
Transcriptomic Diversity of Solanum tuberosum Varieties: A Drive towards Future Analysis of Its Polyploidy Genome †

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.1.1. Potato Cultivars
2.1.2. RNA Extraction and Illumina Sequencing Analysis
2.1.3. Research Data Storage
2.1.4. Computation Services and Analysis Software
2.2. Methods
3. Results
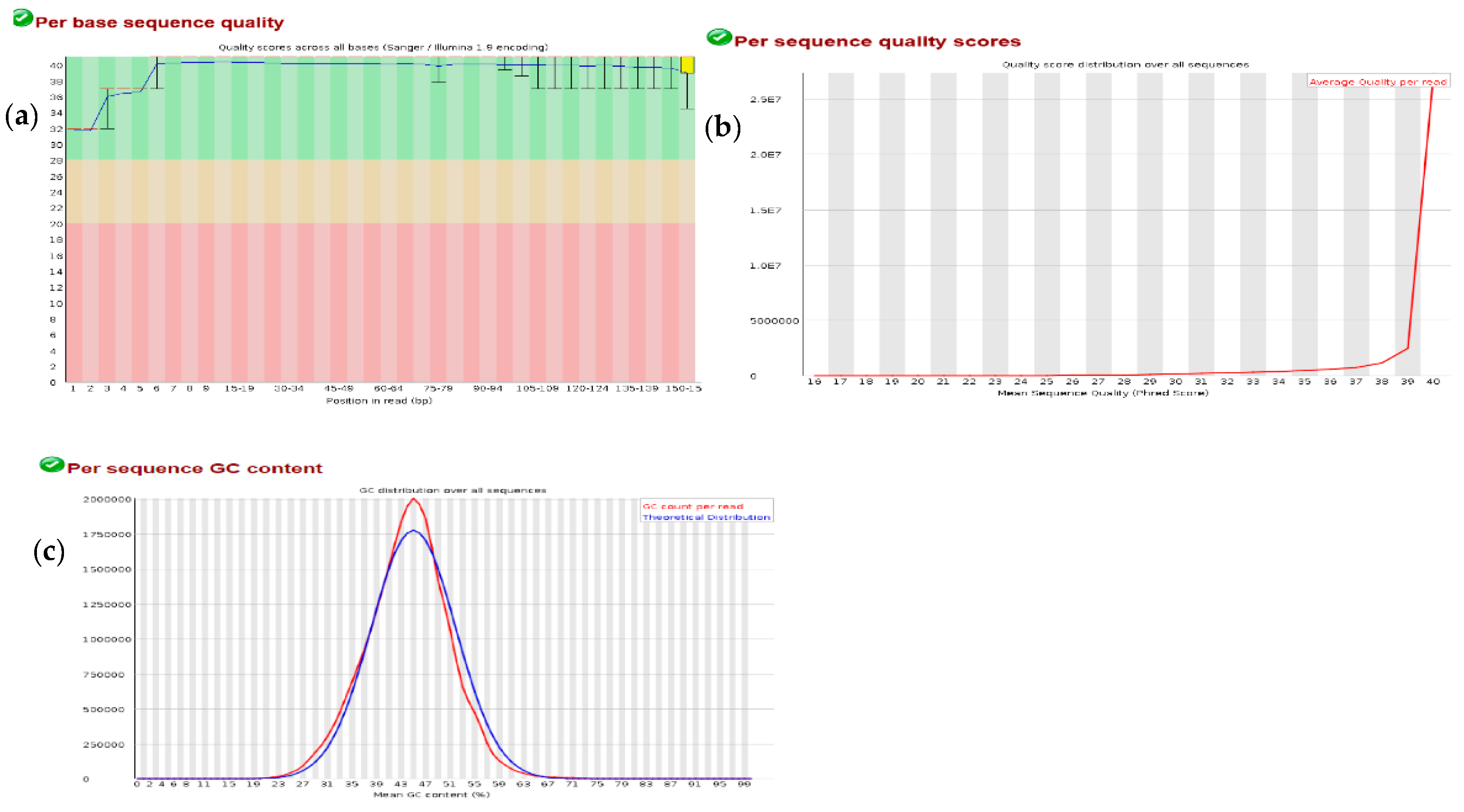
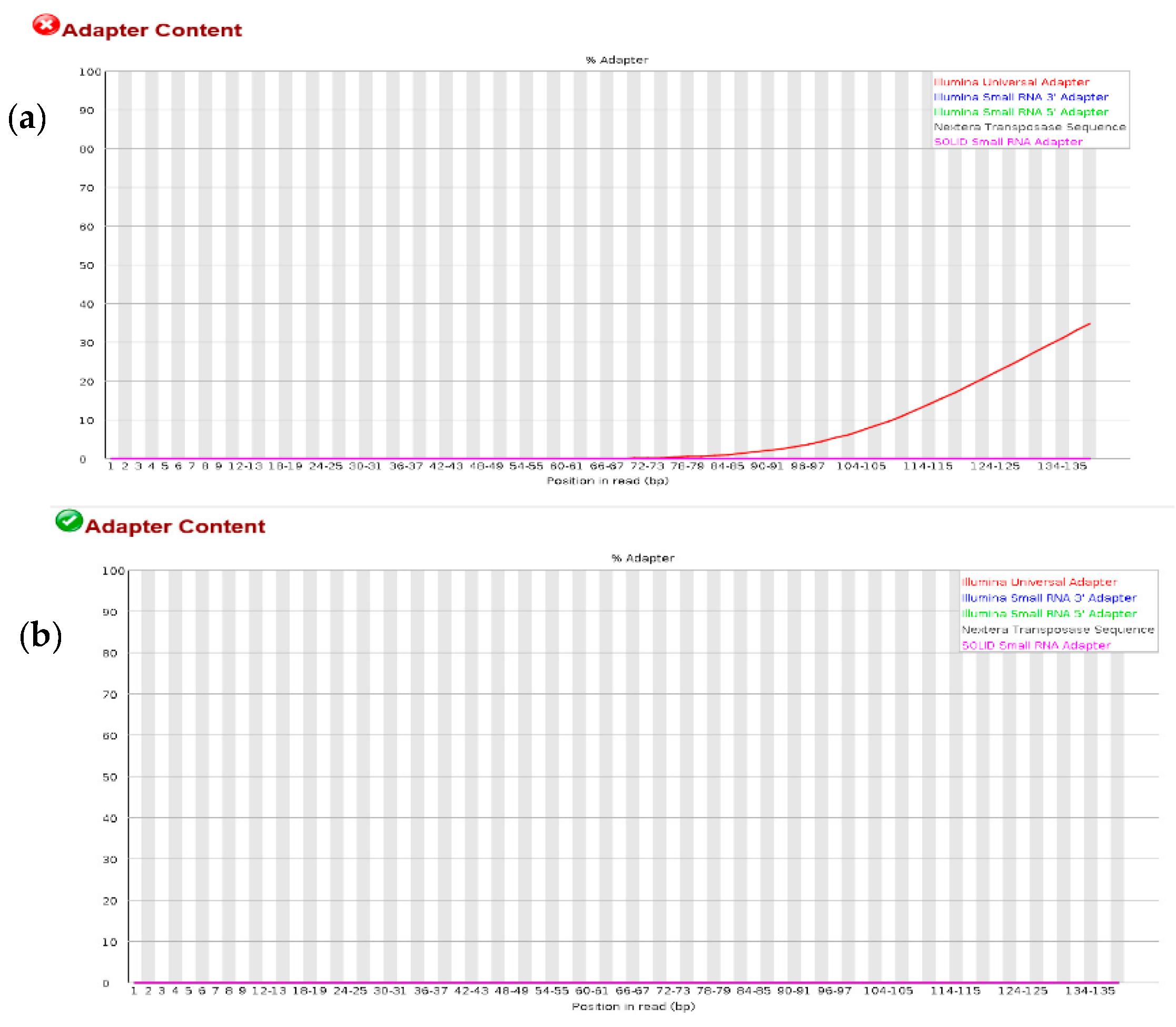
3.1. Quality Check on RNA-Seq Data
3.2. Statistics for Mapping Sequences to Reference Genome
3.3. Top 10 Most Expressed Genes among Potato Varieties
3.4. Number of Similar and Unique Genes among Samples
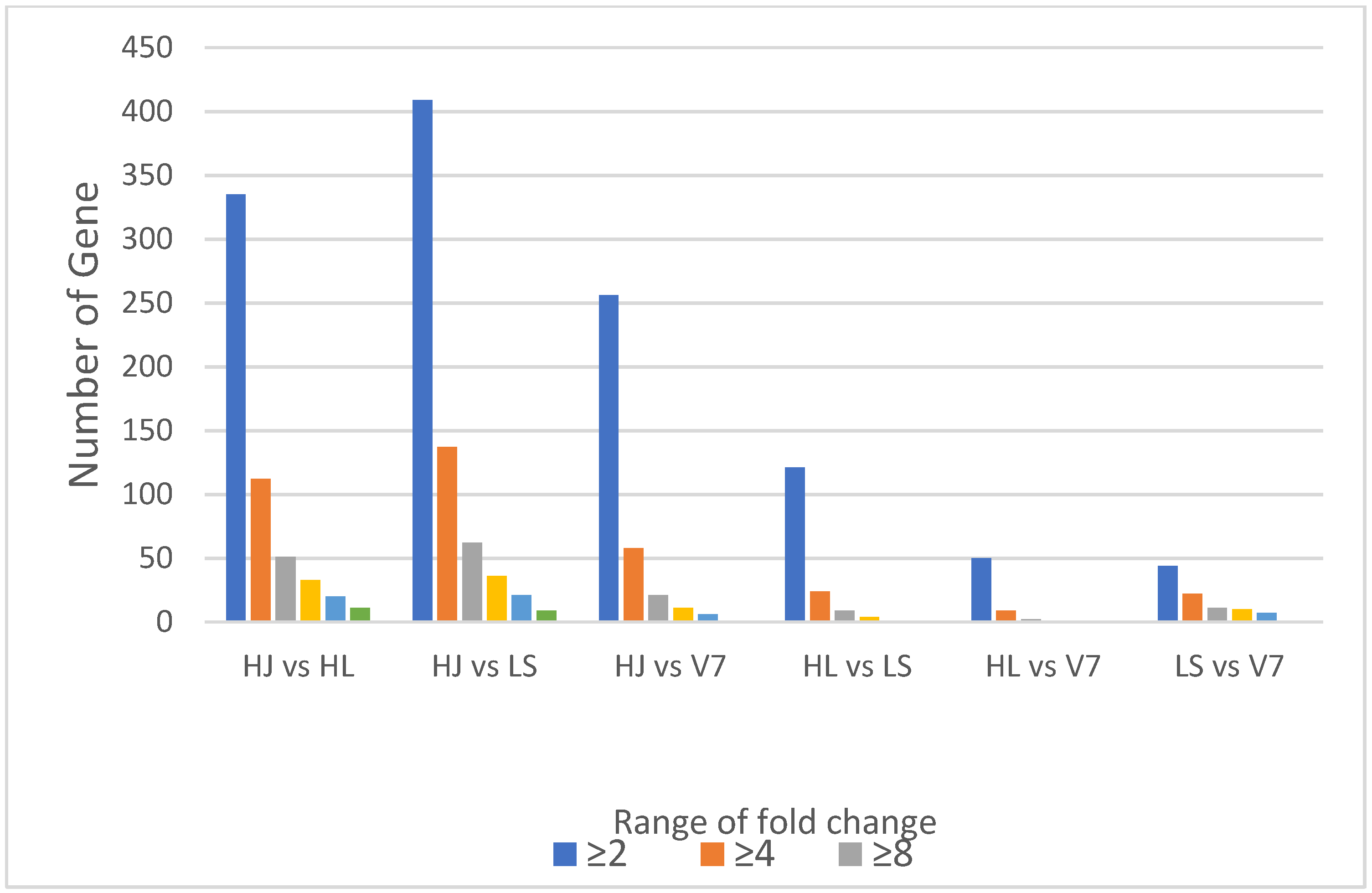
3.5. Fold Change Analysis for Differential Variation in Gene Expression
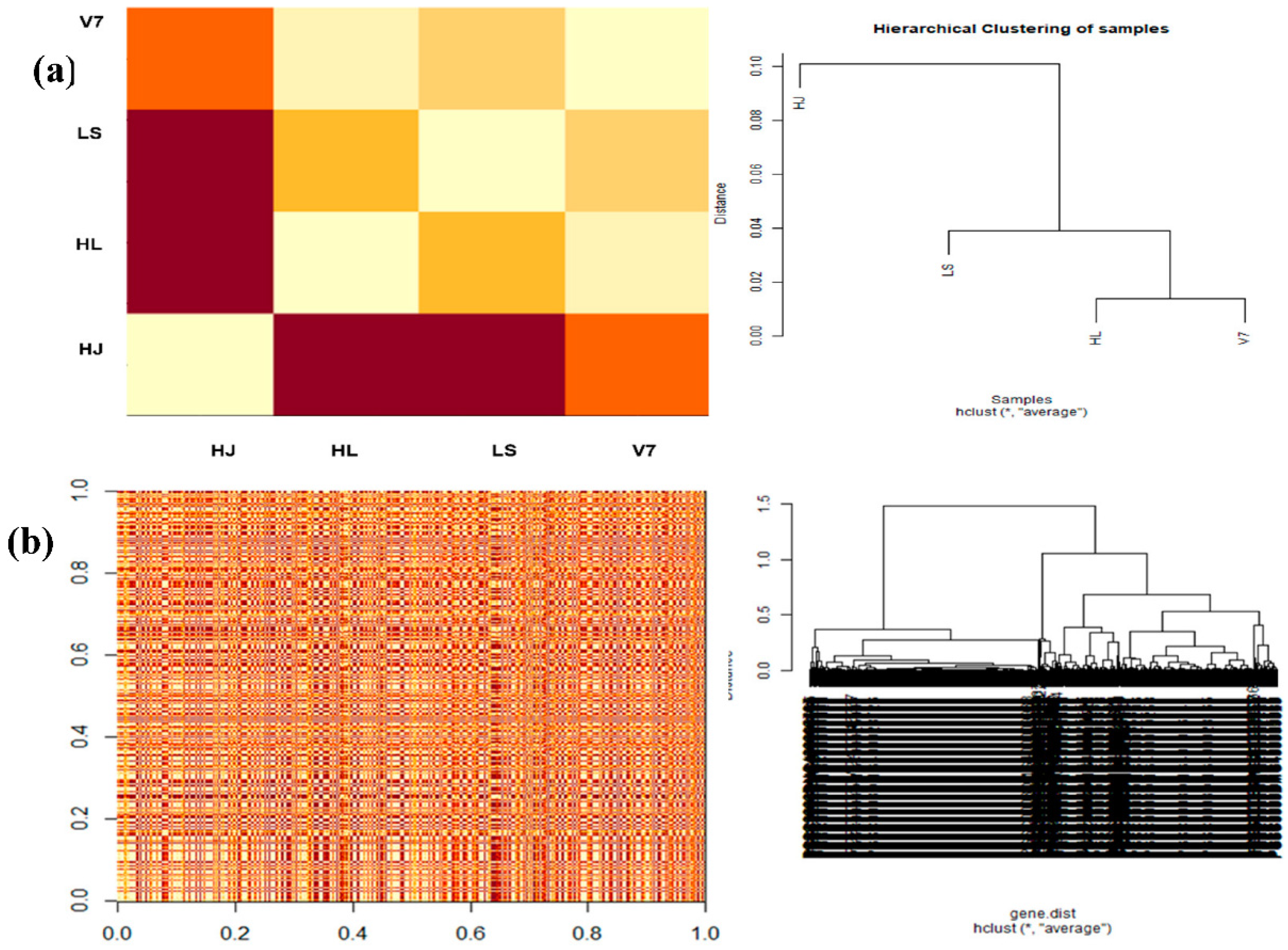
3.6. Estimating Gene and Sample Distance
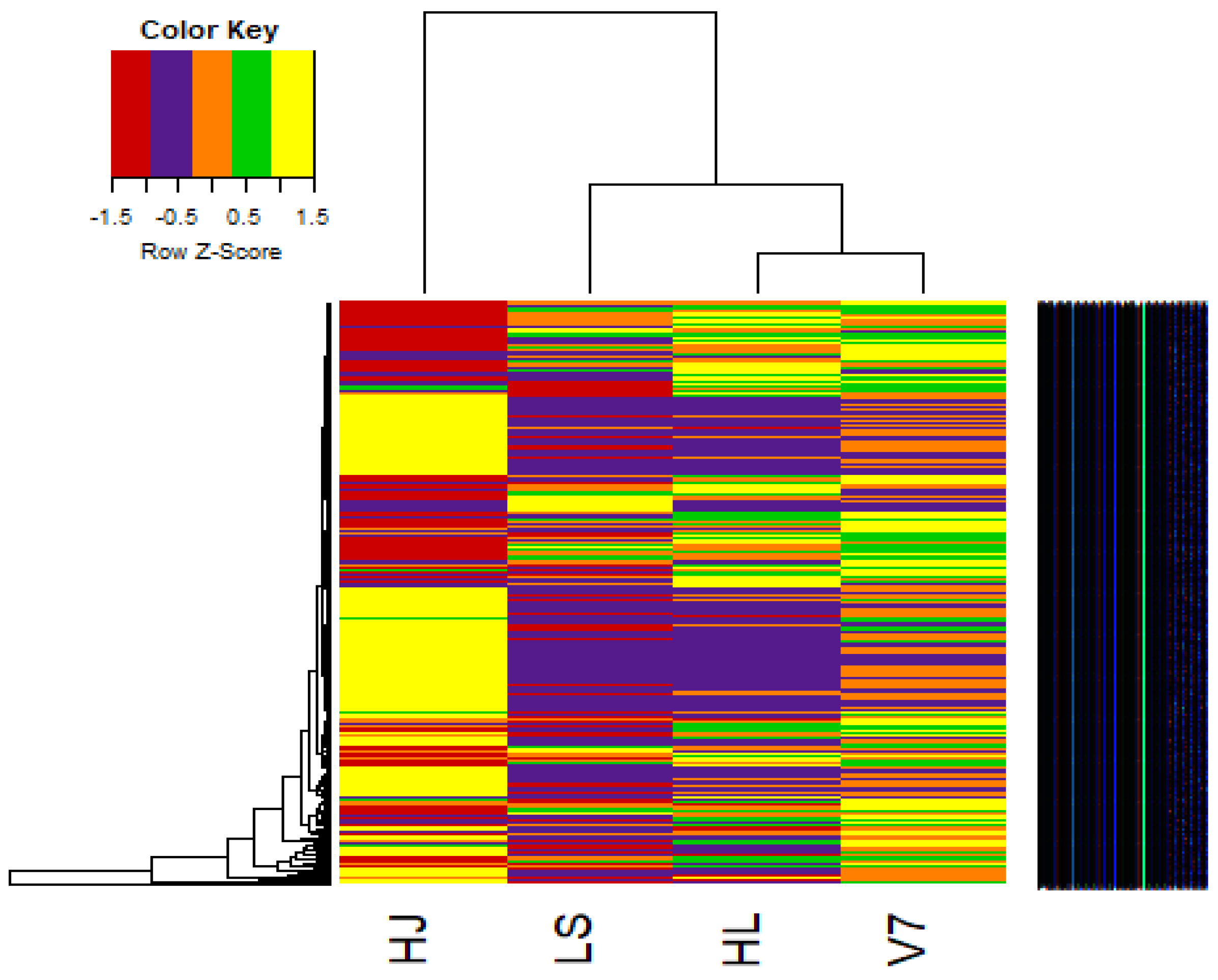
3.7. Heat Maps
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature Count in HJ | Feature Count in HL | Feature Count in LS | Feature Count in V7 | Functions | |
|---|---|---|---|---|---|
| PGSC0003DMT400050381 | 1,237,227 a | 824,318 a | 647,927 a | 1,043,868 a | Ribulose bisphosphate carboxylase small chain 1, chloroplastic |
| PGSC0003DMT400049256 | 769,766 b | 660,312 b | 448,042 b | 812,577 b | Ribulose bisphosphate carboxylase/oxygenase activase |
