
Bacterial Community and Antibiotic Resistance Gene Profiles of Fish Gut Contents and Their Aquaculture Environment in Tianjin, China
 , and
, and 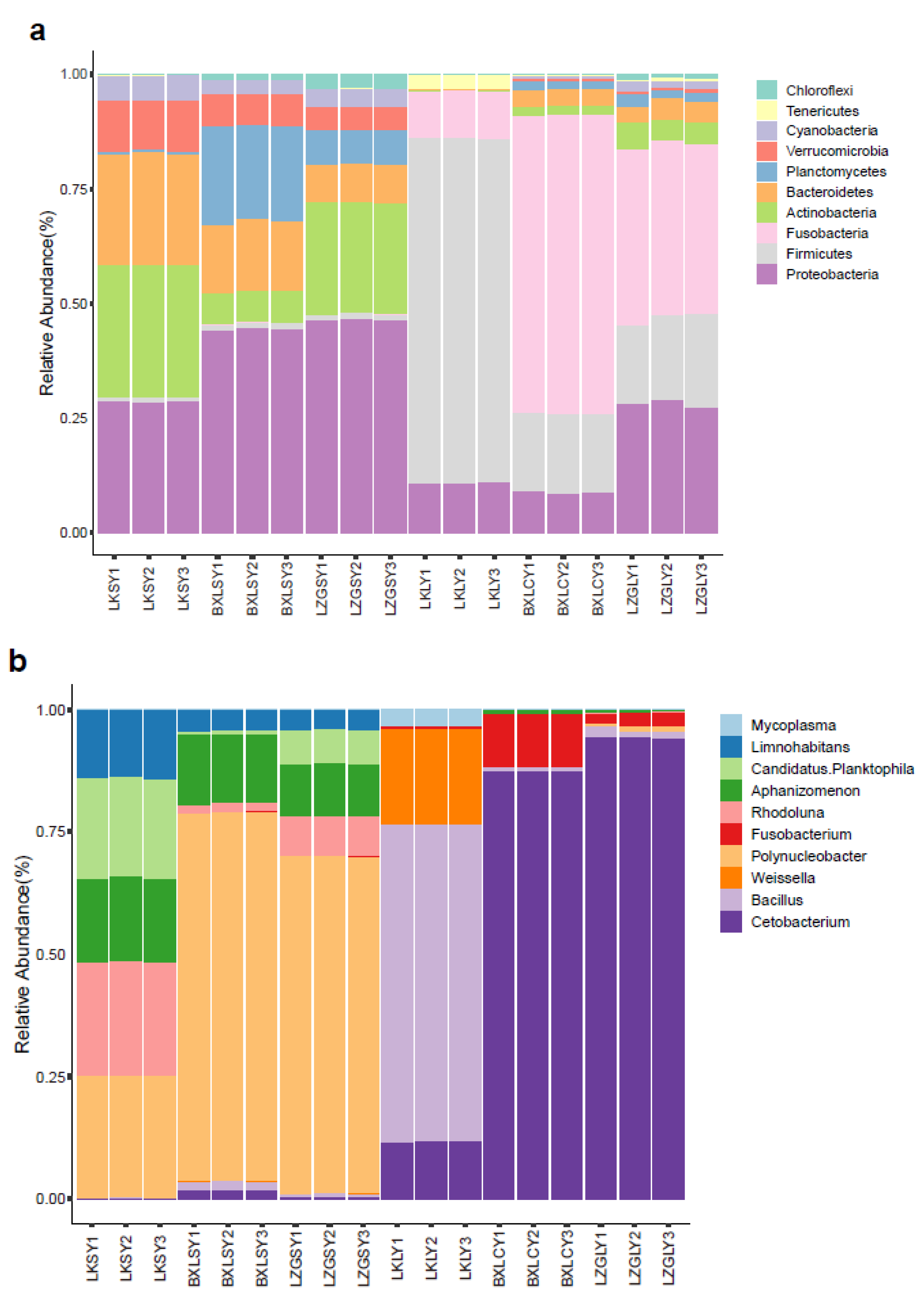{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Antibiotics and Metals Quantification
2.3. DNA Extraction, Library Preparation and Metagenomic Sequencing
2.4. Gene Prediction from Scaftigs
2.5. Taxonomic Classification
2.6. ARGs Analysis
