
Sunlight Degradation of the Aminophosphonate Diethylenetriamine Penta-(Methylenephosphonic Acid)
.png)
 ,
,  , ,
, ,
Abstract
:
1. Introduction
2. Material and Methods
2.1. Chemicals
2.2. UV Treatment of DTPMP
2.3. Actinometry to Determine the Photon Flux in Sunlight with Different Flask Materials
2.4. Sunlight Degradation Experiments
2.5. Analytics
2.6. Data Evaluation
3. Results and Discussion
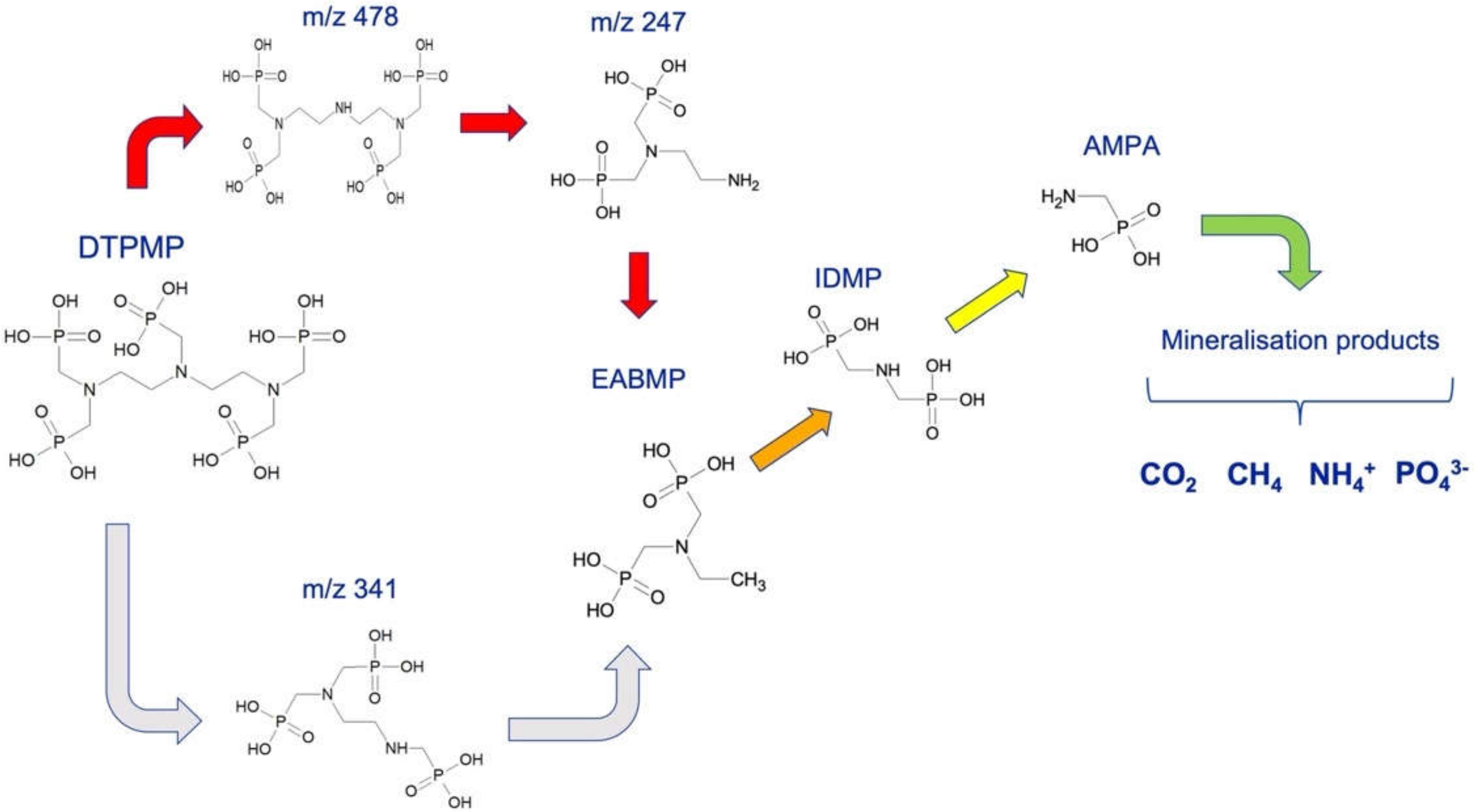
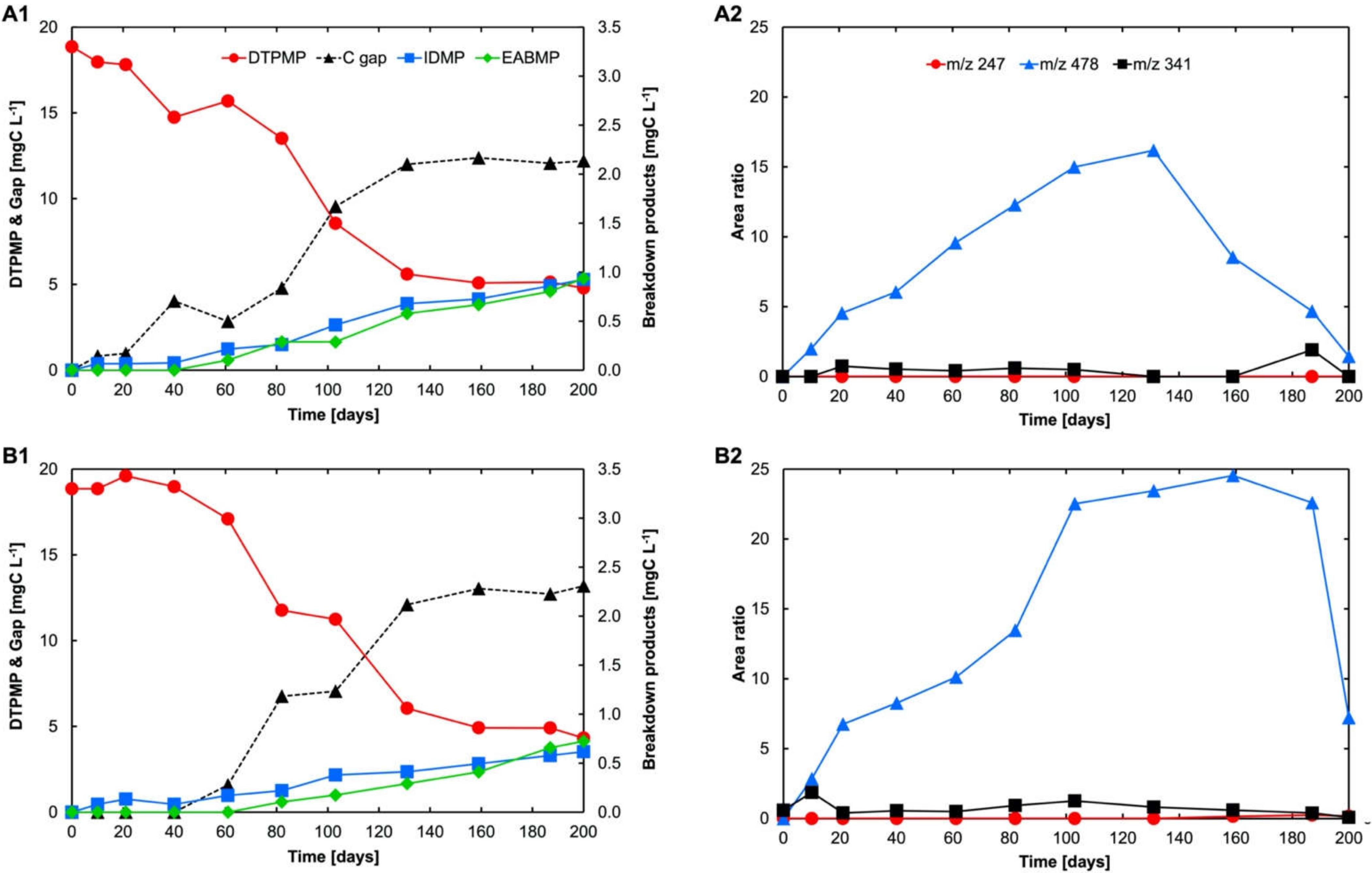
3.1. Degradation Pathway of DTPMP during UV Treatment
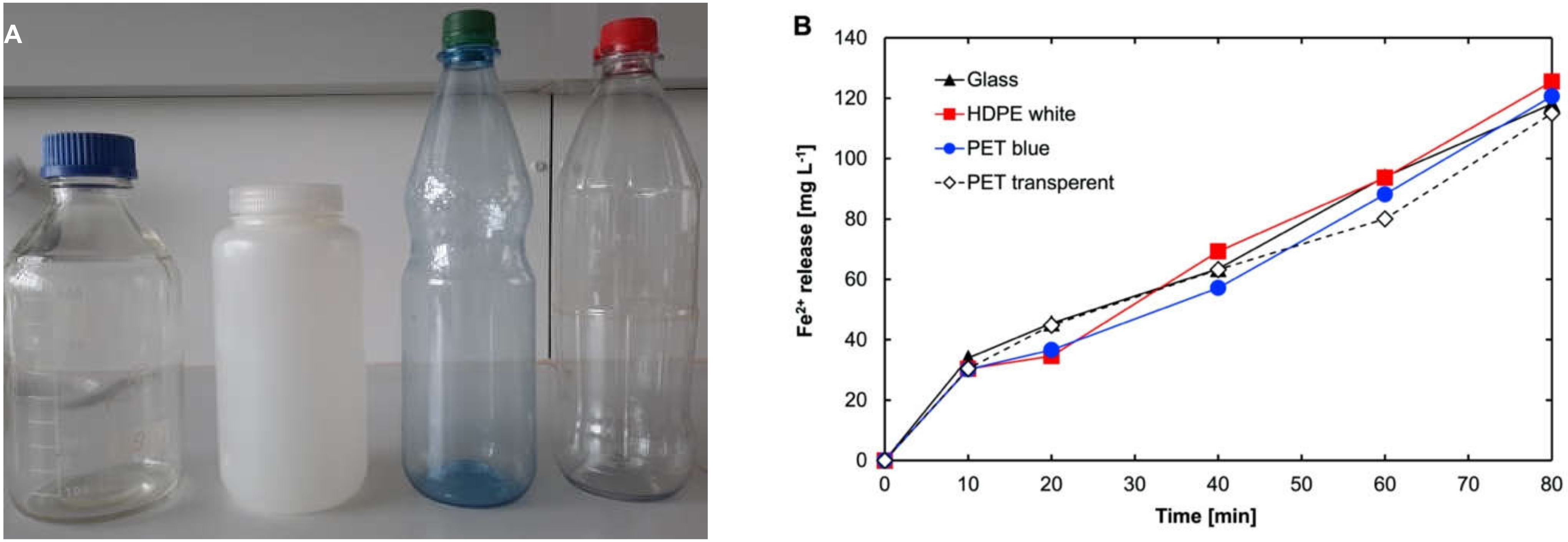
3.2. Actinometry for the Determination of Suitable Material for the Sunlight Degradation Test
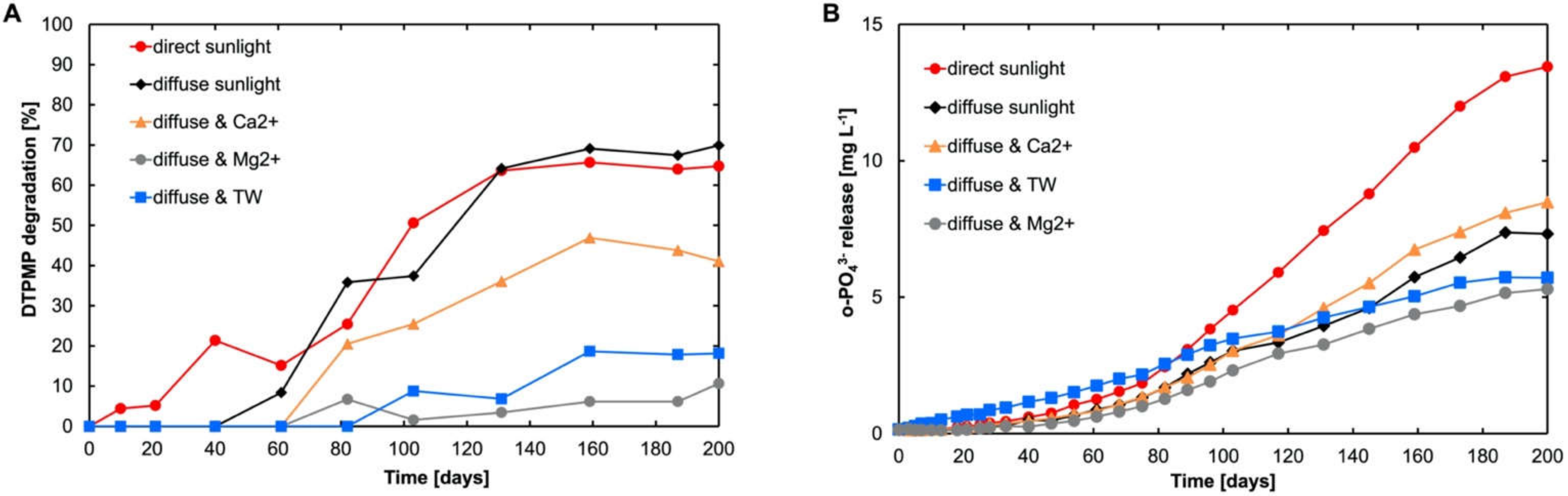
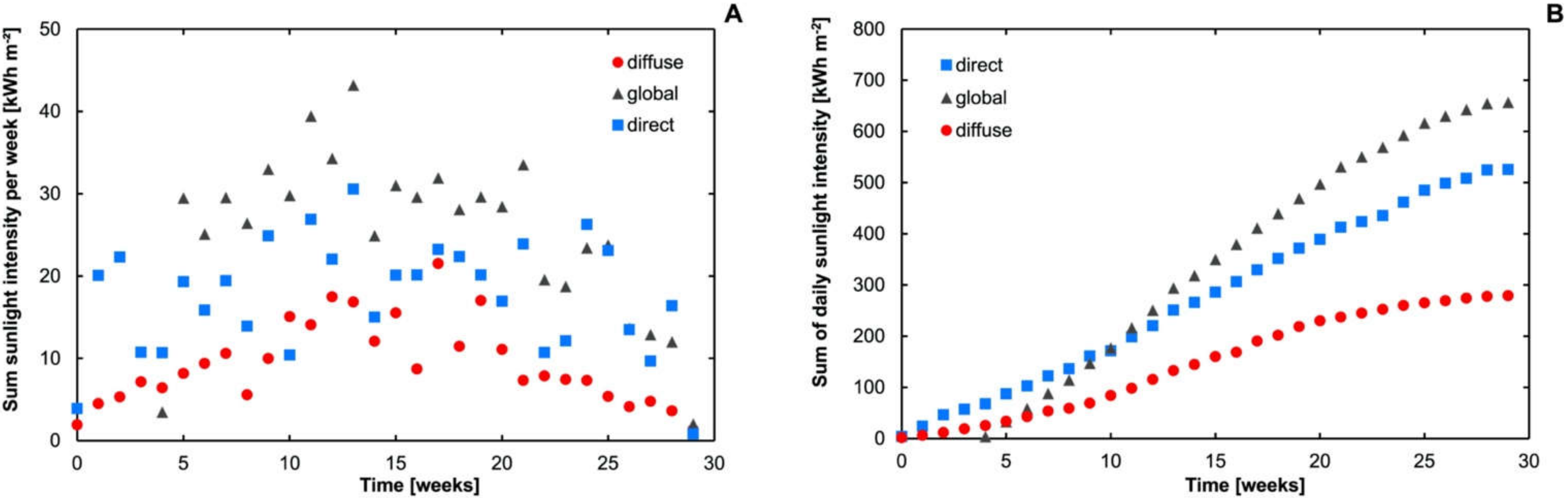
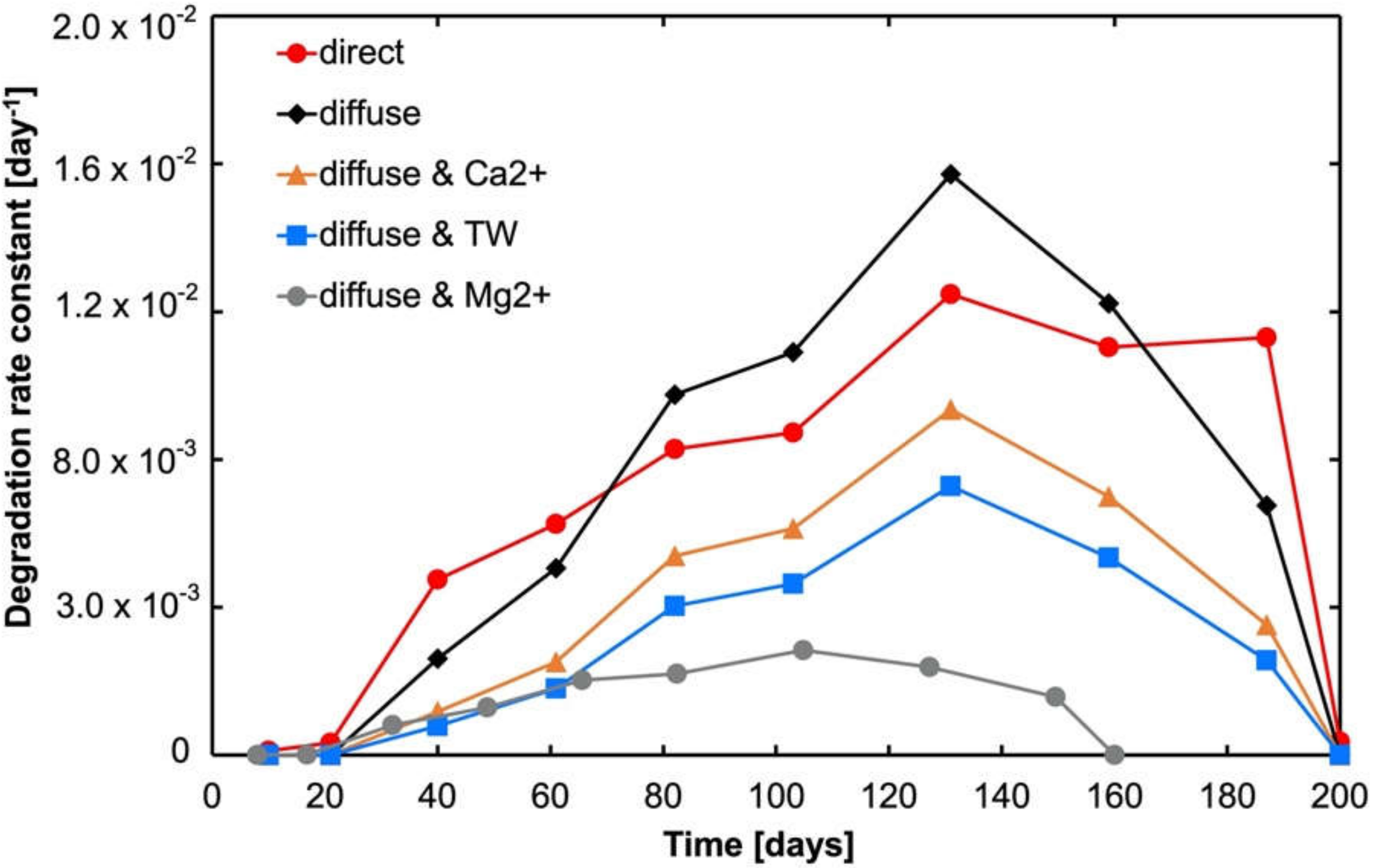
3.3. Influence of Sunlight on the Degradation of DTPMP
3.4. Influence of Sunlight Intensity on DTPMP Degradation Kinetics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Reviewer Broad Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Studnik, H.; Liebsch, S.; Forlani, G.; Wieczorek, D.; Kafarski, P.; Lipok, J. Amino polyphosphonates—Chemical features and practical uses, environmental durability and biodegradation. New Biotechnol. 2015, 32, 1–6. [Google Scholar] [CrossRef]
- Liu, Y.; Dai, Z.; Kan, A.T.; Tomson, M.B.; Zhang, P. Investigation of sorptive interaction between phosphonate inhibitor and barium sulfate for oilfield scale control. J. Petrol. Sci. Eng. 2022, 208, 109425. [Google Scholar] [CrossRef]