| PGSC0003DMT400015740 | 467,111 c | 446,234 c | 373,783 c | 595,895 c | Chlorophyll a-b binding protein 4, chloroplastic |
| PGSC0003DMT400022072 | 391,269 d | 219,183 e | 155,481 h | 282,483 e | Chlorophyll a-b binding protein 13, chloroplastic |
| PGSC0003DMT400062138 | 375,606 e | 133,281 j | 222,351 j | Ribulose bisphosphate carboxylase small chain C, chloroplastic | |
| PGSC0003DMT400021871 | 371,548 f | 194,143 h | 257,340 g | Chloroplast pigment-binding protein CP29 | |
| PGSC0003DMT400057257 | 360,544 g | 418,762 d | 343,654 d | 553,961 d | Photosystem II 10 kDa polypeptide, chloroplastic |
| MSTRG.21453.1 | 351,809 h | 194,605 g | 146,314 1 | 252,948 h | Calmodulin binding protein |
| PGSC0003DMT400050232 | 333,341 i | 173,647 j | 189,848 e | 257,393 f | Chlorophyll a/b binding protein |
| PGSC0003DMT400007189 | 248,230 j | 163,987 g | Oxygen-evolving enhancer protein 1, chloroplastic | ||
| PGSC0003DMT400049574 | 198,114 f | 179,139 f | Chloroplast thiazole biosynthetic protein | ||
| PGSC0003DMT400031351 | 173,794 i | Fructose-bisphosphate aldolase | |||
| PGSC0003DMT400045248 | 235,947 i | Photosystem II 22 kDa protein, chloroplastic |
| Gene | Count | Functions |
|---|---|---|
| MSTRG.392.1 | 358 | Gene of unknown function |
| PGSC0003DMT400057507 | 332 | Conserved gene of unknown function |
| MSTRG.9424.1 | 212 | Gene of unknown function |
| PGSC0003DMT400071789 | 202 | Gene of unknown function |