3. Results
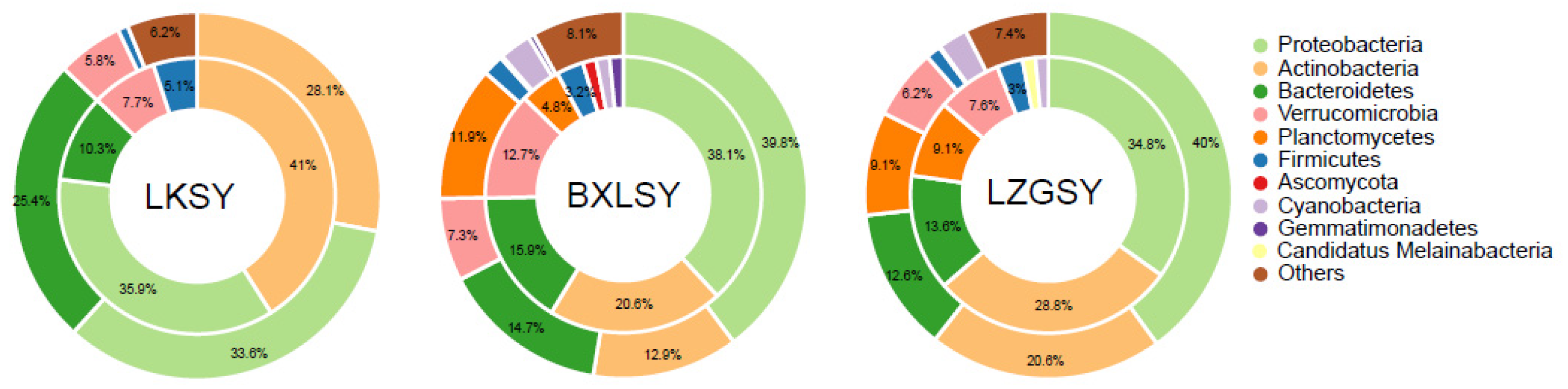
3.1. Bacterial Community Composition
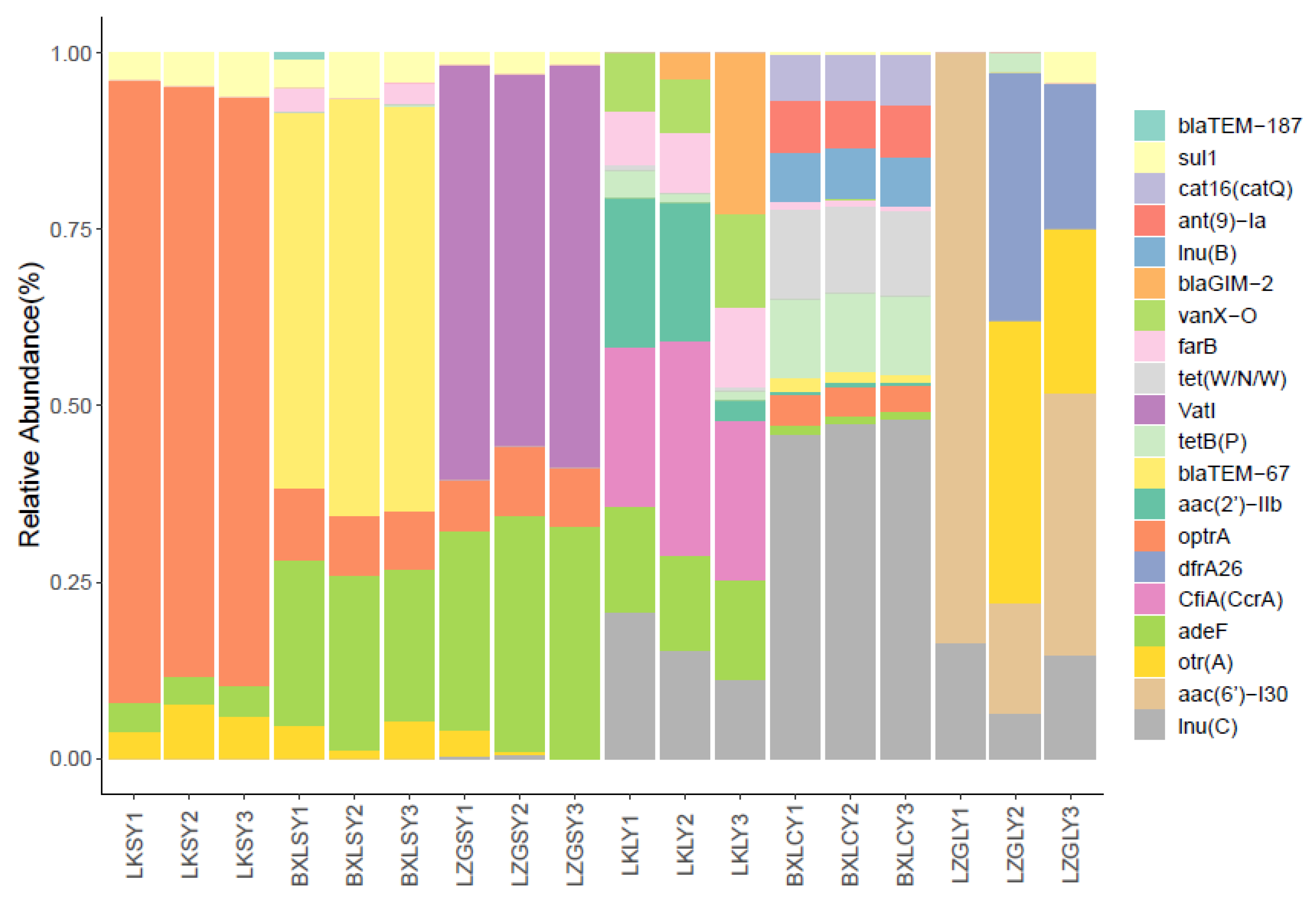
3.2. Diversity and Abundance of ARGs
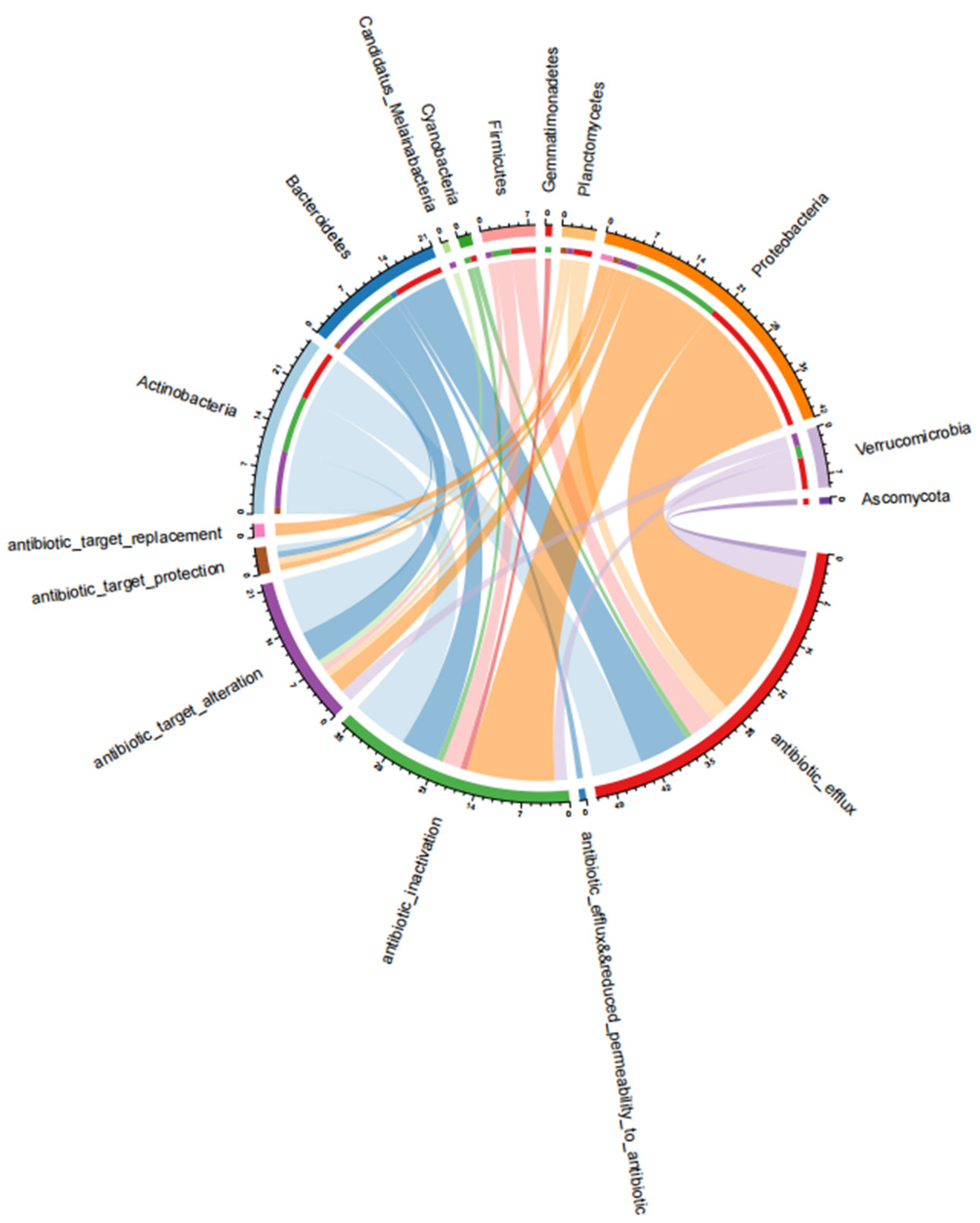
3.3. Antibiotic Resistance Mechanisms of the ARGs
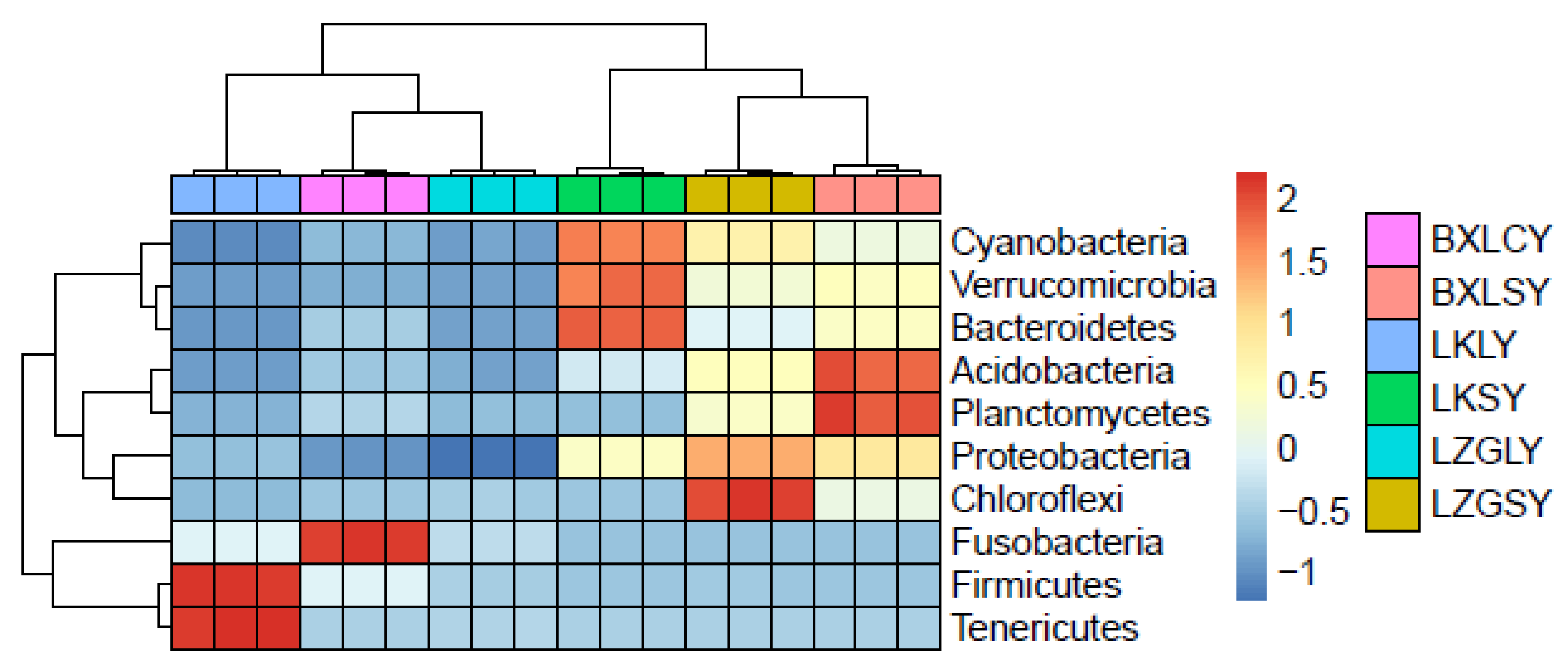
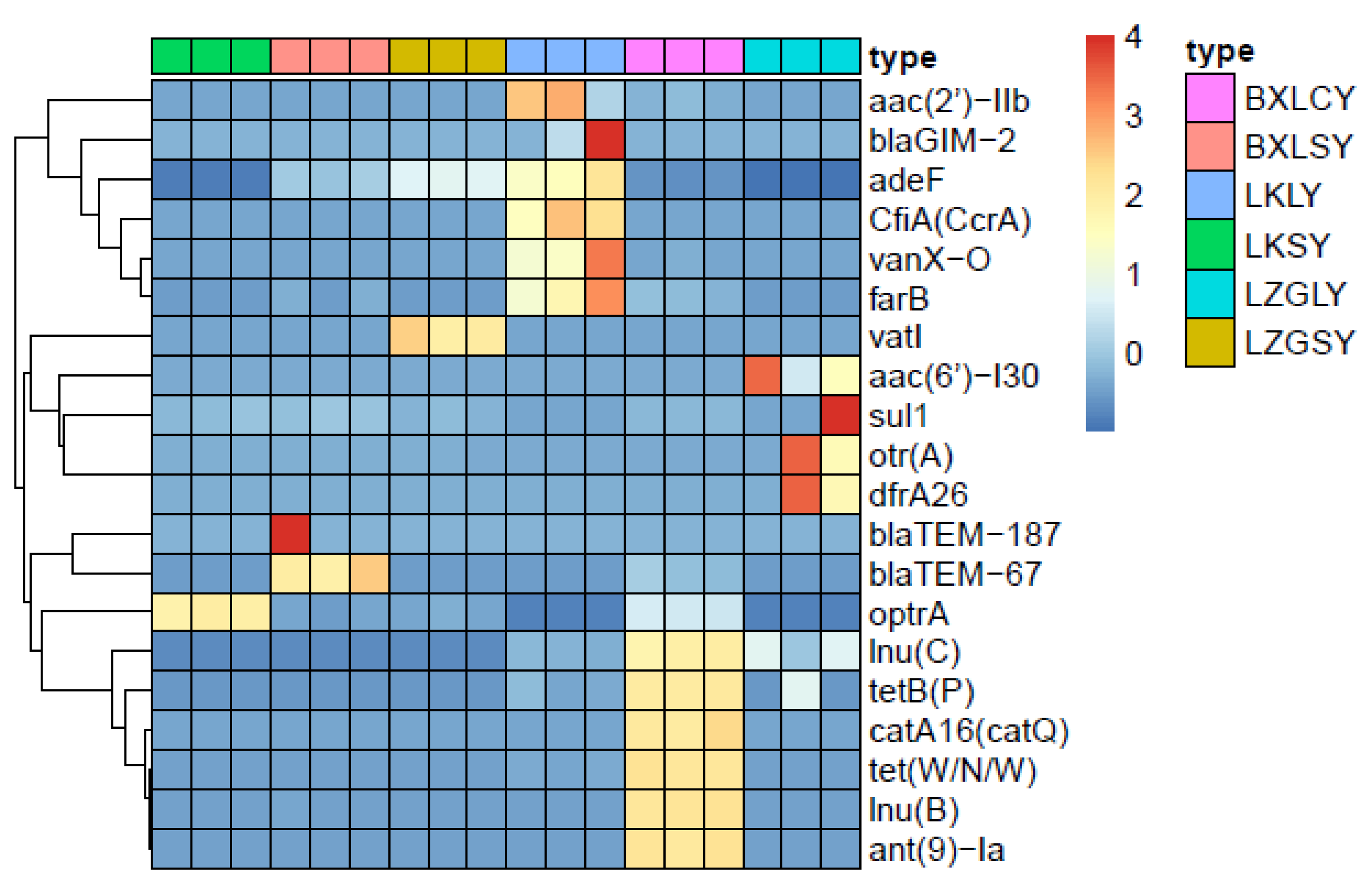
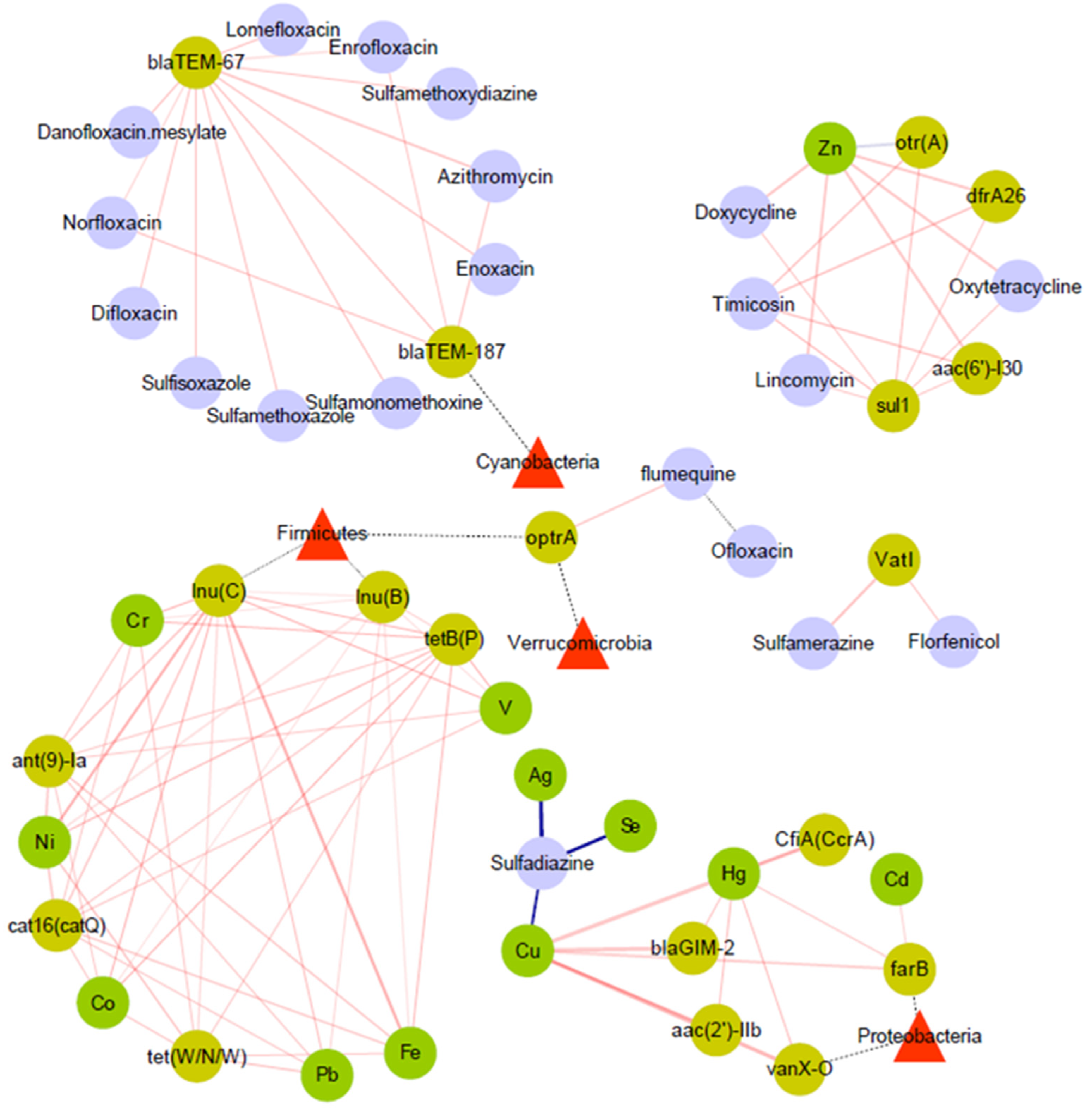
3.4. Relationship between ARGs and Microbial Community