- Mpelwa, M.; Tang, S.F. State of the art of synthetic threshold scale inhibitors for mineral scaling in the petroleum industry: A review. Petrol. Sci. 2019, 16, 830–849. [Google Scholar] [CrossRef] [Green Version]
- Nowack, B. Environmental chemistry of phosphonates. Water Res. 2003, 37, 2533–2546. [Google Scholar] [CrossRef]
- Greenlee, L.F.; Testa, F.; Lawler, D.F.; Freeman, B.D.; Moulin, P. Effect of antiscalant degradation on salt precipitation and solid/liquid separation of RO concentrate. J. Memb. Sci. 2011, 366, 48–61. [Google Scholar] [CrossRef]
- Knepper, T.P. Synthetic chelating agents and compounds exhibiting complexing properties in the aquatic environment. Trends Anal. Chem. 2003, 22, 707–724. [Google Scholar] [CrossRef]
- Neto, D.M.; da Costa, L.S.; de Menezes, F.L.; Fechine, L.M.; Freire, R.M.; Denardin, J.C.; Banobre-López, M.; Vasconcelos, I.F.; Ribeiro, T.S.; Leal, L.K.A.; et al. A novel amino phosphonate-coated magnetic nanoparticle as MRI contrast agent. Appl. Surf. Sci. 2021, 543, 148824. [Google Scholar] [CrossRef]
- Reinhardt, T.; Rott, E.; Schneider, P.A.; Minke, R.; Schönberger, H. Fixed-bed column studies of phosphonate and phosphate adsorption on granular ferric hydroxide (GFH). Process Saf. Environ. 2021, 153, 301–310. [Google Scholar] [CrossRef]