| PGSC0003DMT400066793 | 158 | YABBY1 |
| PGSC0003DMT400074800 | 154 | Mta/sah nucleosidase |
| MSTRG.10422.1 | 104 | Gene of unknown function |
| PGSC0003DMT400021019 | 92 | Glucosyltransferase |
| PGSC0003DMT400037714 | 86 | 3-ketoacyl-CoA synthase |
| MSTRG.11977.1 | 83 | RanGAP1 interacting protein |
| Gene | Count | Functions |
|---|---|---|
| MSTRG.25046.1 | 575 | Gene of unknown function |
| MSTRG.1846.1 | 390 | Glycosyltransferase |
| MSTRG.13022.1 | 380 | Gene of unknown function |
| MSTRG.13023.1 | 257 | Gene of unknown function |
| MSTRG.9940.1 | 137 | Gene of unknown function |
| PGSC0003DMT400028158 | 127 | Lipoxygenase |
| MSTRG.23937.1 | 118 | Gene of unknown function |
| MSTRG.19592.1 | 115 | Gene of unknown function |
| MSTRG.13282.1 | 114 | Gene of unknown function |
| MSTRG.17640.1 | 109 | Gene of unknown function |
| Gene | Count | Functions |
|---|---|---|
| MSTRG.24645.1 | 217 | Gene of unknown function |
| PGSC0003DMT400021314 | 156 | Wound-responsive AP2-like factor 2 |
| MSTRG.9415.1 | 152 | Gene of unknown function |
| PGSC0003DMT400008021 | 145 | Gene of unknown function |
| MSTRG.6112.1 | 140 | Gene of unknown function |
| MSTRG.10787.1 | 137 | Gene of unknown function |
| MSTRG.15651.1 | 135 | Gene of unknown function |
| MSTRG.4193.1 | 130 | Conserved gene of unknown function |