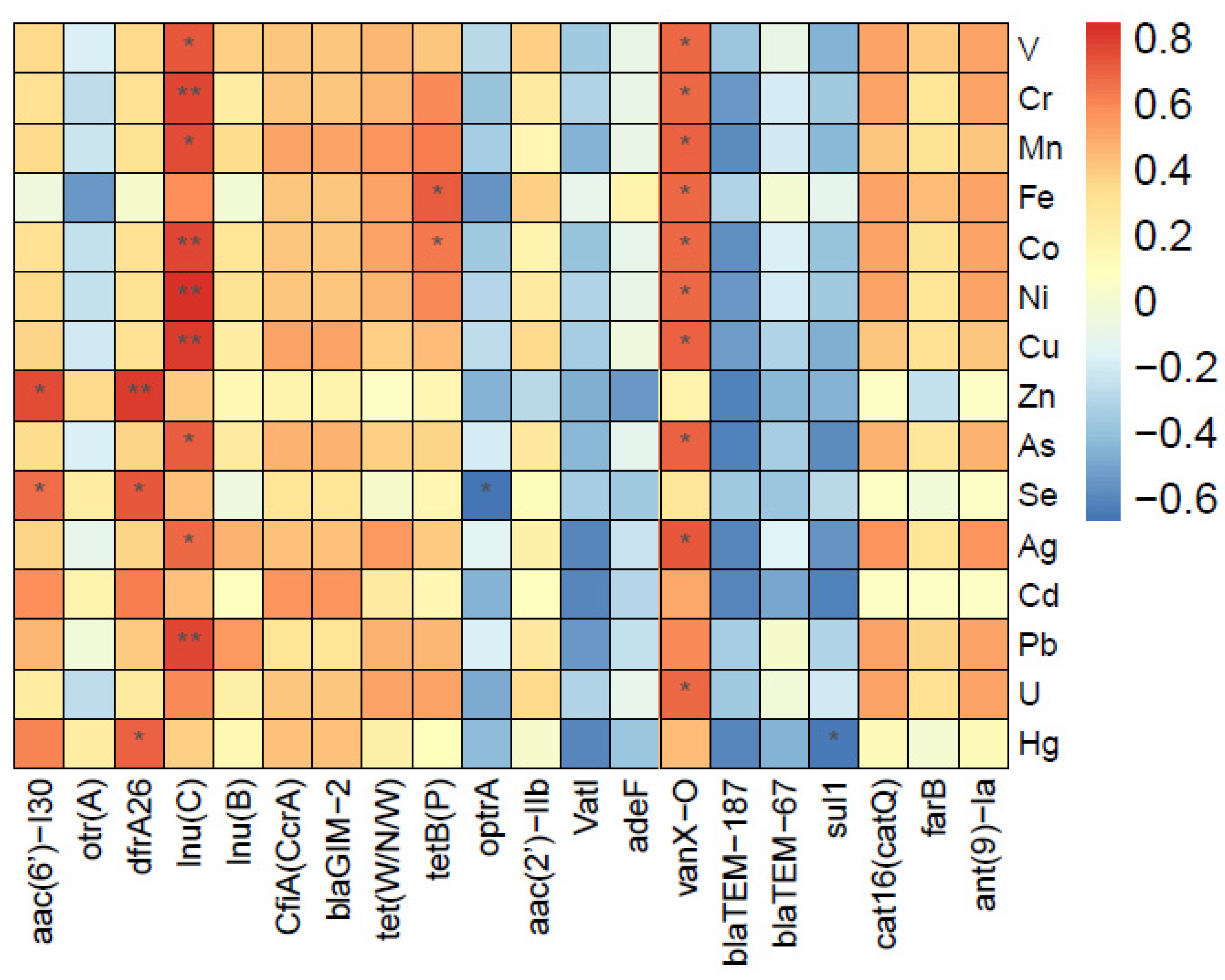
3.5. Relationship between ARGs and Antibiotics
3.6. Associations between ARGs and Heavy Metals
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pruden, A.; Pei, R.; Storteboom, H.; Carlson, K.H. Antibiotic Resistance Genes as Emerging Contaminants: Studies in Northern Colorado. Environ. Sci. Technol. 2006, 40, 7445–7450. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-H.; Lu, J.; Zhang, Y.-X.; Wu, J.; Luo, Y.; Liu, H. Metagenomic analysis of antibiotic resistance genes in coastal industrial mariculture systems. Bioresour. Technol. 2018, 253, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Okyere, A.; Bishoff, D.; Oyaro, M.O.; Ajami, N.J.; Darkoh, C. Analysis of Fish Commonly Sold in Local Supermarkets Reveals the Presence of Pathogenic and Multidrug-Resistant Bacterial Communities. Microbiol. Insights 2018, 11, 1178636118786925. [Google Scholar] [CrossRef]
- The State of World Fisheries and Aquaculture (SOFIA) 2022. Available online: http://www.lmcwater.org.cn/authoritative_opinion/study/202207/t20220701_35915.html (accessed on 1 July 2022).
- Fishery Bureau of Ministry of Agriculture. 2022 China Fishery Statistics Yearbook; China Agriculture Press: Beijing, China, 2022.