- Kuhn, R.; Jensch, R.; Bryant, I.M.; Fischer, T.; Liebsch, S.; Martienssen, M. The influence of selected bivalent metal ions on the photolysis of diethylenetriamine penta(methylenephosphonic acid). Chemosphere 2018, 210, 726–733. [Google Scholar] [CrossRef] [PubMed]
- Rott, E.; Steinmetz, H.; Metzger, J.W. Organophosphonates: A review on environmental relevance, biodegradability and removal in wastewater treatment plants. Sci. Total Environ. 2018, 615, 1176–1191. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Kum, S.; Liu, H. Inland desalination brine disposal: A baseline study from southern California on brine transport infrastructure and treatment potential. ACS EST Engg. 2021, 2, 456–464. [Google Scholar] [CrossRef]
- Armbruster, D.; Rott, E.; Minke, R.; Happel, O. Trace-level determination of phosphonates in liquid and solid phase of wastewater and environmental samples by IC-ESI-MS/MS. Anal. Bioanal. Chem. 2019, 412, 4807–4825. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhang, B.; Shan, C.; Yan, X.; Chen, H.; Pan, B. Occurrence and transformation of phosphonates in textile dyeing wastewater along full-scale combined treatment processes. Water Res. 2020, 184, 116173. [Google Scholar] [CrossRef] [PubMed]
- Nowack, B. The behavior of phosphonates in wastewater treatment plants of Switzerland. Water Res. 1998, 32, 1271–1279. [Google Scholar] [CrossRef]
- Rott, E.; Happel, O.; Armbruster, D.; Minke, R. Behavior of PBTC, HEDP, and aminophosphonates in the process of wastewater treatment. Water 2020, 12, 53. [Google Scholar] [CrossRef] [Green Version]
