreted the results, and wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kedzierska, E.; Dabkowska, L.; Krzanowski, T.; Gibula, E.; Orzelska, J.; Wujec, M. New drugs—From necessity to delivery. Curr. Issues Pharm. Med. Sci. 2018, 31, 69–75. [Google Scholar] [CrossRef] [Green Version]
- Asher, M. 2019 FDA drug approvals. Nat. Rev. Drug Discov. 2020, 19, 79–84. [Google Scholar]
- Coronavirus (COVID-19) Update: FDA Revokes Emergency Use Authorization for Chloroquine and Hydroxychloroquine. Available online: https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-revokes-emergency-use-authorization-chloroquine-and (accessed on 10 April 2020).
- Hernandez, J.J.; Pryszlak, M.; Smith, L.; Yanchus, C.; Kurji, N.; Shahani, V.M.; Molinski, S.V. Giving Drugs a Second Chance: Overcoming Regulatory and Financial Hurdles in Repurposing Approved Drugs As Cancer Therapeutics. Front. Oncol. 2017, 7, 273. [Google Scholar] [CrossRef] [Green Version]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Simsek, M.; Meijer, B.; van Bodegraven, A.A.; de Boer, N.; Mulder, C.J. Finding hidden treasures in old drugs: The challenges and importance of licensing generics. Drug Discov. Today 2018, 23, 17–21. [Google Scholar] [CrossRef]
- Calabrese, E.J.; Hanekamp, J.C.; Hanekamp, Y.N.; Kapoor, R.; Dhawan, G.; Agathokleous, E. Chloroquine commonly induces hormetic dose responses. Sci. Total Environ. 2021, 755, 142436. [Google Scholar] [CrossRef]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef]
- Zheng, Z.; Wu, Y.; Qian, D.; Lian, J. Potential therapeutic drugs for COVID-19 risk prolonging QT interval targeting hERG channel. Authorea 2020, 1–20. [Google Scholar] [CrossRef]
- Szekely, Y.; Lichter, Y.; Abu Shrkihe, B.; Bruck, H.; Oster, H.S.; Viskin, S. Chloroquine-induced torsades de pointes in a patient with coronavirus disease 2019. Heart Rhythm. 2020, 17, 1452–1455. [Google Scholar] [CrossRef] [PubMed]
- Dunlop, J.; Bowlby, M.; Peri, R.; Vasilyev, D.; Arias, R. High-throughput electrophysiology: An emerging paradigm for ion-channel screening and physiology. Nat. Rev. Drug Discov. 2008, 7, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration; US Department of Health and Human Services. Guidance for Industry: E14 Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non-Antiarrhythmic Drugs; Center for Drug Evaluation and Research Food and Drug Administration: Rockville, MD, USA, 2005; pp. 61134–61135. [Google Scholar]
- Kowalska, M.; Nowaczyk, J.; Nowaczyk, A. KV11.1, NaV1.5, and CaV1.2 Transporter Proteins as Antitarget for Drug Cardiotoxicity. Int. J. Mol. Sci. 2020, 21, 8099. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Lou, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. admetSAR 2.0: Web-service for prediction and optimization of chemical ADMET properties. J. Bioinf. 2018, 35, 1067–1069. [Google Scholar] [CrossRef]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. admetSAR: A Comprehensive Source and Free Tool for Assessment of Chemical ADMET Properties. J. Chem. Inf. Model. 2012, 52, 3099–3105. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Ridder, B.J.; Han, X.; Wu, W.W.; Sheng, J.; Tran, P.N.; Wu, M.; Randolph, A.; Johnstone, R.H.; Mirams, G.R.; et al. Assessment of an In Silico Mechanistic Model for Proarrhythmia Risk Prediction Under the Ci PA Initiative. Clin. Pharmacol. Ther. 2019, 105, 466–475. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Zhang, C.; Chen, Y.; Li, X.; Zhuang, S.; Li, W.; Liu, G.; Lee, P.W.; Tang, Y. In silico prediction of chemical aquatic toxicity with chemical category approaches and substructural alerts. Toxicol. Res. 2015, 4, 452–463. [Google Scholar] [CrossRef]