- Liu, Q.; Lai, Z.; Gao, Y.; Wang, C.; Zeng, Y.; Liu, E.; Mai, Y.; Yang, W.; Li, H. Connection between the Gut Microbiota of Largemouth Bass (Micropterus salmoides) and Microbiota of the Pond Culture Environment. Microorganisms 2021, 9, 1770. [Google Scholar] [CrossRef]
- Lloyd, N.A.; Nazaret, S.; Barkay, T. Whole genome sequences to assess the link between antibiotic and metal resistance in three coastal marine bacteria isolated from the mummichog gastrointestinal tract. Mar. Pollut. Bull. 2018, 135, 514–520. [Google Scholar] [CrossRef]
- Zhang, Y.; Gu, A.Z.; Cen, T.; Li, X.; He, M.; Li, D.; Chen, J. Sub-inhibitory concentrations of heavy metals facilitate the horizontal transfer of plasmid-mediated antibiotic resistance genes in water environment. Environ. Pollut. 2018, 237, 74–82. [Google Scholar] [CrossRef]
- Yuan, X.; Zhang, Y.; Sun, C.; Wang, W.; Wu, Y.; Fan, L.; Liu, B. Profile of Bacterial Community and Antibiotic Resistance Genes in Typical Vegetable Greenhouse Soil. Int. J. Environ. Res. Public Health 2022, 19, 7742. [Google Scholar] [CrossRef] [PubMed]
- Di Cesare, A.; Eckert, E.M.; D’Urso, S.; Bertoni, R.; Gillan, D.C.; Wattiez, R.; Corno, G. Co-occurrence of integrase 1, antibiotic and heavy metal resistance genes in municipal wastewater treatment plants. Water Res. 2016, 94, 208–214. [Google Scholar] [CrossRef]
- Yang, Y.; Li, B.; Zou, S.; Fang, H.H.; Zhang, T. Fate of antibiotic resistance genes in sewage treatment plant revealed by metagenomic approach. Water. Res. 2014, 62, 97–106. [Google Scholar] [CrossRef]
- Zhu, Y.-G.; Zhao, Y.; Li, B.; Huang, C.-L.; Zhang, S.-Y.; Si-Yu, Z.; Chen, Y.-S.; Zhang, T.; Gillings, M.; Su, J.-Q. Continental-scale pollution of estuaries with antibiotic resistance genes. Nat. Microbiol. 2017, 2, 16270. [Google Scholar] [CrossRef]
- Di Cesare, F. Functional Metagenomics for Identification of Antibiotic Resistance Genes (ARGs). Methods. Mol. Biol. 2021, 2242, 173–183. [Google Scholar]
- Hu, Y.; Jiang, L.; Sun, X.; Wu, J.; Ma, L.; Zhou, Y.; Lin, K.; Luo, Y.; Cui, C. Risk assessment of antibiotic resistance genes in the drinking water system. Sci. Total Environ. 2021, 800, 149650. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Wang, F.; Mu, R.; Huang, J.; Zhao, R.; Li, X.; Yu, K.; Li, B. Metagenomics analysis revealing the occurrence of antibiotic resistome in salt lakes. Sci. Total Environ. 2021, 790, 148262. [Google Scholar] [CrossRef] [PubMed]
- Nnadozie, C.F.; Odume, O.N. Freshwater environments as reservoirs of antibiotic resistant bacteria and their role in the dissemination of antibiotic resistance genes. Environ. Pollut. 2019, 254, 113067. [Google Scholar] [CrossRef]
- Ding, Q.; Wang, J.F.; Feng, Y.C.; Wu, Z.; Ma, L.L.; Liu, J.; Wang, Y.; Jia, L.; Gao, L.J.; Shao, P.; et al. Determination of 32 kinds of antibiotic residues in fish intestinal content by high performance liquid chromatography-tandem mass spectrometry. J. Food Saf. Qual. 2022, 13, 1141–1149. [Google Scholar]
- Xue, X.; Jia, J.; Yue, X.; Guan, Y.; Zhu, L.; Wang, Z. River contamination shapes the microbiome and antibiotic resistance in sharpbelly (Hemiculter leucisculus). Environ. Pollut. 2021, 268, 115796. [Google Scholar] [CrossRef]
- Liu, X.; Wang, H.; Zhao, H. Propagation of antibiotic resistance genes in an industrial recirculating aquaculture system located at northern China. Environ. Pollut. 2020, 261, 114155. [Google Scholar] [CrossRef]
- Lu, Z.; Na, G.; Gao, H.; Wang, L.; Bao, C.; Yao, Z. Fate of sulfonamide resistance genes in estuary environment and effect of anthropogenic activities. Sci. Total Environ. 2015, 527–528, 429–438. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Mende, D.R.; Waller, A.S.; Sunagawa, S.; Järvelin, A.I.; Chan, M.M.; Arumugam, M.; Raes, J.; Bork, P. Assessment of Metagenomic Assembly Using Simulated Next Generation Sequencing Data. PLoS ONE 2012, 7, e31386. [Google Scholar] [CrossRef]
- Nielsen, H.B.; Almeida, M.; Juncker, A.S.; Rasmussen, S.; Li, J.; Sunagawa, S.; Plichta, D.R.; Gautier, L.; Pedersen, A.G.; Le Chatelier, E.; et al. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat. Biotechnol. 2014, 32, 822–828. [Google Scholar] [CrossRef] [PubMed]
- Qin, N.; Yang, F.; Li, A.; Prifti, E.; Chen, Y.; Shao, L.; Guo, J.; Le Chatelier, E.; Yao, J.; Wu, L.; et al. Alterations of the human gut microbiome in liver cirrhosis. Nature 2014, 513, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jia, H.; Cai, X.; Zhong, H.; Feng, Q.; Sunagawa, S.; Arumugam, M.; Kultima, J.R.; Prifti, E.; Nielsen, T.; et al. An integrated catalog of reference genes in the human gut microbiome. Nat. Biotechnol. 2014, 32, 834–841. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Zeller, G.; Tap, J.; Voigt, A.Y.; Sunagawa, S.; Kultima, J.R.; Costea, P.I.; Amiot, A.; Böhm, J.; Brunetti, F.; Habermann, N.; et al. Potential of fecal microbiota for early-stage detection of colorectal cancer. Mol. Syst. Biol. 2014, 10, 766. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Sunagawa, S.; Coelho, L.P.; Chaffron, S.; Kultima, J.R.; Labadie, K.; Salazar, G.; Djahanschiri, B.; Zeller, G.; Mende, D.R.; Alberti, A.; et al. Ocean Plankton. Structure and function of the global ocean microbiome. Science 2015, 348, 794. [Google Scholar] [CrossRef]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef]