- Boels, L.; Tervahauta, T.; Witkamp, G.J. Adsorptive removal of nitrilotris(methylenephosphonic acid) antiscalant from membrane concentrates by iron-coated waste filtration sand. J. Hazard. Mater. 2010, 182, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Lesueur, C.; Pfeffer, M.; Fuerhacker, M. Photodegradation of phosphonates in water. Chemosphere 2005, 59, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.X.; Zhang, T.Y.; Dao, G.H.; Xu, Z.B.; Wu, Y.H.; Hu, H.Y. Assessment and mechanisms of microalgae growth inhibition by phosphonates: Effects of intrinsic toxicity and complexation. Water Res. 2020, 186, 116333. [Google Scholar] [CrossRef] [PubMed]
- Schindler, D.W.; Carpenter, S.R.; Chapra, S.C.; Hecky, R.E.; Orihel, D.M. Reducing phosphorus to curb lake eutrophication is a success. Environ. Sci. Technol. 2016, 50, 8923–8929. [Google Scholar] [CrossRef] [PubMed]
- Rott, E.; Minke, R.; Bali, U.; Steinmetz, H. Removal of phosphonates from industrial wastewater with UV/Fe(II), Fenton and UV/Fenton treatment. Water Res. 2017, 122, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Wang, S.; Ye, Y.; Pan, B. Highly efficient removal of phosphonates from water by a combine Fe(II)/UV/co-precipitation process. Water Res. 2019, 153, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.; Wang, W.-L.; Xu, Z.-B.; Wu, Q.-Y.; Hu, H.