- Dow, G.S.; Milner, E.; Bathurst, I.; Bhonsle, J.; Caridha, D.; Gardner, S.; Gerena, L.; Kozar, M.; Lanteri, C.; Mannila, A.; et al. Central nervous system exposure of next generation quinoline methanols is reduced relative to mefloquine after intravenous dosing in mice. Malar. J. 2011, 10, 150. [Google Scholar] [CrossRef] [Green Version]
- Thompson, A.J.; Lochner, M.; Lummis, S.C.R. The antimalarial drugs quinine, chloroquine and mefloquine are antagonists at 5-HT3 receptors. Br. J. Pharmacol. 2007, 151, 666–677. [Google Scholar] [CrossRef]
- Weiss, S.; Benwell, K.; Cliffe, I.; Gillespie, R.; Knight, A.; Lerpiniere, J.; Misra, A.; Pratt, R.; Revell, D.; Upton, R.; et al. Discovery of nonxanthine adenosine A2A receptor antagonists for the treatment of Parkinson’s disease. Neurology 2003, 61, S101–S106. [Google Scholar] [CrossRef]
- Maxwell, N.M.; Nevin, R.L.; Stahl, S.; Block, J.; Shugarts, S.; Wu, A.H.B.; Dominy, S.; Solano-Blanco, M.A.; Kappelman-Culver, S.; Lee-Messer, C.; et al. Prolonged neuropsychiatric effects following management of chloroquine intoxication with psychotropic polypharmacy. Clin. Case Rep. 2015, 3, 379–387. [Google Scholar] [CrossRef]
- ARALEN. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/006002s044lbl.pdf (accessed on 10 April 2020).
- Hassanipour, M.; Shirzadian, A.; Boojar, M.M.-A.; Abkhoo, A.; Abkhoo, A.; Delazar, S.; Amiri, S.; Rahimi, N.; Ostadhadi, S.; Dehpour, A.-R. Possible involvement of nitrergic and opioidergic systems in the modulatory effect of acute chloroquine treatment on pentylenetetrazol induced convulsions in mice. Brain Res. Bull. 2016, 121, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Yamamoto, H.; Atta, M.S.M.; Mahmoud, S.; Oh-Hashi, K.; Kiuchi, K. Chloroquine inhibits glutamate-induced death of a neuronal cell line by reducing reactive oxygen species through sigma-1 receptor. J. Neurochem. 2011, 119, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Ben-Zvi, I.; Kivity, S.; Langevitz, P.; Shoenfeld, Y. Hydroxychloroquine: From Malaria to Autoimmunity. Clin. Rev. Allergy Immunol. 2011, 42, 145–153. [Google Scholar] [CrossRef]
- Li, G.; Sun, J.; Li, Y.; Shi, Y.; Zhao, J.; Zhang, T.Y.; Zhang, X. Enantiomers of Chloroquine and Hydroxychloroquine Exhibit Different Activities Against SARS-CoV-2 in vitro, Evidencing S-Hydroxychloroquine as a Potentially Superior Drug for COVID-19. bioRxiv 2020. [Google Scholar] [CrossRef]
- Zhan, X.; Dowell, S.; Shen, Y.; Lee, D.L. Chloroquine to fight COVID-19: A consideration of mechanisms and adverse effects? Heliyon 2020, 6, e04900. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Liu, L.; Shu, Y.-Q.; Jing, P.; Lu, Y.; Zhang, X.-X.; Zong, X.-G.; Guo, L.-J.; Li, C.-J. Blockade of HCN2 Channels Provides Neuroprotection Against Ischemic Injury via Accelerating Autophagic Degradation in Hippocampal Neurons. Neurosci. Bull. 2020, 36, 875–894. [Google Scholar] [CrossRef]
- Jankelson, L.; Karam, G.; Becker, M.L.; Chinitz, L.A.; Tsai, M. QT prolongation, torsades de pointes and sudden death with short courses of chloroquine or hydroxychloroquine as used in COVID-19: A systematic review. Heart Rhythm. 2020, 17, 1472–1479. [Google Scholar] [CrossRef] [PubMed]
| Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).




{kind=link}