| MSTRG.4684.1 | 130 | Conserved gene of unknown function |
| PGSC0003DMT400037122 | 123 | CRT binding factor 3 |
| Gene | Count | Functions |
|---|---|---|
| MSTRG.519.1 | 652 | Gene of unknown function |
| MSTRG.9306.1 | 392 | Gene of unknown function |
| MSTRG.82.1 | 291 | Late blight resistance protein |
| MSTRG.3637.1 | 175 | Gene of unknown function |
| MSTRG.24469.1 | 132 | Gene of unknown function |
| MSTRG.18347.1 | 105 | Pentatricopeptide repeat-containing protein |
| MSTRG.15759.1 | 104 | Gene of unknown function |
| MSTRG.17428.1 | 104 | Gene of unknown function |
| MSTRG.23833.1 | 102 | Gene of unknown function |
| MSTRG.6288.1 | 100 | Gene of unknown function |
References
- Camire, M.E. Potatoes and Human Health. Adv. Potato Chem. Technol. 2016, 8398, 685–704. [Google Scholar] [CrossRef]
- Hirsch, C.N.; Hirsch, C.; Felcher, K.; Coombs, J.; Zarka, D.; Van Deynze, A.; De Jong, W.; Veilleux, R.E.; Jansky, S.; Bethke, P.; et al. Retrospective View of North American Potato (Solanum tuberosum L.) Breeding in the 20th and 21st Centuries. G3: Genes|Genomes|Genetics 2013, 3, 1003–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Upadhyay, C.P. Biotechnological improvement of nutritional and therapeutic value of cultivated potato. Front. Biosci. 