-Y. UV/chlorine oxidation of the phosphonate antiscalant 1-Hydroxyethane-1, 1-diphosphonic acid (HEDP) used for reverse osmosis processes: Organic phosphorus removal and scale inhibition properties changes. J. Environ. Manage. 2019, 237, 180–186. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, S.; Li, H.; Qian, J.; Lv, L.; Pan, B. Degradation of phosphonates in Co(II)/peroxymonosulfate process: Performance and mechanism. Water Res. 2021, 202, 117397. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.H.; Chang, T.F.M.; Chen, C.Y.; Sone, M.; Hsu, Y.J. Mechanistic insight into photodegradation of organic dyes using heterostructure photocatalysts. Catalysts 2019, 9, 430. [Google Scholar] [CrossRef] [Green Version]
- Alulema-Pullupaxi, P.; Fernández, L.; Debut, A.; Santacruz, C.P.; Villacis, W.; Fierro, C.; Espinoza-Montero, P.J. Photoelectrocatalytic degradation of glyphosate on titanium dioxide synthesized by sol-gel/spin-coating on boron doped diamond (TiO2/BDD) as a photoanode. Chemosphere 2021, 278, 130488. [Google Scholar] [CrossRef]
- Huang, Y.; Li, Z.; Yao, K.; Chen, C.; Deng, C.; Fang, Y.; Li, R.; Tian, H. Suppressing toxic intermediates during photocatalytic degradation glyphosate by controlling adsorption modes. Appl. Catal. B-Environ. 2021, 299, 120671. [Google Scholar] [CrossRef]
- Tang, Q.Y.; Yang, M.J.; Yang, S.Y.; Xu, Y.H. Enhanced photocatalytic degradation of glyphosate over 2D CoS/BiOBr heterojunctions under visible light irradiation. J. Hazard. Mater. 2021, 407, 124798. [Google Scholar] [CrossRef]
