
Traumatic Brain Injury and Secondary Neurodegenerative Disease

 ,
,  ,
, 
Abstract
1. Introduction
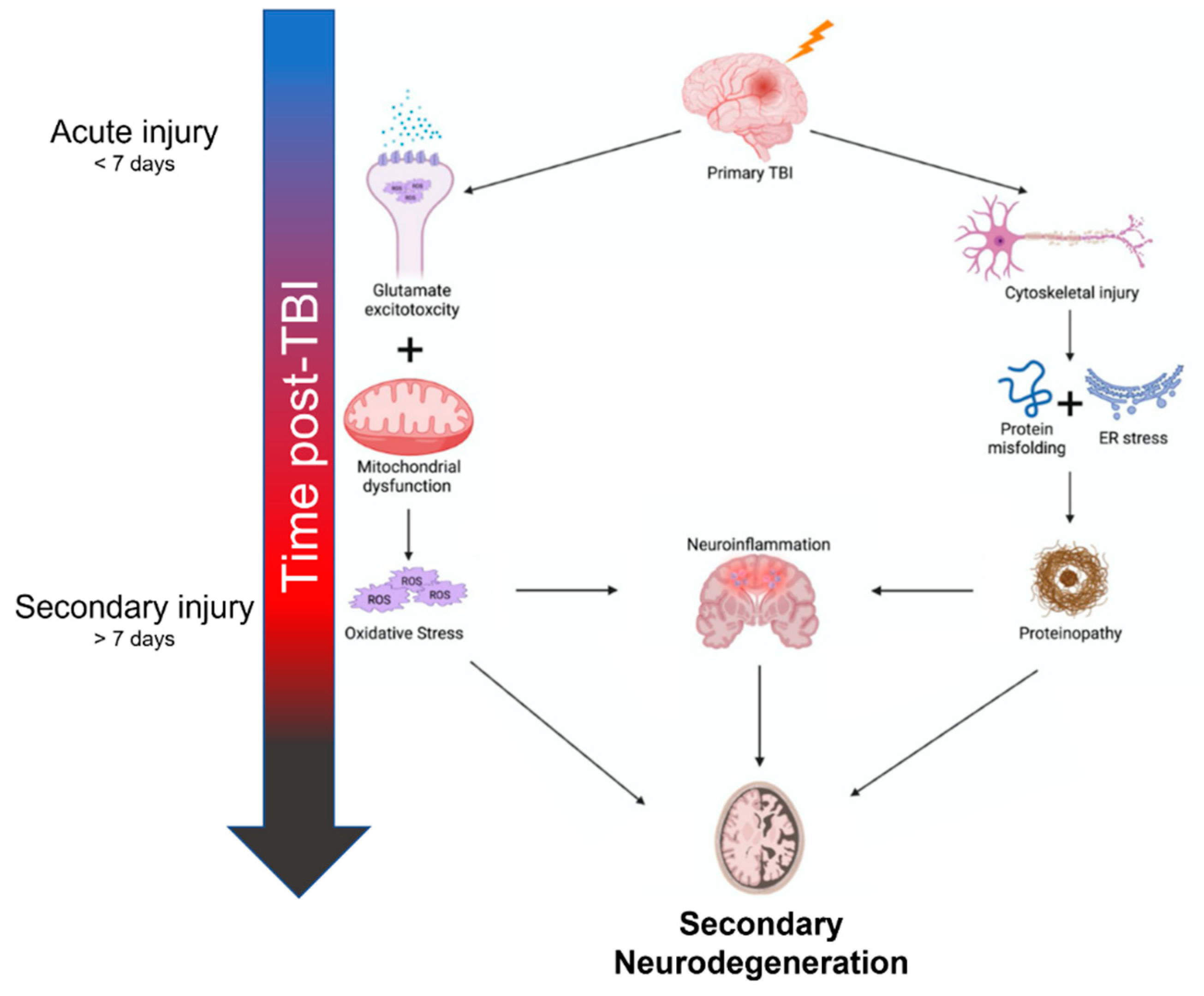
2. Mechanisms of Neurodegeneration after TBI
3. Imaging
4. Cognitive and Behavioral Manifestations
5. Therapeutic Targets
5.1. Oxidative Stress
5.2. Cell-Based Therapies
5.3. Aggregation-Prone Proteins
5.4. Neuroinflammation

{kind=link}
| Therapeutic Target | Outcomes & Mechanism of Action | References |
|---|---|---|
| Oxidative Stress | ||
| PEG-SOD | Catalyzes the degradation of superoxide radicals; combined with PEG molecules to increase in vivo half-life | [64] |
| Polyphenols & Flavonoids | Water-soluble antioxidants; directly react with ROS in addition to stimulating the Nrf2-ARE pathway | [65,66,67,68] |
| Mitoquinone | Acts as a renewable antioxidant to reduce mitochondrial ROS | [70] |
| Cell-based Therapies | ||
| Neural stem cells | Improves motor recovery and cognition by replacing neurons lost to neurodegeneration | [73,74,75] |
| Mesenchymal stromal stem cells | Reduces proinflammatory mediators and improved functional recovery. IL-10 overexpression alters microglial polarization in favor of anti-inflammatory processes | [76,77] |
| Aggregation-prone Proteins | ||
| Tau | Preventing pathologic accumulation via immunization against phosphorylated tau improves neurocognitive outcomes | [84,85,86] |
| Amyloid-beta protein | Aβ may be targeted through enhanced clearance (immunization), decreased production (α-secretase overexpression), or decreased aggregation (Aβ binding molecules). | [87,88,89,90] |
| Neuroinflammation | ||
| Minocycline | Attenuates microglial activation and improves functional outcomes | [92,93,94] |
| Doxycycline | Decreases MMP-9 activity and preserves blood-brain-barrier integrity | [95] |
| Hydroxychloroquine/chloroquine | Attenuates microglial activation and preserves blood-brain-barrier integrity | [96,97] |
| PPAR agonists | Attenuates microglial activation and mitochondrial dysfunction, decreases TBI lesion sizes | [98,99,100] |
| Cannabinoid 2 receptor agonist | Prevents neuronal degeneration and preserves blood-brain-barrier integrity | [101] |
| Inflammasomes | Decreases pro-inflammatory mediators such as caspase-1, IL-18, & IL-1β. Can also reduce pyroptotic cell death by inhibiting gasdermin D cleavage. | [102,103,104] |
| Interferon-beta | Attenuates neuroinflammation, decreases lesion volume, and improves long-term functional outcomes | [105] |
6. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Fesharaki-Zadeh, A. Chronic Traumatic Encephalopathy: A Brief Overview. Front. Neurol. 2019, 10, 713. [Google Scholar] [CrossRef] [PubMed]
- Raymont, V.; Thayanandan, T. What do we know about the risks of developing dementia after traumatic brain injury? Minerva Med. 2021, 112, 288–297. [Google Scholar] [CrossRef]
- Crane, P.K.; Gibbons, L.E.; Dams-O’Connor, K.; Trittschuh, E.; Leverenz, J.B.; Keene, C.D.; Sonnen, J.; Montine, T.J.; Bennett, D.A.; Leurgans, S.; et al. Association between Traumatic Brain Injury and Late Life Neurodegenerative Conditions and Neuropathological Findings. JAMA Neurol. 2016, 73, 1062–1069. [Google Scholar] [CrossRef]
- Delic, V.; Beck, K.D.; Pang, K.C.H.; Citron, B.A. Biological links between traumatic brain injury and Parkinson’s disease. Acta Neuropathol. Commun. 2020, 8, 45. [Google Scholar] [CrossRef]
- Ramos-Cejudo, J.; Wisniewski, T.; Marmar, C.; Zetterberg, H.; Blennow, K.; de Leon, M.J.; Fossati, S. Traumatic Brain Injury and Alzheimer’s Disease: The Cerebrovascular Link. eBioMedicine 2018, 28, 21–30. [Google Scholar] [CrossRef]
- VanItallie, T.B. Traumatic brain injury (TBI) in collision sports: Possible mechanisms of transformation into chronic traumatic encephalopathy (CTE). Metabolism 2019, 100, 153943. [Google Scholar] [CrossRef]
- Werner, C.; Engelhard, K. Pathophysiology of traumatic brain injury. Br. J. Anaesth. 2007, 99, 4–9. [Google Scholar] [CrossRef]
- Gaetz, M. The neurophysiology of brain injury. Clin. Neurophysiol. 2004, 115, 4–18. [Google Scholar] [CrossRef]
- Johnstone, V.P.; Shultz, S.R.; Yan, E.B.; O’Brien, T.J.; Rajan, R. The acute phase of mild traumatic brain injury is characterized by a distance-dependent neuronal hypoactivity. J. Neurotrauma. 2014, 31, 1881–1895. [Google Scholar] [CrossRef]