2018, 10, 217–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gavrilenko, T.; Antonova, O.; Shuvalova, A.; Krylova, E.; Alpatyeva, N.; Spooner, D.M.; Novikova, L. Genetic diversity and origin of cultivated potatoes based on plastid microsatellite polymorphism. Genet. Resour. Crop. Evol. 2013, 60, 1997–2015. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, F.; Wang, L.; Leach, L.; Luo, Z. Orthogonal contrast based models for quantitative genetic analysis in autotetraploid species. New Phytol. 2018, 220, 332–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szcześniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, N.; Zhang, F.; Wu, J.; Chen, Y.; Hu, X.; Fang, O.; Leach, L.J.; Wang, D.; Luo, Z. A highly robust and optimized sequence-based approach for genetic polymorphism discovery and genotyping in large plant populations. Theor. Appl. Genet. 2016, 129, 1739–1757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Potato Genome Sequencing Consortium. Genome sequence and analysis of the tuber crop potato. Nat. Cell Biol. 2011, 475, 189–195. [Google Scholar] [CrossRef] [Green Version]
- Bourke, P.M.; Voorrips, R.E.; Visser, R.G.F.; Maliepaard, C. Tools for Genetic Studies in Experimental Populations of Polyploids. Front. Plant Sci. 2018, 9, 513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa-Silva, J.; Domingues, D.; Lopes, F.M. RNA-Seq differential expression analysis: An extended review and a software tool. PLoS ONE 2017, 