- Lv, Y.R.; He, R.K.; Chen, Z.Y.; Li, X.; Xu, Y.H. Fabrication of hierarchical copper sulfide/bismuth tungstate p-n heterojunction with two-dimensional (2D) interfacial coupling for enhanced visible-light photocatalytic degradation of glyphosate. J. Colloid Interface Sci. 2020, 560, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, R.; Tóth, E.; Geppert, H.; Fischer, T.; Liebsch, S.; Martienssen, M. Identification of the complete degradation pathway of ethylendiaminetetra(methylenephosphonic acid) in aquatic solution. CLEAN Air Soil Water 2017, 45, 1500774. [Google Scholar] [CrossRef]
- Kuhn, R.; Jensch, R.; Bryant, I.M.; Fischer, T.; Liebsch, S.; Martiensen, M. Photodegradation of ethylenediaminetetra(methylenephosphonic acid)—Effect of the system configuration. J. Photochem. Photobiol. A Chem. 2020, 388, 112192. [Google Scholar] [CrossRef]
- Stookey, L.L. Ferrozine: A new spectrophotometer reagent for iron. Anal. Chem. 1970, 42, 779–781. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, H.J.; Braslavsky, S.E.; Schmidt, R. Chemical actinometry (IUPAC technical report). Pure Appl. Chem. 2004, 76, 2105–2146. [Google Scholar] [CrossRef]
- Dudok de Wit, T.; Watermann, J. Solar forcing of the terrestrial atmosphere, C.R. Geoscience 2010, 342, 259–272. [Google Scholar] [CrossRef] [Green Version]