- Risbrough, V.B.; Vaughn, M.N.; Friend, S.F. Role of Inflammation in Traumatic Brain Injury–Associated Risk for Neuropsychiatric Disorders: State of the Evidence and Where Do We Go from Here. Biol. Psychiatry 2022, 91, 438–448. [Google Scholar] [CrossRef]
- Mayer, A.R.; Quinn, D.K.; Master, C.L. The spectrum of mild traumatic brain injury: A review. Neurology 2017, 89, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Saxena, S. ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 2017, 13, 477–491. [Google Scholar] [CrossRef]
- Freire, M.A.M. Pathophysiology of neurodegeneration following traumatic brain injury. West Indian Med. J. 2012, 61, 751–755. [Google Scholar]
- Cruz-Haces, M.; Tang, J.; Acosta, G.; Fernandez, J.; Shi, R. Pathological correlations between traumatic brain injury and chronic neurodegenerative diseases. Transl. Neurodegener. 2017, 6, 20. [Google Scholar] [CrossRef]
- Khatri, N.; Thakur, M.; Pareek, V.; Kumar, S.; Sharma, S.; Datusalia, A.K. Oxidative Stress: Major Threat in Traumatic Brain Injury. CNS Neurol. Disord. Drug Targets 2018, 17, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.-I.; Jou, M.-J. Oxidative stress caused by mitochondrial calcium overload. Ann. N. Y. Acad. Sci. 2010, 1201, 183–188. [Google Scholar] [CrossRef]
- Ma, Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, H. Targeting the NF-E2-Related Factor 2 Pathway: A Novel Strategy for Traumatic Brain Injury. Mol. Neurobiol. 2018, 55, 1773–1785. [Google Scholar] [CrossRef]
- Egawa, J.; Pearn, M.L.; Lemkuil, B.P.; Patel, P.M.; Head, B.P. Membrane lipid rafts and neurobiology: Age-related changes in membrane lipids and loss of neuronal function. J. Physiol. 2016, 594, 4565–4579. [Google Scholar] [CrossRef] [PubMed]
- Head, B.P.; Peart, J.N.; Panneerselvam, M.; Yokoyama, T.; Pearn, M.L.; Niesman, I.R.; Bonds, J.A.; Schilling, J.M.; Miyanohara, A.; Headrick, J.; et al. Loss of Caveolin-1 Accelerates Neurodegeneration and Aging. PLoS ONE 2010, 5, e15697. [Google Scholar] [CrossRef]
- Webster, K.M.; Wright, D.; Sun, M.; Semple, B.D.; Ozturk, E.; Stein, D.G.; O’Brien, T.; Shultz, S.R. Progesterone treatment reduces neuroinflammation, oxidative stress and brain damage and improves long-term outcomes in a rat model of repeated mild traumatic brain injury. J. Neuroinflamm. 2015, 12, 238. [Google Scholar] [CrossRef]
- Johnson, V.E.; Stewart, W.; Smith, D.H. Axonal pathology in traumatic brain injury. Exp. Neurol. 2013, 246, 35–43. [Google Scholar] [CrossRef]
- Jang, S.H. Diagnostic Problems in Diffuse Axonal Injury. Diagnostics 2020, 10, 117. [Google Scholar] [CrossRef]
- Lozano, D.; Schimmel, S.J.; Acosta, S. Neuroinflammation in traumatic brain injury: A chronic response to an acute injury. Brain Circ. 2017, 3, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Pearn, M.L.; Niesman, I.R.; Egawa, J.; Sawada, A.; Almenar-Queralt, A.; Shah, S.B.; Duckworth, J.L.; Head, B.P. Pathophysiology Associated with Traumatic Brain Injury: Current Treatments and Potential Novel Therapeutics. Cell. Mol. Neurobiol. 2017, 37, 571–585. [Google Scholar] [CrossRef]
- O’Brien, W.T.; Pham, L.; Symons, G.F.; Monif, M.; Shultz, S.R.; McDonald, S.J. The NLRP3 inflammasome in traumatic brain injury: Potential as a biomarker and therapeutic target. J. Neuroinflamm. 2020, 17, 104–112. [Google Scholar] [CrossRef]
- Gerakis, Y.; Hetz, C. Emerging roles of ER stress in the etiology and pathogenesis of Alzheimer’s disease. FEBS J. 2018, 285, 995–1011. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.Z.A.; Zhao, D.; Khan, S.H.; Yang, L. Unfolded Protein Response Pathways in Neurodegenerative Diseases. J. Mol. Neurosci. 2015, 57, 529–537. [Google Scholar] [CrossRef]
- Washington, P.M.; Villapol, S.; Burns, M.P. Polypathology and Dementia After Brain Trauma: Does Brain Injury Trigger Distinct Neurodegenerative Diseases, or Should They Be Classified Together as Traumatic Encephalopathy? Exp. Neurol. 2016, 275 Pt 3, 381–388. [Google Scholar] [CrossRef]
- Uryu, K.; Chen, X.-H.; Martinez, D.; Browne, K.D.; Johnson, V.E.; Graham, D.I.; Lee, V.M.-Y.; Trojanowski, J.Q.; Smith, D.H. Multiple proteins implicated in neurodegenerative diseases accumulate in axons after brain trauma in humans. Exp. Neurol. 2007, 208, 185–192. [Google Scholar] [CrossRef]