- Villar, E.; Farrant, G.K.; Follows, M.; Garczarek, L.; Speich, S.; Audic, S.; Bittner, L.; Blanke, B.; Brum, J.R.; Brunet, C.; et al. Environmental characteristics of Agulhas rings affect interocean plankton transport. Science 2015, 348, 1261447. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Huson, Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, F.H.; Fåk, F.; Nookaew, I.; Tremaroli, V.; Fagerberg, B.; Petranovic, D.; Bäckhed, F.; Nielsen, J. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat. Commun. 2012, 3, 1245. [Google Scholar] [CrossRef] [PubMed]
- Jia, B.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017, 45, D566–D573. [Google Scholar] [CrossRef]
- Martínez, J.L.; Coque, T.M.; Baquero, F. What is a resistance gene? Ranking risk in resistomes. Nat. Rev. Microbiol. 2015, 13, 116–123. [Google Scholar] [CrossRef]
- McArthur, A.G.; Waglechner, N.; Nizam, F.; Yan, A.; Azad, M.A.; Baylay, A.J.; Bhullar, K.; Canova, M.J.; De Pascale, G.; Ejim, L.; et al. The Comprehensive Antibiotic Resistance Database. Antimicrob. Agents Chemother. 2013, 57, 3348–3357. [Google Scholar] [CrossRef] [PubMed]
- Xiao, K.-Q.; Li, B.; Ma, L.; Bao, P.; Zhou, X.; Zhang, T.; Zhu, Y.-G. Metagenomic profiles of antibiotic resistance genes in paddy soils from South China. FEMS Microbiol. Ecol. 2016, 92, fiw023. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, B.; Ju, F.; Zhang, T. Exploring Variation of Antibiotic Resistance Genes in Activated Sludge over a Four-Year Period through a Metagenomic Approach. Environ. Sci. Technol. 2013, 47, 10197–10205. [Google Scholar] [CrossRef]
- Huijbers, P.M.; Graat, E.A.; Haenen, A.P.; van Santen, M.G.; van Essen-Zandbergen, A.; Mevius, D.J.; van Duijkeren, E.; van Hoek, A.H. Extended-spectrum and AmpC β-lactamase-producing Escherichia coli in broilers and people living and/or working on broiler farms: Prevalence, risk factors and molecular characteristics. J. Antimicrob. Chemother. 2014, 69, 2669–2675. [Google Scholar] [CrossRef]
- Overdevest, I.; Willemsen, I.; Rijnsburger, M.; Eustace, A.; Xu, L.; Hawkey, P.; Heck, M.; Savelkoul, P.; Vandenbroucke-Grauls, C.; van der Zwaluw, K.; et al. Extended-spectrum β-lactamase genes of Escherichia coli in chicken meat and humans, The Netherlands. Emerg. Infect. Dis. 2011, 17, 1216–1222. [Google Scholar] [CrossRef]
- Voets, G.M.; Fluit, A.C.; Scharringa, J.; Schapendonk, C.; Munckhof, T.V.D.; Hall, M.A.L.-V.; Stuart, J.C. Identical plasmid AmpC beta-lactamase genes and plasmid types in E. coli isolates from patients and poultry meat in the Netherlands. Int. J. Food Microbiol. 2013, 167, 359–362. [Google Scholar] [CrossRef]
- Miranda, C.D.; Tello, A.; Keen, P.L. Mechanisms of antimicrobial resistance in finfish aquaculture environments. Front. Microbiol. 2013, 4, 233. [Google Scholar] [CrossRef] [PubMed]
- Van Bambeke, F.; Balzi, E.; Tulkens, P.M. Antibiotic efflux pumps. Biochem. Pharmacol. 2000, 60, 457–470. [Google Scholar] [CrossRef]
- Pantosti, A.; Sanchini, A.; Monaco, M. Mechanisms of antibiotic resistance in Staphylococcus aureus. Futur. Microbiol. 2007, 2, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Mazhar, S.H.; Li, X.; Rashid, A.; Su, J.; Xu, J.; Brejnrod, A.D.; Su, J.-Q.; Wu, Y.; Zhu, Y.-G.; Zhou, S.G.; et al. Co-selection of antibiotic resistance genes, and mobile genetic elements in the presence of heavy metals in poultry farm environments. Sci. Total Environ. 2021, 755, 142702. [Google Scholar] [CrossRef]
- Hazards, E.; Panel, o.B.; Koutsoumanis, K.; Allende, A.; Álvarez-Ordóñez, A.; Bolton, D.; Bover-Cid, S.; Chemaly, M.; Davies, R.; de Cesare, A.; et al. Role played by the environment in the emergence and spread of antimicrobial resistance (AMR) through the food chain. EFSA J. 2021, 19, e06651. [Google Scholar]
- Xiong, W.; Sun, Y.; Zhang, T.; Ding, X.; Li, Y.; Wang, M.; Zeng, Z. Antibiotics, Antibiotic Resistance Genes, and Bacterial Community Composition in Fresh Water Aquaculture Environment in China. Microb. Ecol. 2015, 70, 425–432. [Google Scholar] [CrossRef]
- Liu, P.; Jia, S.; He, X.; Zhang, X.; Ye, L. Different impacts of manure and chemical fertilizers on bacterial community structure and antibiotic resistance genes in arable soils. Chemosphere 2017, 188, 455–464. [Google Scholar] [CrossRef]
- Lin, S.-M.; Zhou, X.-M.; Zhou, Y.-L.; Kuang, W.-M.; Chen, Y.-J.; Luo, L.; Dai, F.-Y. Intestinal morphology, immunity and microbiota response to dietary fibers in largemouth bass, Micropterus salmoide. Fish Shellfish Immunol. 2020, 103, 135–142. [Google Scholar] [CrossRef]
- Ruzauskas, M.; Armalytė, J.; Lastauskienė, E.; Šiugždinienė, R.; Klimienė, I.; Mockeliūnas, R.; Bartkienė, E. Microbial and Antimicrobial Resistance Profiles of Microbiota in Common Carps (Cyprinus carpio) from Aquacultured and Wild Fish Populations. Animals 2021, 11, 929. [Google Scholar] [CrossRef]