- Jaisi, D.P.; Li, H.; Wallace, A.F.; Paudel, P.; Sun, M.; Balakrishna, A.; Lerch, R.N. Mechanisms of bond cleavage during manganese oxide and UV degradation of glyphosate: Results from phosphate oxygen isotopes and molecular simulations. J. Agric. Food Chem. 2016, 64, 8474–8482. [Google Scholar] [CrossRef] [PubMed]
- Sandy, E.H.; Blake, R.E.; Chang, S.J.; Jun, Y.; Yu, C. Oxygen isotope signature of glyphosate and phosphonoacetate: Tracing source and cycling of phosphonates. J. Hazard. Mater. 2013, 260, 947–954. [Google Scholar] [CrossRef]
- Kuhn, R.; Bryant, I.M.; Jensch, R.; Liebsch, S.; Martienssen, M. Photolysis of hexamethylenediaminetetra(methylenephosphonic acid) (HDTMP) using manganese and hydrogen peroxide. Emerg. Contam. 2020, 6, 10–19. [Google Scholar] [CrossRef]
- Nowack, B.; Stone, A.T. Homogeneous and heterogeneous oxidation of nitrilotrismethylenephosphonic acid (NTMP) in the presence of manganese (II, III) and molecular oxygen. J. Phys. Chem. B 2002, 106, 6227–6233. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Glass | HDPE | PETblue | PETtransparent | UV Lamp | |
|---|---|---|---|---|---|
| Reaction rate (mol s−1) | 3.6 × 10−7 | 4.2 × 10−7 | 3.9 × 10−7 | 3.4 × 10−7 | 1.5 × 10−7 |
| Ratio UV vs. Sun (-) | 42.5 | 36.7 | 39.3 | 45.3 | - |
| Test Condition | DTPMP (%) | o-PO43− (%) | o-PO43− (mg P L−1) |
|---|---|---|---|
| Direct sunlight | 74.6 | 16.2 | 4.4 |
| Diffuse sunlight | 77.1 | 8.9 | 2.4 |
| Diffuse sunlight and Ca2+ | 54.7 | 10.3 | 2.8 |
| Diffuse sunlight and Mg2+ | 30.7 | 6.4 | 1.7 |
| Diffuse sunlight and tap water | 48.5 | 6.9 | 1.9 |