- Johnson, V.E.; Stewart, W.; Smith, D.H. Widespread τ and amyloid-β pathology many years after a single traumatic brain injury in humans. Brain Pathol. 2012, 22, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, W.L.; MacKinnon, M.-A.; Stewart, J.E.; Graham, D.I. Stereology of cerebral cortex after traumatic brain injury matched to the Glasgow Outcome Score. Brain 2010, 133, 139–160. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.H.; Doyle, D.; Ford, I.; Gennarelli, T.A.; Graham, D.I.; McLellan, D.R. Diffuse axonal injury in head injury: Definition, diagnosis and grading. Histopathology 1989, 15, 49–59. [Google Scholar] [CrossRef]
- Hofman, P.A.; Verhey, F.R.; Wilmink, J.T.; Rozendaal, N.; Jolles, J. Brain lesions in patients visiting a memory clinic with postconcussional sequelae after mild to moderate brain injury. J. Neuropsychiatry Clin. Neurosci. 2002, 14, 176–184. [Google Scholar] [CrossRef]
- Bigler, E.D.; Tate, D.F. Brain volume, intracranial volume, and dementia. Investig. Radiol. 2001, 36, 539–546. [Google Scholar] [CrossRef]
- Mamere, A.E.; Saraiva, L.A.L.; Matos, A.L.M.; Carneiro, A.A.O.; Santos, A.C. Evaluation of Delayed Neuronal and Axonal Damage Secondary to Moderate and Severe Traumatic Brain Injury Using Quantitative MR Imaging Techniques. Am. J. Neuroradiol. 2009, 30, 947–952. [Google Scholar] [CrossRef] [PubMed]
- Graham, N.S.; Sharp, D.J. Understanding neurodegeneration after traumatic brain injury: From mechanisms to clinical trials in dementia. J. Neurol. Neurosurg. Psychiatry 2019, 90, 1221–1233. [Google Scholar] [CrossRef]
- Bigler, E.D. Anterior and middle cranial fossa in traumatic brain injury: Relevant neuroanatomy and neuropathology in the study of neuropsychological outcome. Neuropsychology 2007, 21, 515–531. [Google Scholar] [CrossRef]
- De La Plata, C.D.M.; Garces, J.; Kojori, E.S.; Grinnan, J.; Krishnan, K.; Pidikiti, R.; Spence, J.; Devous, M.D.; Moore, C.; McColl, R.; et al. Deficits in Functional Connectivity of Hippocampal and Frontal Lobe Circuits After Traumatic Axonal Injury. Arch. Neurol. 2011, 68, 74–84. [Google Scholar]
- Veeramuthu, V.; Narayanan, V.; Kuo, T.L.; Delano-Wood, L.; Chinna, K.; Bondi, M.W.; Waran, V.; Ganesan, D.; Ramli, N. Diffusion Tensor Imaging Parameters in Mild Traumatic Brain Injury and Its Correlation with Early Neuropsychological Impairment: A Longitudinal Study. J. Neurotrauma 2015, 32, 1497–1509. [Google Scholar] [CrossRef]
- Farbota, K.D.; Bendlin, B.B.; Alexander, A.L.; Rowley, H.A.; Dempsey, R.J.; Johnson, S.C. Longitudinal diffusion tensor imaging and neuropsychological correlates in traumatic brain injury patients. Front. Hum. Neurosci. 2012, 6, 160. [Google Scholar] [CrossRef] [PubMed]
- Hellyer, P.J.; Leech, R.; Ham, T.E.; Bonnelle, V.; Sharp, D.J. Individual prediction of white matter injury following traumatic brain injury. Ann. Neurol. 2013, 73, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Holleran, L.; Kim, J.H.; Gangolli, M.; Stein, T.; Alvarez, V.; McKee, A.; Brody, D.L. Axonal disruption in white matter underlying cortical sulcus tau pathology in chronic traumatic encephalopathy. Acta Neuropathol. 2017, 133, 367–380. [Google Scholar] [CrossRef]
- Palacios, E.M.; Sala-Llonch, R.; Junque, C.; Roig, T.; Tormos, J.M.; Bargallo, N.; Vendrell, P. White matter integrity related to functional working memory networks in traumatic brain injury. Neurology 2012, 78, 852–860. [Google Scholar] [CrossRef]
- Wang, J.Y.; Bakhadirov, K.; Abdi, H.; Devous, M.D.; De La Plata, C.D.M.; Moore, C.; Madden, C.J.; Diaz-Arrastia, R. Longitudinal changes of structural connectivity in traumatic axonal injury. Neurology 2011, 77, 818–826. [Google Scholar] [CrossRef] [PubMed]
- Sidaros, A.; Engberg, A.W.; Sidaros, K.; Liptrot, M.G.; Herning, M.; Petersen, P.; Paulson, O.B.; Jernigan, T.L.; Rostrup, E. Diffusion tensor imaging during recovery from severe traumatic brain injury and relation to clinical outcome: A longitudinal study. Brain 2008, 131, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Stern, R.A.; Adler, C.H.; Chen, K.; Navitsky, M.; Luo, J.; Dodick, D.W.; Alosco, M.L.; Tripodis, Y.; Goradia, D.D.; Martin, B.; et al. Tau Positron-Emission Tomography in Former National Football League Players. N. Engl. J. Med. 2019, 380, 1716–1725. [Google Scholar] [CrossRef]