12, e0190152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trivedi, U.H.; Cézard, T.; Bridgett, S.; Montazam, A.; Nichols, J.; Blaxter, M.; Gharbi, K.; Cezard, T. Quality control of next-generation sequencing data without a reference. Front. Genet. 2014, 5, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Su, X.; Wang, A.; Xu, J.; Ning, K. QC-Chain: Fast and Holistic Quality Control Method for Next-Generation Sequencing Data. PLoS ONE 2013, 8, e60234. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Sims, D.; Sudbery, I.; Ilott, N.E.; Heger, A.; Ponting, C.P. Sequencing depth and coverage: Key considerations in genomic analyses. Nat. Rev. Genet. 2014, 15, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Munir, S.; Khan, M.R.G.; Song, J.; Munir, S.; Zhang, Y.; Ye, Z.; Wang, T. Genome-wide identification, characterization and expression analysis of calmodulin-like (CML) proteins in tomato (Solanum lycopersicum). Plant Physiol. Biochem. 2016, 102, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Hardigan, M.A.; Laimbeer, F.P.E.; Newton, L.; Crisovan, E.; Hamilton, J.; Vaillancourt, B.; Wiegert-Rininger, K.; Wood, J.; Douches, D.S.; Farré, E.M.; et al. Genome diversity of tuber-bearing Solanum uncovers complex evolutionary history and targets of domestication in the cultivated potato. Proc. Natl. Acad. Sci. USA 2017, 114, E9999–E10008. [Google Scholar] [CrossRef] [PubMed] [Green Version]