- Wang, S.-T.; Meng, X.-Z.; Dai, Y.-F.; Zhang, J.-H.; Shen, Y.; Xu, X.-Y.; Wang, R.-Q.; Li, J.-L. Characterization of the intestinal digesta and mucosal microbiome of the grass carp (Ctenopharyngodon idella). Comp. Biochem. Physiol. Part D Genom. Proteom. 2021, 37, 100789. [Google Scholar] [CrossRef]
- Kallscheuer, N.; Jogler, C. The bacterial phylum Planctomycetes as novel source for bioactive small molecules. Biotechnol. Adv. 2021, 53, 107818. [Google Scholar] [CrossRef] [PubMed]
- Kuebutornye, F.K.A.; Abarike, E.D.; Lu, Y. A review on the application of Bacillus as probiotics in aquaculture. Fish Shellfish Immunol. 2019, 87, 820–828. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Yang, R.; Wu, Q.; Ding, Y.; Wang, Z.; Zhang, J.; Lei, T.; Wu, S.; Zhang, F.; Zhang, W.; et al. First report of the optrA-carrying multidrug resistance genomic island in Campylobacter jejuni isolated from pigeon meat. Int. J. Food. Microbiol. 2021, 354, 109320. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Gao, J.; Dai, H.; Wang, Z.; Zhao, Y.; Cui, Y. Higher spreading risk of antibacterial biocide and heavy metal resistance genes than antibiotic resistance genes in aerobic granular sludge. Environ. Res. 2022, 212, 113356. [Google Scholar] [CrossRef] [PubMed]
- Du, B.; Yang, Q.; Wang, R.; Wang, R.; Wang, Q.; Xin, Y. Evolution of Antibiotic Resistance and the Relationship between the Antibiotic Resistance Genes and Microbial Compositions under Long-Term Exposure to Tetracycline and Sulfamethoxazole. Int. J. Environ. Res. Public Health 2019, 16, 4681. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xin, Z.; Zhang, Y.; Chen, J.; Yan, J.; Li, H.; Hu, H. Long-term manure application increased the levels of antibiotics and antibiotic resistance genes in a greenhouse soil. Appl. Soil Ecol. 2017, 121, 193–200. [Google Scholar] [CrossRef]
- Pan, Z.; Yang, S.; Zhao, L.; Li, X.; Weng, L.; Sun, Y.; Li, Y. Temporal and spatial variability of antibiotics in agricultural soils from Huang-Huai-Hai Plain, northern China. Chemosphere 2021, 272, 129803. [Google Scholar] [CrossRef]
- Liu, C.; Chen, Y.; Li, X.; Zhang, Y.; Ye, J.; Huang, H.; Zhu, C. Temporal effects of repeated application of biogas slurry on soil antibiotic resistance genes and their potential bacterial hosts. Environ. Pollut. 2020, 258, 113652. [Google Scholar] [CrossRef]
- Rahman, M.M.; Shan, J.; Yang, P.; Shang, X.; Xia, Y.; Yan, X. Effects of long-term pig manure application on antibiotics, abundance of antibiotic resistance genes (ARGs), anammox and denitrification rates in paddy soils. Environ. Pollut. 2018, 240, 368–377. [Google Scholar] [CrossRef]
- Song, J.; Shen, Q.; Wang, L.; Qiu, G.; Shi, J.; Xu, J.; Brookes, P.C.; Liu, X. Effects of Cd, Cu, Zn and their combined action on microbial biomass and bacterial community structure. Environ. Pollut. 2018, 243, 510–518. [Google Scholar] [CrossRef]
- Wang, X.; Lan, B.; Fei, H.; Wang, S.; Zhu, G. Heavy metal could drive co-selection of antibiotic resistance in terrestrial subsurface soils. J. Hazard. Mater. 2021, 411, 124848. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Wang, M.; Zhong, X.; Liu, P.; Xie, X.; Wangxiao, J.; Sun, Y. Dissemination of resistance genes in duck/fish polyculture ponds in Guangdong Province: Correlations between Cu and Zn and antibiotic resistance genes. Environ. Sci. Pollut. Res. 2019, 26, 8182–8193. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Lv, Z.; Shen, Y.; Liu, D.; Fu, Y.; Zhou, L.; Liu, W.; Chen, K.; Ye, H.; Xia, X.; et al. Metagenomic insights into differences in environmental resistome profiles between integrated and monoculture aquaculture farms in China. Environ. Int. 2020, 144, 106005. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Q.; Zhou, X.; Liu, Y.; Ding, Q.; Wu, Z.; Deng, J.; Zuo, J.; Yuan, L.; Shao, P.; Cheng, B.; et al. Bacterial Community and Antibiotic Resistance Gene Profiles of Fish Gut Contents and Their Aquaculture Environment in Tianjin, China. Aquac. J. 2022, 2, 269-284. https://doi.org/10.3390/aquacj2040016
Wang Q, Zhou X, Liu Y, Ding Q, Wu Z, Deng J, Zuo J, Yuan L, Shao P, Cheng B, et al. Bacterial Community and Antibiotic Resistance Gene Profiles of Fish Gut Contents and Their Aquaculture Environment in Tianjin, China. Aquaculture Journal. 2022; 2(4):269-284. https://doi.org/10.3390/aquacj2040016
Chicago/Turabian StyleWang, Qiushui, Xin Zhou, Yue Liu, Qi Ding, Zan Wu, Jie Deng, Jia Zuo, Liyan Yuan, Peng Shao, Bo Cheng, and et al. 2022. "Bacterial Community and Antibiotic Resistance Gene Profiles of Fish Gut Contents and Their Aquaculture Environment in Tianjin, China" Aquaculture Journal 2, no. 4: 269-284. https://doi.org/10.3390/aquacj2040016
APA StyleWang, Q., Zhou, X., Liu, Y., Ding, Q., Wu, Z., Deng, J., Zuo, J., Yuan, L., Shao, P., Cheng, B., & Gao, L. (2022). Bacterial Community and Antibiotic Resistance Gene Profiles of Fish Gut Contents and Their Aquaculture Environment in Tianjin, China. Aquaculture Journal, 2(4), 269-284. https://doi.org/10.3390/aquacj2040016