- Hong, Y.T.; Veenith, T.; Dewar, D.; Outtrim, J.G.; Mani, V.; Williams, C.; Pimlott, S.; Hutchinson, P.J.; Tavares, A.; Canales, R.; et al. Amyloid imaging with carbon 11-labeled Pittsburgh compound B for traumatic brain injury. JAMA Neurol. 2014, 71, 23–31. [Google Scholar] [CrossRef]
- Mielke, M.M.; Savica, R.; Wiste, H.J.; Weigand, S.D.; Vemuri, P.; Knopman, D.S.; Lowe, V.J.; Roberts, R.O.; Machulda, M.M.; Geda, Y.E.; et al. Head trauma and in vivo measures of amyloid and neurodegeneration in a population-based study. Neurology 2014, 82, 70–76. [Google Scholar] [CrossRef]
- Scott, G.; Ramlackhansingh, A.F.; Edison, P.; Hellyer, P.; Cole, J.; Veronese, M.; Leech, R.; Greenwood, R.J.; Turkheimer, F.E.; Gentleman, S.M.; et al. Amyloid pathology and axonal injury after brain trauma. Neurology 2016, 86, 821–828. [Google Scholar] [CrossRef]
- Chen, S.T.; Siddarth, P.; Merrill, D.A.; Martinez, J.; Emerson, N.D.; Liu, J.; Wong, K.-P.; Satyamurthy, N.; Giza, C.C.; Huang, S.-C.; et al. FDDNP-PET Tau Brain Protein Binding Patterns in Military Personnel with Suspected Chronic Traumatic Encephalopathy1. J. Alzheimer’s Dis. 2018, 65, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Leavitt, M.J.; Bernick, C.B.; Leger, G.C.; Rabinovici, G.; Banks, S.J. A Systematic Review of Positron Emission Tomography of Tau, Amyloid Beta, and Neuroinflammation in Chronic Traumatic Encephalopathy: The Evidence to Date. J. Neurotrauma 2018, 35, 2015–2024. [Google Scholar] [CrossRef]
- Pierre, K.; Dyson, K.; Dagra, A.; Williams, E.; Porche, K.; Lucke-Wold, B. Chronic Traumatic Encephalopathy: Update on Current Clinical Diagnosis and Management. Biomedicines 2021, 9, 415. [Google Scholar] [CrossRef] [PubMed]
- Barrio, J.R.; Small, G.W.; Wong, K.P.; Huang, S.C.; Liu, J.; Merrill, D.A.; Giza, C.C.; Fitzsimmons, R.P.; Omalu, B.; Bailes, J.; et al. In Vivo characterization of chronic traumatic encephalopathy using [F-18]FDDNP PET brain imaging. Proc. Natl. Acad. Sci. USA 2015, 112, E2039–E2047. [Google Scholar] [CrossRef] [PubMed]
- Olsen, A.; Babikian, T.; Bigler, E.D.; Caeyenberghs, K.; Conde, V.; Dams-O’Connor, K.; Dobryakova, E.; Genova, H.; Grafman, J.; Håberg, A.K.; et al. Toward a global and reproducible science for brain imaging in neurotrauma: The ENIGMA adult moderate/severe traumatic brain injury working group. Brain Imaging Behav. 2021, 15, 526–554. [Google Scholar] [CrossRef] [PubMed]
- Amyot, F.; Arciniegas, D.B.; Brazaitis, M.P.; Curley, K.C.; Diaz-Arrastia, R.; Gandjbakhche, A.; Herscovitch, P.; Hinds, S.R., 2nd; Manley, G.T.; Pacifico, A.; et al. A Review of the Effectiveness of Neuroimaging Modalities for the Detection of Traumatic Brain Injury. J. Neurotrauma 2015, 32, 1693–1721. [Google Scholar] [CrossRef]
- Christensen, J.; Wright, D.K.; Yamakawa, G.R.; Shultz, S.R.; Mychasiuk, R. Repetitive Mild Traumatic Brain Injury Alters Glymphatic Clearance Rates in Limbic Structures of Adolescent Female Rats. Sci. Rep. 2020, 10, 6254. [Google Scholar]
- Piantino, J.; Schwartz, D.L.; Luther, M.; Newgard, C.; Silbert, L.; Raskind, M.; Pagulayan, K.; Kleinhans, N.; Iliff, J.; Peskind, E. Link between Mild Traumatic Brain Injury, Poor Sleep, and Magnetic Resonance Imaging: Visible Perivascular Spaces in Veterans. J. Neurotrauma 2021, 38, 2391–2399. [Google Scholar] [CrossRef]
- Kaur, J.; Davoodi-Bojd, E.; Fahmy, L.M.; Zhang, L.; Ding, G.; Hu, J.; Zhang, Z.; Chopp, M.; Jiang, Q. Magnetic Resonance Imaging and Modeling of the Glymphatic System. Diagnostics 2020, 10, 344. [Google Scholar] [CrossRef]
- Taoka, T.; Naganawa, S. Glymphatic imaging using MRI. J. Magn. Reson. Imaging 2020, 51, 11–24. [Google Scholar] [CrossRef]
- Gupta, R.K.; Prasad, S. Age-Dependent Alterations in the Interactions of NF-κB and N-myc with GLT-1/EAAT2 Promoter in the Pericontusional Cortex of Mice Subjected to Traumatic Brain Injury. Mol. Neurobiol. 2016, 53, 3377–3388. [Google Scholar] [CrossRef] [PubMed]
- MacFarlane, M.P.; Glenn, T.C. Neurochemical cascade of concussion. Brain Inj. 2015, 29, 139–153. [Google Scholar] [CrossRef] [PubMed]
- Esopenko, C.; Levine, B. Aging, Neurodegenerative Disease, and Traumatic Brain Injury: The Role of Neuroimaging. J. Neurotrauma 2015, 32, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Sen, N. Traumatic brain injury: A risk factor for neurodegenerative diseases. Rev. Neurosci. 2016, 27, 93–100. [Google Scholar] [CrossRef]