| Software | Stage of Analysis | |
|---|---|---|
| 1 | Trim Galore! version 0.4.2 and Cutadapt version 1.16 | Trim adapter sequences |
| FastQC version 0.11.5 | Perform sequence reading quality checks | |
| 2 | HISAT2 version 2.1.0 | Mapping sequence reads to reference genome |
| 3 | SAMtools version 1.8 | i. Convert. sam file to .bam files ii. estimating genome coverage and percentage mapped to reference genome |
| 4 | StringTie version 1.3.3 | i. Assembling transcripts from individual samples ii. Merged assembled transcriptomes from all samples |
| 5 | GffCompare version 0.10.2 | Compare the assembled transcriptome to reference genome files. |
| 6 | Subread version 1.5.3 (featurecount) | Generate the number of reads mapped to each gene for each sample. |
| 7 | R version 3.5.0 | Analysis of reads mapped to each gene and generating informative graphs. |
| Potato Varieties | Number of Paired Reads | Number Mapped to Reference Genome | % Mapped to Reference Genome |
|---|---|---|---|
| HJ | 81,938,945 | 73,194,049 | 89.33 |
| HL | 69,099,623 | 61,511,499 | 89.02 |
| LS | 61,462,784 | 53,486,309 | 87.02 |
| V7 | 83,537,644 | 74,357,905 | 89.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ezeorba, T.P.C.; Okeke, E.S.; Okagu, I.U.; Nweze, E.J.; Asomadu, R.O.; Ezeorba, W.F.C.; Chukwuma, I.F.; Ononiwu, C.P.; Ezema, C.A.; Okorigwe, E.M.; et al. Transcriptomic Diversity of Solanum tuberosum Varieties: A Drive towards Future Analysis of Its Polyploidy Genome. Biol. Life Sci. Forum 2022, 11, 46. https://doi.org/10.3390/IECPS2021-12058