| Treatment Condition | A | Ea*weff (kJ2 mol−1 cm−2) | R2 | RSS * |
|---|---|---|---|---|
| Direct sunlight | 2.1 × 10−4 | 2.8 × 104 | 0.97 | 313.9 |
| Diffuse sunlight | 2.7 × 10−4 | 2.0 × 104 | 0.99 | 163.4 |
| Diffuse sunlight and Ca2+ | 1.8 × 10−4 | 2.3 × 104 | 0.99 | 98.9 |
| Diffuse sunlight and Mg2+ | 0.4 × 10−4 | 1.4 × 104 | 0.96 | 57.1 |
| Diffuse sunlight and tap water | 1.4 × 10−4 | 2.4 × 104 | 0.99 | 45.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuhn, R.; Jensch, R.; Fischer, T.; Keuler, K.; Bryant, I.M.; Martienssen, M. Sunlight Degradation of the Aminophosphonate Diethylenetriamine Penta-(Methylenephosphonic Acid). Solar 2022, 2, 141-157. https://doi.org/10.3390/solar2020009
Kuhn R, Jensch R, Fischer T, Keuler K, Bryant IM, Martienssen M. Sunlight Degradation of the Aminophosphonate Diethylenetriamine Penta-(Methylenephosphonic Acid). Solar. 2022; 2(2):141-157. https://doi.org/10.3390/solar2020009
Chicago/Turabian StyleKuhn, Ramona, Robert Jensch, Thomas Fischer, Klaus Keuler, Isaac Mbir Bryant, and Marion Martienssen. 2022. "Sunlight Degradation of the Aminophosphonate Diethylenetriamine Penta-(Methylenephosphonic Acid)" Solar 2, no. 2: 141-157. https://doi.org/10.3390/solar2020009
APA StyleKuhn, R., Jensch, R., Fischer, T., Keuler, K., Bryant, I. M., & Martienssen, M. (2022). Sunlight Degradation of the Aminophosphonate Diethylenetriamine Penta-(Methylenephosphonic Acid). Solar, 2(2), 141-157. https://doi.org/10.3390/solar2020009