- Borgen, I.M.; Røe, C.; Brunborg, C.; Tenovuo, O.; Azouvi, P.; Dawes, H.; Majdan, M.; Ranta, J.; Rusnak, M.; Wiegers, E.J.; et al. Care transitions in the first 6 months following traumatic brain injury: Lessons from the CENTER-TBI study. Ann. Phys. Rehabil. Med. 2021, 64, 101458. [Google Scholar] [CrossRef]
- Abdelmalik, P.A.; Draghic, N.; Ling, G.S.F. Management of moderate and severe traumatic brain injury. Transfusion 2019, 59, 1529–1538. [Google Scholar] [CrossRef]
- Kemper, B.; Von Wild, K. Neuropsychological fields in early neurotrauma rehabilitation. Zentralbl. Neurochir. 1999, 60, 168–171. [Google Scholar]
- Prince, C.; Bruhns, M.E. Evaluation and Treatment of Mild Traumatic Brain Injury: The Role of Neuropsychology. Brain Sci. 2017, 7, 105. [Google Scholar] [CrossRef]
- Sheinerman, K.S.; Umansky, S.R. Early detection of neurodegenerative diseases: Circulating brain-enriched microRNA. Cell Cycle. 2013, 12, 1–2. [Google Scholar] [CrossRef]
- Tartaglia, M.C.; Hazrati, L.-N.; Davis, K.D.; Green, R.E.A.; Wennberg, R.; Mikulis, D.; Ezerins, L.J.; Keightley, M.; Tator, C. Chronic traumatic encephalopathy and other neurodegenerative proteinopathies. Front. Hum. Neurosci. 2014, 8, 30. [Google Scholar] [CrossRef]
- McKee, A.C.; Stein, T.; Nowinski, C.J.; Stern, R.; Daneshvar, D.; Alvarez, V.E.; Lee, H.-S.; Hall, G.; Wojtowicz, S.M.; Baugh, C.; et al. The spectrum of disease in chronic traumatic encephalopathy. Brain 2013, 136, 43–64. [Google Scholar] [CrossRef] [PubMed]
- McKee, A.C.; Cantu, R.C.; Nowinski, C.J.; Hedley-Whyte, E.T.; Gavett, B.; Budson, A.E.; Santini, V.E.; Lee, H.-S.; Kubilus, C.A.; Stern, R. Chronic Traumatic Encephalopathy in Athletes: Progressive Tauopathy After Repetitive Head Injury. J. Neuropathol. Exp. Neurol. 2009, 68, 709–735. [Google Scholar] [CrossRef]
- Deb, S.; Burns, J. Neuropsychiatric consequences of traumatic brain injury: A comparison between two age groups. Brain Inj. 2007, 21, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.; Rosenberg, P.; Miles, Q.S.; Patadia, D.; Treiber, K.; Bertrand, M.; Norton, M.; Steinberg, M.; Tschanz, J.; Lyketsos, C. Neuropsychiatric Symptoms in Dementia Patients with and without a History of Traumatic Brain Injury. J. Neuropsychiatry Clin. Neurosci. 2010, 22, 166–172. [Google Scholar] [CrossRef]
- Jordan, B.D. The clinical spectrum of sport-related traumatic brain injury. Nat. Rev. Neurol. 2013, 9, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Bray, M.J.C.; Richey, L.N.; Bryant, B.R.; Krieg, A.; Jahed, S.; Tobolowsky, W.; LoBue, C.; Peters, M.E. Traumatic brain injury alters neuropsychiatric symptomatology in all-cause dementia. Alzheimer’s Dement. 2021, 17, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Padmakumar, S.; Kulkarni, P.; Ferris, C.F.; Bleier, B.S.; Amiji, M.M. Traumatic brain injury and the development of parkinsonism: Understanding pathophysiology, animal models, and therapeutic targets. Biomed. Pharmacother. 2022, 149, 112812. [Google Scholar] [CrossRef]
- Anderson, K.E. Behavioral disturbances in Parkinson’s disease. Dialog. Clin. Neurosci. 2004, 6, 323–332. [Google Scholar] [CrossRef]
- Srinivasan, G.; Brafman, D.A. The Emergence of Model Systems to Investigate the Link Between Traumatic Brain Injury and Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 813544. [Google Scholar] [CrossRef]
- Bature, F.; Guinn, B.; Pang, D.; Pappas, Y. Signs and symptoms preceding the diagnosis of Alzheimer’s disease: A systematic scoping review of literature from 1937 to 2016. BMJ Open 2017, 7, e015746. [Google Scholar] [CrossRef] [PubMed]
- Muizelaar, J.P.; Marmarou, A.; Young, H.F.; Choi, S.C.; Wolf, A.; Schneider, R.L.; Kontos, H.A. Improving the outcome of severe head injury with the oxygen radical scavenger polyethylene glycol-conjugated superoxide dismutase: A Phase II trial. J. Neurosurg. 1993, 78, 375–382. [Google Scholar] [CrossRef]
- Fang, J.; Wang, H.; Zhou, J.; Dai, W.; Zhu, Y.; Zhou, Y.; Wang, X.; Zhou, M.-L. Baicalin provides neuroprotection in traumatic brain injury mice model through Akt/Nrf2 pathway. Drug Des. Dev. Ther. 2018, 12, 2497–2508. [Google Scholar] [CrossRef]
- Shi, Z.; Qiu, W.; Xiao, G.; Cheng, J.; Zhang, N. Resveratrol Attenuates Cognitive Deficits of Traumatic Brain Injury by Activating p38 Signaling in the Brain. Med Sci. Monit. 2018, 24, 1097–1103. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, C.; Peng, W.; Xia, Z.; Gan, P.; Huang, W.; Shi, Y.; Fan, R. Hydroxysafflor yellow A exerts antioxidant effects in a rat model of traumatic brain injury. Mol. Med. Rep. 2016, 14, 3690–3696. [Google Scholar] [CrossRef]