Ezeorba TPC, Okeke ES, Okagu IU, Nweze EJ, Asomadu RO, Ezeorba WFC, Chukwuma IF, Ononiwu CP, Ezema CA, Okorigwe EM, et al. Transcriptomic Diversity of Solanum tuberosum Varieties: A Drive towards Future Analysis of Its Polyploidy Genome. Biology and Life Sciences Forum. 2022; 11(1):46. https://doi.org/10.3390/IECPS2021-12058
Chicago/Turabian StyleEzeorba, Timothy P. C., Emmanuel S. Okeke, Innocent U. Okagu, Ekene J. Nweze, Rita O. Asomadu, Wisdom F. C. Ezeorba, Ifeoma F. Chukwuma, Chidinma P. Ononiwu, Chinonso A. Ezema, Ekezie M. Okorigwe, and et al. 2022. "Transcriptomic Diversity of Solanum tuberosum Varieties: A Drive towards Future Analysis of Its Polyploidy Genome" Biology and Life Sciences Forum 11, no. 1: 46. https://doi.org/10.3390/IECPS2021-12058
APA StyleEzeorba, T. P. C., Okeke, E. S., Okagu, I. U., Nweze, E. J., Asomadu, R. O., Ezeorba, W. F. C., Chukwuma, I. F., Ononiwu, C. P., Ezema, C. A., Okorigwe, E. M., Nwanelo, V. O., & Joshua, P. E. (2022). Transcriptomic Diversity of Solanum tuberosum Varieties: A Drive towards Future Analysis of Its Polyploidy Genome. Biology and Life Sciences Forum, 11(1), 46. https://doi.org/10.3390/IECPS2021-12058