- Xu, J.; Wang, H.; Ding, K.; Zhang, L.; Wang, C.; Li, T.; Wei, W.; Lu, X. Luteolin provides neuroprotection in models of traumatic brain injury via the Nrf2–ARE pathway. Free Radic. Biol. Med. 2014, 71, 186–195. [Google Scholar] [CrossRef]
- Ismail, H.; Shakkour, Z.; Tabet, M.; Abdelhady, S.; Kobaisi, A.; Abedi, R.; Nasrallah, L.; Pintus, G.; Al-Dhaheri, Y.; Mondello, S.; et al. Traumatic Brain Injury: Oxidative Stress and Novel Anti-Oxidants Such as Mitoquinone and Edaravone. Antioxidants 2020, 9, 943. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, H.; Shen, R.; Fang, J.; Yang, Y.; Dai, W.; Zhu, Y.; Zhou, M. Mitochondrial-targeted antioxidant MitoQ provides neuroprotection and reduces neuronal apoptosis in experimental traumatic brain injury possibly via the Nrf2-ARE pathway. Am. J. Transl. Res. 2018, 10, 1887–1899. [Google Scholar]
- Bonilla, C.; Zurita, M. Cell-Based Therapies for Traumatic Brain Injury: Therapeutic Treatments and Clinical Trials. Biomedicines 2021, 9, 669. [Google Scholar] [CrossRef]
- Xiong, Y.; Mahmood, A.; Chopp, M. Current understanding of neuroinflammation after traumatic brain injury and cell-based therapeutic opportunities. Chin. J. Traumatol. 2018, 21, 137–151. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Acosta, S.; Tuazon, J.P.; Xu, K.; Nguyen, H.; Lippert, T.; Liska, M.G.; Semechkin, A.; Garitaonandia, I.; Gonzalez, R.; et al. Human parthenogenetic neural stem cell grafts promote multiple regenerative processes in a traumatic brain injury model. Theranostics 2019, 9, 1029–1046. [Google Scholar] [CrossRef]
- Xiong, L.-L.; Hu, Y.; Zhang, P.; Zhang, Z.; Li, L.-H.; Gao, G.-D.; Zhou, X.-F.; Wang, T.-H. Neural Stem Cell Transplantation Promotes Functional Recovery from Traumatic Brain Injury via Brain Derived Neurotrophic Factor-Mediated Neuroplasticity. Mol. Neurobiol. 2018, 55, 2696–2711. [Google Scholar] [CrossRef]
- Haus, D.L.; López-Velázquez, L.; Gold, E.M.; Cunningham, K.M.; Perez, H.; Anderson, A.J.; Cummings, B.J. Transplantation of human neural stem cells restores cognition in an immunodeficient rodent model of traumatic brain injury. Exp. Neurol. 2016, 281, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Liu, Y.; Yan, K.; Chen, L.; Chen, X.-R.; Li, P.; Chen, F.-F.; Jiang, X.-D. Anti-inflammatory and immunomodulatory mechanisms of mesenchymal stem cell transplantation in experimental traumatic brain injury. J. Neuroinflamm. 2013, 10, 571. [Google Scholar] [CrossRef]
- Peruzzaro, S.T.; Andrews, M.M.M.; Al-Gharaibeh, A.; Pupiec, O.; Resk, M.; Story, D.; Maiti, P.; Rossignol, J.; Dunbar, G.L. Transplantation of mesenchymal stem cells genetically engineered to overexpress interleukin-10 promotes alternative inflammatory response in rat model of traumatic brain injury. J. Neuroinflamm. 2019, 16, 2. [Google Scholar] [CrossRef]
- Bird, S.M.; Sohrabi, H.R.; Sutton, T.A.; Weinborn, M.; Rainey-Smith, S.R.; Brown, B.; Patterson, L.; Taddei, K.; Gupta, V.; Carruthers, M.; et al. Cerebral amyloid-β accumulation and deposition following traumatic brain injury—A narrative review and meta-analysis of animal studies. Neurosci. Biobehav. Rev. 2016, 64, 215–228. [Google Scholar] [CrossRef]
- Ojo, J.O.; Mouzon, B.; Algamal, M.; Leary, P.; Lynch, C.; Abdullah, L.; Evans, J.; Mullan, M.; Bachmeier, C.; Stewart, W.; et al. Chronic Repetitive Mild Traumatic Brain Injury Results in Reduced Cerebral Blood Flow, Axonal Injury, Gliosis, and Increased T-Tau and Tau Oligomers. J. Neuropathol. Exp. Neurol. 2016, 75, 636–655. [Google Scholar] [CrossRef]
- Ikonomovic, M.D.; Abrahamson, E.E.; Carlson, S.W.; Graham, S.H.; Dixon, C.E. Novel therapies for combating chronic neuropathological sequelae of TBI. Neuropharmacology 2019, 145, 160–176. [Google Scholar] [CrossRef]
- Wu, Y.; Wu, H.; Zeng, J.; Pluimer, B.; Dong, S.; Xie, X.; Guo, X.; Ge, T.; Liang, X.; Feng, S.; et al. Mild traumatic brain injury induces microvascular injury and accelerates Alzheimer-like pathogenesis in mice. Acta Neuropathol. Commun. 2021, 9, 74. [Google Scholar] [CrossRef]
- Shin, M.-K.; Vázquez-Rosa, E.; Koh, Y.; Dhar, M.; Chaubey, K.; Cintrón-Pérez, C.J.; Barker, S.; Miller, E.; Franke, K.; Noterman, M.F.; et al. Reducing acetylated tau is neuroprotective in brain injury. Cell 2021, 184, 2715–2732.e23. [Google Scholar] [CrossRef] [PubMed]
- Albayram, O.; Kondo, A.; Mannix, R.; Smith, C.; Tsai, C.-Y.; Colin, S.; Herbert, M.K.; Qiu, J.; Monuteaux, M.; Driver, J.; et al. Cis P-tau is induced in clinical and preclinical brain injury and contributes to post-injury sequelae. Nat. Commun. 2017, 8, 1000. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.-L.; Hu, W.; Tung, Y.C.; Liu, F.; Gong, C.-X.; Iqbal, K. Tau passive immunization blocks seeding and spread of Alzheimer hyperphosphorylated Tau-induced pathology in 3 × Tg-AD mice. Alzheimer’s Res. Ther. 2018, 10, 13. [Google Scholar] [CrossRef]
- Braczynski, A.K.; Schulz, J.B.; Bach, J.-P. Vaccination strategies in tauopathies and synucleinopathies. J. Neurochem. 2017, 143, 467–488. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.P.; Kondo, A.; Albayram, O.; Herbert, M.K.; Liu, H.; Zhou, X.Z. Potential of the Antibody Against cis-Phosphorylated Tau in the Early Diagnosis, Treatment, and Prevention of Alzheimer Disease and Brain Injury. JAMA Neurol. 2016, 73, 1356–1362. [Google Scholar] [CrossRef]
- Mockett, B.G.; Richter, M.; Abraham, W.C.; Müller, U.C. Therapeutic Potential of Secreted Amyloid Precursor Protein APPsα. Front. Mol. Neurosci. 2017, 10, 30. [Google Scholar] [CrossRef]
- Corrigan, F.; Vink, R.; Blumbergs, P.C.; Masters, C.L.; Cappai, R.; Heuvel, C.V.D. Evaluation of the effects of treatment with sAPPα on functional and histological outcome following controlled cortical impact injury in mice. Neurosci. Lett. 2012, 515, 50–54. [Google Scholar] [CrossRef]
- Corrigan, F.; Vink, R.; Blumbergs, P.C.; Masters, C.L.; Cappai, R.; Heuvel, C.V.D. sAPPα rescues deficits in amyloid precursor protein knockout mice following focal traumatic brain injury. J. Neurochem. 2012, 122, 208–220. [Google Scholar] [CrossRef]
- Cohen, A.D.; Ikonomovic, M.D.; Abrahamson, E.E.; Paljug, W.R.; DeKosky, S.T.; Lefterov, I.M.; Koldamova, R.P.; Shao, L.; Debnath, M.L.; Mason, N.S.; et al. Anti-Amyloid Effects of Small Molecule Aβ-Binding Agents in PS1/APP Mice. Lett. Drug Des. Discov. 2009, 6, 437. [Google Scholar] [CrossRef]
- Celorrio, M.; Shumilov, K.; Payne, C.; Vadivelu, S.; Friess, S.H. Acute minocycline administration reduces brain injury and improves long-term functional outcomes after delayed hypoxemia following traumatic brain injury. Acta Neuropathol. Commun. 2022, 10, 10. [Google Scholar] [CrossRef]
- Whitney, K.; Nikulina, E.; Rahman, S.N.; Alexis, A.; Bergold, P.J. Delayed dosing of minocycline plus N-acetylcysteine reduces neurodegeneration in distal brain regions and restores spatial memory after experimental traumatic brain injury. Exp. Neurol. 2021, 345, 113816. [Google Scholar] [CrossRef]
- Bye, N.; Habgood, M.D.; Callaway, J.K.; Malakooti, N.; Potter, A.; Kossmann, T.; Morganti-Kossmann, M.C. Transient neuroprotection by minocycline following traumatic brain injury is associated with attenuated microglial activation but no changes in cell apoptosis or neutrophil infiltration. Exp. Neurol. 2007, 204, 220–233. [Google Scholar] [CrossRef]
- Robinson, B.D.; Isbell, C.L.; Melge, A.R.; Lomas, A.M.; Shaji, C.A.; Mohan, C.G.; Huang, J.H.; Tharakan, B. Doxycycline prevents blood–brain barrier dysfunction and microvascular hyperpermeability after traumatic brain injury. Sci. Rep. 2022, 12, 5415. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Wang, X.; Chen, X.; Fang, Y.; Chen, K.; Peng, W.; Wang, Z.; Guo, K.; Tan, X.; Liang, F.; et al. Hydroxychloroquine attenuates neuroinflammation following traumatic brain injury by regulating the TLR4/NF-κB signaling pathway. J. Neuroinflamm. 2022, 19, 71. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Li, R.; Cui, C.-M.; Gao, J.-L.; Sun, L.-Q.; Wang, Y.-C.; Wang, K.-J.; Tian, Y.-X.; Cui, J.-Z. Chloroquine exerts neuroprotection following traumatic brain injury via suppression of inflammation and neuronal autophagic death. Mol. Med. Rep. 2015, 12, 2323–2328. [Google Scholar] [CrossRef]
- Yonutas, H.M.; Sullivan, P.G. Targeting PPAR isoforms following CNS injury. Curr. Drug Targets 2013, 14, 733–742. [Google Scholar] [CrossRef]
- Strosznajder, A.K.; Wójtowicz, S.; Jeżyna, M.J.; Sun, G.Y.; Strosznajder, J.B. Recent Insights on the Role of PPAR-β/δ in Neuroinflammation and Neurodegeneration, and Its Potential Target for Therapy. Neuromol. Med. 2021, 23, 86–98. [Google Scholar] [CrossRef]
- Sauerbeck, A.; Gao, J.; Readnower, R.; Liu, M.; Pauly, J.R.; Bing, G.; Sullivan, P.G. Pioglitazone attenuates mitochondrial dysfunction, cognitive impairment, cortical tissue loss, and inflammation following traumatic brain injury. Exp. Neurol. 2011, 227, 128–135. [Google Scholar] [CrossRef]
- Amenta, P.S.; Jallo, J.I.; Tuma, R.F.; Elliott, M.B. A cannabinoid type 2 receptor agonist attenuates blood-brain barrier damage and neurodegeneration in a murine model of traumatic brain injury. J. Neurosci. Res. 2012, 90, 2293–2305. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Zhong, W.; Gu, Y.; Li, Y. Emerging Mechanisms and Targeted Therapy of Pyroptosis in Central Nervous System Trauma. Front. Cell Dev. Biol. 2022, 10, 832114. [Google Scholar] [CrossRef]
- Irrera, N.; Russo, M.; Pallio, G.; Bitto, A.; Mannino, F.; Minutoli, L.; Altavilla, D.; Squadrito, F. The Role of NLRP3 Inflammasome in the Pathogenesis of Traumatic Brain Injury. Int. J. Mol. Sci. 2020, 21, 6204. [Google Scholar] [CrossRef]
- Ismael, S.; Nasoohi, S.; Ishrat, T. MCC950, the Selective Inhibitor of Nucleotide Oligomerization Domain-Like Receptor Protein-3 Inflammasome, Protects Mice against Traumatic Brain Injury. J. Neurotrauma 2018, 35, 1294–1303. [Google Scholar] [CrossRef]
- Barrett, J.P.; Henry, R.; Shirey, K.A.; Doran, S.J.; Makarevich, O.D.; Ritzel, R.; Meadows, V.A.; Vogel, S.N.; Faden, A.I.; Stoica, B.A.; et al. Interferon-β Plays a Detrimental Role in Experimental Traumatic Brain Injury by Enhancing Neuroinflammation That Drives Chronic Neurodegeneration. J. Neurosci. 2020, 40, 2357–2370. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zheng, J.; Xu, S.; Fang, Y.; Wu, Y.; Zeng, J.; Shao, A.; Shi, L.; Lu, J.; Mei, S.; et al. Mer regulates microglial/macrophage M1/M2 polarization and alleviates neuroinflammation following traumatic brain injury. J. Neuroinflamm. 2021, 18, 2. [Google Scholar] [CrossRef] [PubMed]
- Nathalie, M.; Polineni, S.P.; Chin, C.N.; Fawcett, D.; Clervius, H.; Maria, Q.S.; Legnay, F.; Rego, L.; Mahavadi, A.K.; Jermakowicz, W.J.; et al. Targeting Microglial Polarization to Improve TBI Outcomes. CNS Neurol. Disord. Drug Targets 2021, 20, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Chen, C.; Fan, S.; Wu, S.; Yang, F.; Fang, Z.; Fu, H.; Li, Y. Omega-3 polyunsaturated fatty acid attenuates the inflammatory response by modulating microglia polarization through SIRT1-mediated deacetylation of the HMGB1/NF-κB pathway following experimental traumatic brain injury. J. Neuroinflamm. 2018, 15, 116. [Google Scholar] [CrossRef]
- Chio, C.-C.; Lin, M.-T.; Chang, C.-P. Microglial activation as a compelling target for treating acute traumatic brain injury. Curr. Med. Chem. 2015, 22, 759–770. [Google Scholar] [CrossRef] [PubMed]

| Neurotrauma-Related Disease | Key Behavioral Features | References |
|---|---|---|
| Chronic traumatic encephalopathy | Paranoia, mood swings, apathy, impulsivity, depression, and suicidality | [70,71,72] |
| Unclassified dementia | Anxiety, apathy, and possibly agitation/disinhibition | [73,74,75,76] |
| Parkinson’s disease | Motivational decline and slowed thinking | [3,77,78] |
| Alzheimer’s disease | Depression, cognitive impairment, memory loss | [79,80] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dodd, W.S.; Panther, E.J.; Pierre, K.; Hernandez, J.S.; Patel, D.; Lucke-Wold, B. Traumatic Brain Injury and Secondary Neurodegenerative Disease. Trauma Care 2022, 2, 510-522. https://doi.org/10.3390/traumacare2040042
Dodd WS, Panther EJ, Pierre K, Hernandez JS, Patel D, Lucke-Wold B. Traumatic Brain Injury and Secondary Neurodegenerative Disease. Trauma Care. 2022; 2(4):510-522. https://doi.org/10.3390/traumacare2040042
Chicago/Turabian StyleDodd, William S., Eric J. Panther, Kevin Pierre, Jairo S. Hernandez, Devan Patel, and Brandon Lucke-Wold. 2022. "Traumatic Brain Injury and Secondary Neurodegenerative Disease" Trauma Care 2, no. 4: 510-522. https://doi.org/10.3390/traumacare2040042
APA StyleDodd, W. S., Panther, E. J., Pierre, K., Hernandez, J. S., Patel, D., & Lucke-Wold, B. (2022). Traumatic Brain Injury and Secondary Neurodegenerative Disease. Trauma Care, 2(4), 510-522. https://doi.org/10.3390/traumacare2040042